



-
It is thought that intrauterine environment may impact the conceptus (i.e., the embryo/fetus and associated extra-embryonic membranes) during 'critical periods' of rapid cell division, thereby altering the expression of the fetal genome. Increasing evidence[1-2]suggests that complex interactions between genes and the environment play a major role in many common human diseases, such as diabetes, through the involvement of epigenetic factors, which occur without alterations in the DNA sequence.
DNA methylation at promoter CpG islands has been associated with gene repression and is a well-studied epigenetic marker in the context of tumor suppressor genes and cancer[3]. A recent study has shown that intrauterine growth retardation can lead to T2D due to epigenetic silencing of Pdx1, a key transcription factor that regulates INS expression and beta cell differentiation. Another study[4] compared rats that were subjected to intrauterine growth restriction (IUGR) to normal rats at 7 weeks of age-prior to the onset of diabetes; this study revealed changes in DNA methylation at a number of novel loci that were not limited to canonical CpG islands or promoters. However, the role of these modifications is unknown. Some research[5-6] has indicated that low birth weight is an independent risk factor for developing type 2 diabetes. Therefore, can epigenetic changes be linked to differences in birth weight? Thus, this study aims to explore the global DNA methylation analysis of infants of different birth weights.
This study was approved by the Ethics Committee of the Peking Union Medical College Hospital (S-002). Written informed consent was obtained from all subjects prior to participation.
Cord blood samples and placenta samples were obtained from women attending the Maternity Unit of the Peking Union Medical College Hospital between July 2009 and January 2010. Demographic data regarding maternal medical history, age at delivery, parity, body mass index pre-pregnancy, pregnancy complications, newborn gender, gestational age, birth weight, and body length were recorded. Inclusion criteria include single fetus full-term newborns, maternal health, and no smoking or drinking history. Birth weight was used to identify cases and controls: low birth weight (LBW, birth weight < 3, 000 g), normal birth weight (NBW, birth weight ≥ 3, 000 g and < 4, 000 g), and high birth weight (HBW, birth weight ≥ 4, 000 g). We consider NBW as control group, and LBW, HBW as cases. Gestational age at delivery, gender, mother's age and pre-pregnancy BMI were matched, resulting in the inclusion of 9 cases in the study. The demographic characteristics of the 9 subjects are provided in Table 1.
No. Gender Gestational
Age(W)Birth
Weight(g)Length
(cm)Head
Circum-ference(cm)Placental
Weight(g)L1 M 37 2, 210 45 29 490 L2 F 37 2, 380 46 32 545 L3 M 39+1 2, 520 47 33.6 500 N1 M 38+6 3, 250 49 33.5 540 N2 M 38+6 3, 250 51 34 660 N3 F 39+5 3, 260 49 35 655 H1 M 41+5 4, 410 51 35 850 H2 M 38 4, 510 52 34.8 955 H3 F 40+3 4, 775 52 32.5 1, 150 P(Lvs.N) 0.0006 0.0074 0.0428 P(Hvs.N) 0.00007 0.00079 0.0088 Note.L: low birth weight infant, N: normal birth weight infant, H: high birth weight infant, M: male, F: female. Table 1. Demographic Characteristics of the Infants
DNA was extracted from cord blood samples and placenta samples. DNA samples from the cases and controls were interrogated using the Illumina Infinium® Human Methylation27 BeadChip. Then We used the Methylation Module of Illumina Genome Studio software for differential methylation analysis. The differentially methylated gene sets were used to identify enriched KEGG pathways with the functional enrichment analysis software package Web-based Gene Set Analysis Toolkit (WebGestalt). Glucose metabolism is a complex process that involves many metabolic pathways, such as lipid metabolism, fatty acid metabolism, glycerolipid metabolism, carbohydrate metabolism, glycolysis/ gluconeogenesis, mTOR signaling, and adipocytokine signaling. This study focused on the methylation level of the genes involved in these pathways. To understand the interactions and dependencies among the critical pathways, we generated a network model based on the relationships in the KEGG database.
Characteristics of the subjects are summarized in Table 1. Nine healthy full-term infants were enrolled. All infants were divided into three groups according to birth weight: low birth weight (LBW, birth weight < 3, 000 g), normal birth weight (NBW, birth weight ≥ 3, 000 g and < 4, 000 g), and high birth weight (HBW, birth weight ≥ 4, 000 g). Every group contained 2 (66.7%) male infants and 1 (33.3%) female infant. Overall, the gestational age was consistent between the groups. However, significant differences in length, and placental weight were observed among the groups.
Overall, 6, 428 (23.3%) differentially methylated regions (DMRs) were found in the cord blood of the HBW group; while 7, 791 (28.3%), 3, 753 (13.6%), and 2, 668 (9.67%) DMRs were identified in the placental DNA of the HBW group, the cord blood of the LBW group, and the placental DNA of the LBW group, respectively. Most of the differentially methylated regions were hypermethylated and were located in CpG islands (Supplement Table 1, available in www.besjournal.com). The DMRs were located in 5, 430 (37.2%), 6, 328 (43.3%), 3, 321 (22.7%), and 2, 459 (16.8%) genes of the cord blood of the HBW group, the placental DNA of the HBW group, the cord blood of the LBW group, and the placental DNA of the LBW group, respectively (Supplement Table 1, available in www.besjournal.com).
Item HBW LBW Cord blood Placenta Cord blood Placenta DMRs 6, 428 7, 791 3, 753 2, 668 The number of located genes 5, 430 6, 328 3, 321 2, 459 Percent of total probes 23.3% 28.3% 13.6% 9.67% Up-regulation in methylation 5, 604 6, 404 3, 216 1, 894 CpG island 4, 707 6, 219 2, 092 1, 842 Non-CpG i 897 185 1, 124 52 Down-regulation in methylation 824 1, 387 537 774 CpG island 398 653 331 459 Non-CpG i 426 734 206 315 Note.DMRs, differentially methylated regions. Table Supplement Table 1. The probe Numbers of the Methylation Change in HBW Group and LBW Group
According to KEGG analysis, differentially methylated genes in HBW and LBW groups were enriched in hundreds of pathways compared to NBW group. We identified more than ten pathways related to glucose and lipid metabolism (Table 2).
Pathway LBW HBW Cord Blood Placenta Cord Blood Placenta Hyper Hypo Hyper Hypo Hyper Hypo Hyper Hypo Metabolism pathway 167 25 129 39 314 37 350 72 Lipidmetabolism Fatty acid metabolism 0 0 0 0 10 0 18 0 Glycerolipid metabolism 0 0 0 0 14 0 16 0 Carbohydrate metabolism Glycolysis/Gluconeogenesis 13 0 0 0 17 0 20 0 MAPK signaling pathway 44 9 37 16 87 13 105 25 Insulin signaling pathway 31 6 20 7 53 9 51 14 Type 2 DM pathway 10 0 7 0 16 0 14 7 PPAR signaling pathway 11 0 9 0 16 5 22 0 MODY 0 0 0 0 9 0 8 0 mTOR signaling pathway 10 0 0 0 16 5 15 0 Oxidation Phosphorylation pathway 18 0 19 0 32 0 41 0 TCA cycle 0 0 0 0 8 0 13 0 Adipocytokine signaling pathway 0 0 0 0 23 0 23 7 Table 2. Differentially methylation genes numbers in the pathways in HBW and LBW groups
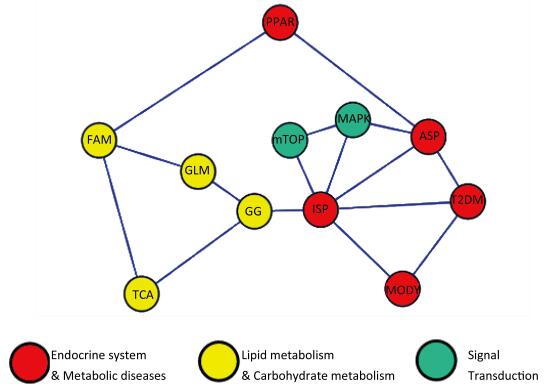
A network model demonstrating the relationships among the pathways was drawn using CytoScape software. The oxidation phosphorylation pathway was excluded from the network model. Meanwhile, we found that the insulin signaling pathway (ISP), adipocytokine signaling pathway (ASP) and MAPK signaling pathways were located in the core of network (Figure 1).
Figure 1. Pathway network model. ASP: Adipocytokine signaling pathway, GG: Glycolysis/Gluconeogenesis, GLM: Glycerolipid metabolism, FAM: Fatty acid metabolism, MAPK: MAPK signaling pathway, MODY: Maturity onset diabetes of the young, mTOR: mTOR signaling pathway, PPAR: PPAR signaling pathway, ISP: Insulin signaling pathway, TCA: TCA cycle
Intrauterine life playing an important role in determining a child's susceptibility to obesity and type 2 diabetes. However, the molecular mechanisms linking fetal life and the long-term increased risk of obesity and type 2 diabetes are poorly understood. Epigenetic changes such as DNA methylation could be one of the molecular mechanisms involved. This study investigated the methylation state of approximately 14, 000 CGIs in the promoter regions of genes spread throughout the genome of different birth weight groups. Many findings demonstrate the central importance of AKT signaling to insulin sensitivity. In our study, the AKT locus is hypermethylated in the cord blood of the high birth weight group. Then, we spectulated that the expression of AKT may be inhibited in high birth weight infants in our study.
Activated Akt is required for insulin-stimulated glucose transport via several mechanisms. In this study, we found that the PI3K/AKT locus was noticeably hypomethylated in the high birth weight group. We therefore speculated that this signal is inhibited, resulting in decreased glucose transport, increased hepatic gluconeogenesis, increased glycogen synthesis, and increased lipolysis. Thus, epigenetic regulation of the insulin signaling pathway could influence glucose metabolism in high birth weight infants.
Activation of p38 MAPK pathways results in fatty acid oxidation and glucose uptake in skeletal muscle, and inhibition of gluconeogenesis in liver[7]. P38 MAPK plays a stimulatory role in hepatic gluconeogenesis and may contribute to the unrestrained hepatic gluconeogenesis in both type 1 and type 2 diabetes[8-9]. One study revealed that epigenetic changes in p38 MAPK are involved in the development of diabetic complications[10]. In the current study, genes encoding p38 MAPK signaling components are hypermethylated in the experimental groups. Thus, in the HBW and LBW groups, the p38 MAPK pathway and hepatic gluconeogenesis may be inhibited, potentially altering glucose metabolism.
Leptin is involved in glucose metabolism. In this study, the LEP CpG island is generally unmethylated in placental tissue in the HBW group, confirming our speculation that the level of leptin in the cord blood of the high birth weight group is higher than that of the control group (unpublished result). Higher DNA methylation levels were observed at the LEPR promoter in the placenta and cord blood of the high birth weight group, indicating that LEPR expression may be inhibited. Thus, the LEP pathway is inhibited in the high birth weight group. Our study reveals that glucose metabolic genes are epigenetically regulated in infants of different birth weights. However, the number of infants involved in this study was limited, and further experiments describing the expression of the genes should be performed.
HTML
-
We thank all the participants for their participation and contribution.
 Quick Links
Quick Links
 DownLoad:
DownLoad: