



-
Blood flow infection is a common clinical emergency that poses a significant threat to patient safety, with a notable high mortality rate[1]. Staphylococcus aureus (SA), Pseudomonas aeruginosa (PA), and Acinetobacter baumannii (AB) account for a substantial proportion of bloodstream infections (BSI) caused by bacteria and are common causes of death[2-4]. In recent years, owing to the application of various types of antibiotics, immunosuppressive drugs, invasive surgery, and other treatment methods, the number of drug-resistant bacteria and infection opportunities has significantly increased, leading to an upward trend in the incidence of BSI[5-6]. Blood culture remains the gold standard for separating and detecting BSI in clinical practice; however, its extended detection cycle and low positivity rate hinder its ability to fulfill the requirements for early rapid diagnosis and treatment of BSI in clinical practice[7]. In the early stages of infection, treatment is often limited to broad-spectrum antibiotics, which can exacerbate the development of multiple bacterial resistance and increase the cost of treatment, thereby complicating the therapeutic process[8]. At present, the different molecular biology techniques used for diagnosing BSI include quantitative polymerase chain reaction (qPCR), multiple qPCR, and isothermal amplification[9-12]. Although these methods have reduced the diagnostic time to a certain extent, they exhibit inadequate sensitivity and are unable to swiftly identify samples with low bacterial concentrations (< 10 CFU/mL) in the patient’s blood[13]. Previously, we reported a recombinase-aided polymerase chain reaction (PCR) (RAP) assay combined with recombinase-aided amplification (RAA) with qPCR and a multiple recombinase-aided PCR (M-RAP) assay that were used for the rapid and highly sensitive detection of multiple respiratory viruses[14-15]. Human mannan-binding lectin protein (M1 protein) has a strong ability to bind to various pathogens in vitro[16]. Moreover, RAP coupled with enrichment using M1 protein-conjugated magnetic beads (M1 beads) is a rapid and highly sensitive assay for detecting Epstein-Barr virus (EBV) in the blood[17]. In this study, M-RAP combined with the M1 bead enrichment method was developed to detect SA, PA, and AB in patients with BSI and compared with qPCR to evaluate its clinical performance.
-
Standard strains of SA (ATCC 29213), PA (ATCC 27853), AB (ATCC 19606), and M1 proteins were provided by the Institute of Infectious Diseases of the Chinese Center for Disease Control and Prevention. Other samples of clinical strains were provided by the Microbiology Department of Hebei Provincial People’s Hospital. Plasma samples from 30 patients with BSI and plasma samples from 30 patients with negative blood cultures were all provided by the Laboratory of Hebei Provincial People’s Hospital, including 10 samples of SA, 10 samples of PA, and 10 samples of AB. These blood culture-positive specimens were examined by mass spectrometry in the Hebei Provincial People’s Hospital. The study was performed in accordance with national ethics regulations and approved by the institutional review board of Hebei Provincial People’s Hospital. After obtaining informed consent, whole blood samples were collected from healthy volunteers and placed in heparin anticoagulant tubes. After centrifugation, plasma was obtained and stored in a −80 °C refrigerator for future use. The nucleic acids of the strains were extracted from the genomic deoxyribonucleic acid (DNA) following the guidelines outlined in the FastPure® Microbiome DNA Isolation Kit instruction manual.
-
A pair of outer primers was used for the RAA reaction, while a pair of inner primers was used for qPCR. Data on the staphylococcal protein A (SpA) gene of SA, gyrase B (GyrB) gene of PA, and outer membrane protein A (OmpA) gene of AB were downloaded from the National Center for Biotechnology Information. These genes were located in highly conserved regions that could meet species-level identification requirements[18-20]. The qPCR primer-probe set was selected based on the information from the literature[21-23]. Sequence alignment was performed using the Vector NTI 11.5.1. The length of the RAA primers was generally 30–35 bp. An excessively short primer may impede the formation of a nucleic acid-protein complex between the recombinant enzyme and primer. Alternatively, an overly long primer may facilitate the formation of a stable primer dimer structure[24]. Following the design principle of the RAA primer, AmplifX and Oligo7 software were used to analyze and evaluate the GC content, primer dimers, hairpin structures, tertiary structures, and other indicators. The sequence information is presented in Table 1. All primers and probes were synthesized by Sangon Biotech (Shanghai, China). Fragments (300 bp, 425 bp, and 276 bp) from the SpA, GyrB, and OmpA genes of SA, PA, and AB, respectively, that were cloned into the pUC57 vector were provided by TsingKe Biotech Corp. (Beijing, China). The recombinant plasmid DNA was quantified using the Qubit® dsDNA HS Assay Kit (Thermo Fisher Scientific, MA, United States) and Qubit2.0 fluorescence quantitative meter (Life Technologies, United States). The plasmid copy numbers were calculated using the following formula: plasmid copy number (copy number/μL) = [6.02 × 1023 × plasmid concentration (ng/μL) × 10-9] / [plasmid length × 660][25]. Finally, the standard plasmids were prepared in 1 × TE buffer in a 10-fold concentration gradient, and the concentration used ranged from 1 to 105 copies/μL. All plasmids were kept at a temperature of –20 °C until use.
Region Primer/probe Sequence (5′–3′) Source SpA SA-RAA-F
SA-RAA-R
SA-qPCR-F
SA-qPCR-R
SA-qPCR-PaACCTGGTAAAGAAGATGGTAACGGAGTACA
CTGCACCTAAGGCTAATGATAATCCACCAA
TACATGTCGTTAAACCTGGTG
TACAGTTGTACCGATGAATGG
FAM-CAAACGGCACTACTGCTGACAAAATTGCTGCA-BHQ1This paper
This paper
[21]
[21]
[21]GyrB PA-RAA-F
PA-RAA-R
PA-qPCR-F
PA-qPCR-R
PA-qPCR-PaAAGGAAGAAGGGGTTTCTGCGGCGGAAGTG
CCGCCTTCGTACTTGAACAGCTCCTCCTTG
CCTGACCATCCGTCGCCACAAC
CGCAGCAGGATGCCGACGCC
VIC-CCGTGGTGGTAGACCTGTTCCCAGACC-BHQ1This paper
This paper
[22]
[22]
[22]OmpA AB-RAA-F
AB-RAA-R
AB-qPCR-F
AB-qPCR-R
AB-qPCR-PaGTACTTTAGGTAACGCTGGTGTTGGTGCTT
TGTTAGTATCAAAGAACACACGAAGTTCCA
TCTTGGTGGTCACTTGAAGC
ACTCTTGTGGTTGTGGAGCA
ROX-AAGTTGCTCCAGTTGAACCAACTCCA-BHQ2This paper
This paper
[23]
[23]
[23]Note. aProbe modifications: FAM, 6-carboxyfuorescein; VIC, 6-phosphoramidite; ROX, 5-carboxy-X-rhodamine; BHQ, black hole quencher; SA, Staphylococcus aureus; PA, Pseudomonas aeruginosa; AB, Acinetobacter baumannii; SpA, staphylococcal protein A; GyrB, gyrase B; OmpA, outer membrane protein A; RAA, recombinase-aided amplification; qPCR, quantitative polymerase chain reaction. Table 1. Primer and probe sequences used in this study
-
Based on previous studies conducted in our laboratory[26], the optimal volume ratio of RAA for qPCR was 1:4. Docosane (JSNEB, Hong Kong, China) solidified at room temperature and melted above 44 °C, creating an impermeable barrier that exhibited the best blocking effect when 30 μL was added. M-RAP was performed in two stages in a single tube: the first stage involved the use of an upper RAA system, while the second stage involved the use of a lower qPCR system separated by a thermally removable dodecane barrier in the middle. The probes for SA, PA, and AB were labeled with FAM, VIC, and ROX fluorophores, respectively, so that they could be detected simultaneously in the same RAP reaction system for blood-infected bacteria.
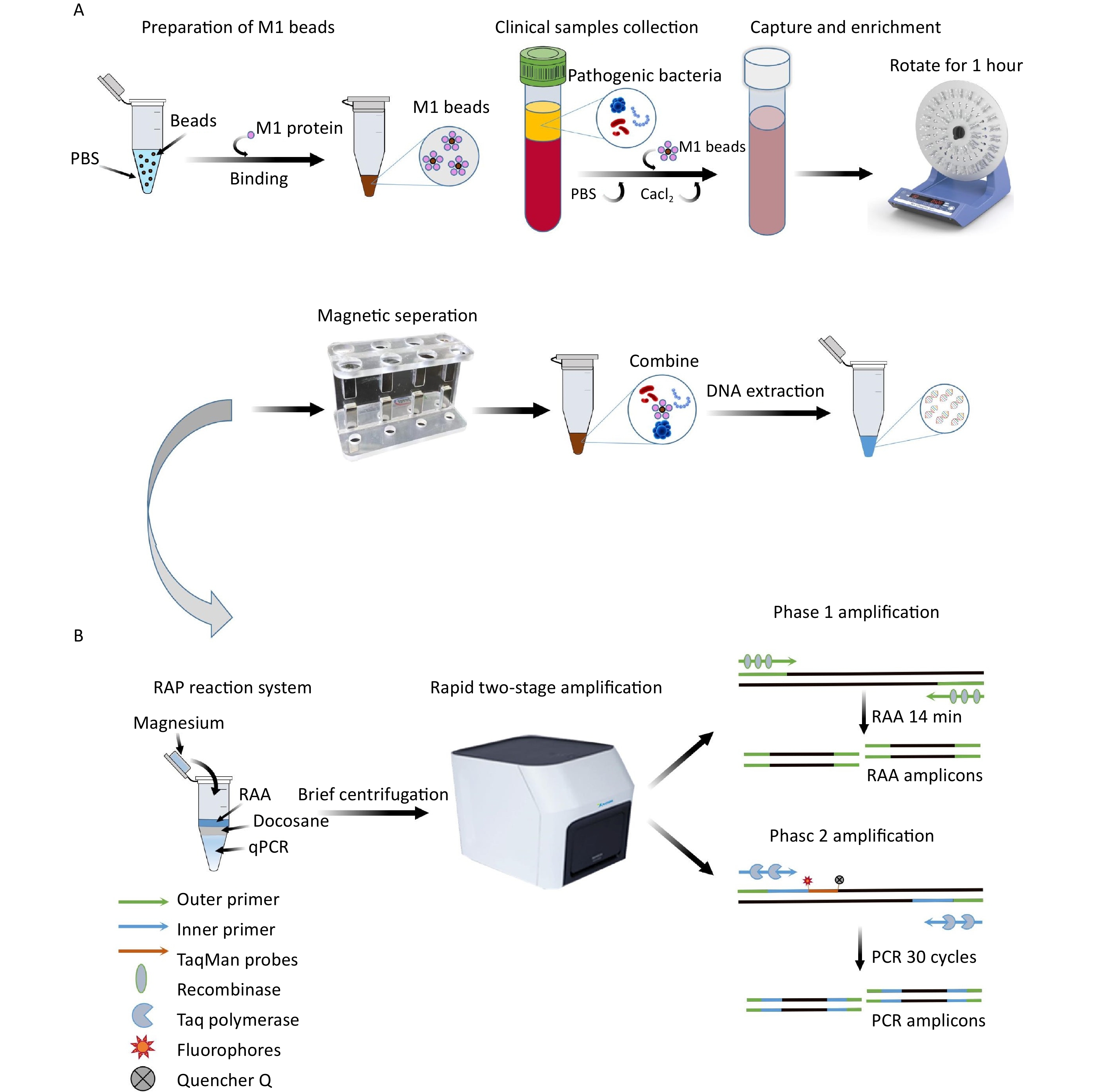
The total reaction volume of M-RAP was 50 μL, including 10 μL of RAA in the upper layer of the reaction system (Amp-Future, Changzhou, China). The RAA mixture consisted of 5.88 μL of buffer, 0.92 μL of DEPC water, 1 μL of magnesium acetate (140 mmol/L), 0.2 μL of RAA forward and reverse primers (10 μmol/L), and 1 μL of DNA template. The 40 μL of lower qPCR (Entrans qPCR Probe Set V2, ABclonal, Wuhan, China) comprised 12.5 μL of buffer, 20.7 μL of DEPC water, 0.5 μL of PCR forward and reverse primer (10 μmol/L), 0.5 μL of PCR probe (10 μmol/L), 0.6 μL of dNTP (10 mmol/L), 0.5 μL of Taq DNA polymerase, and 1.2 μL of MgCl2 (50 mmol/L). First, 40 μL of qPCR mixture was added to the reaction unit tube; then, 30 μL of docosane was added to solidify the mixture on the upper layer of the qPCR system after 1 min at 60 °C and 30 s at 4 °C. Then, 8 μL of RAA mixture and 1 μL of DNA template were added to the solid surface. Finally, 1 μL of magnesium acetate was added to the inner surface of the tube cover for instantaneous centrifugation. The M-RAP reaction conditions were 39 °C for 14 min and 95 °C for 10 min, followed by 30 cycles at 95 °C for 15 s and 60 °C for 1 min. Positive standard plasmids with concentration gradients of 1–105 copies/μL for SA, PA, and AB were used as templates to determine the reaction time (10, 12, 14, and 16 min) of the upper RAA; then, the second stage of qPCR amplification was performed to determine the optimal reaction time of RAA. The M-RAP detection principle is shown in Figure 1B.
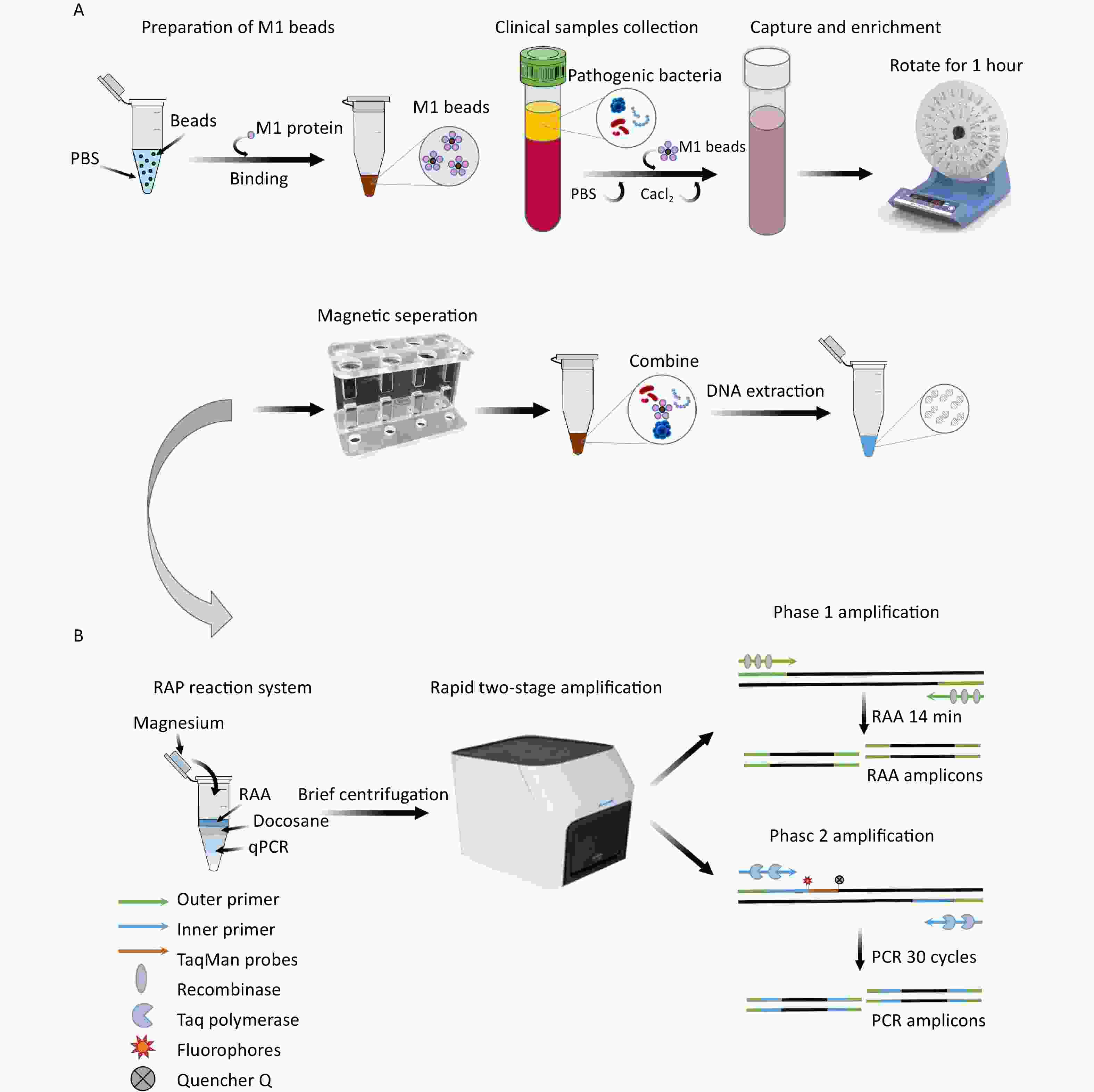
Figure 1. The enrichment process of M1 beads (A) and the detection principle of M-RAP (B). M1 protein, recombinant human mannan-binding lectin protein; M1 beads, M1 protein conjugated magnetic beads; RAP, recombinase-aided PCR; PCR, polymerase chain reaction.
-
To evaluate the sensitivity and repeatability of the M-RAP methods, recombinant plasmids with SA, PA, and AB concentrations of 1–105 copies/μL and the diluted nucleic acids of the standard strain with concentrations of 10−6–100 ng/μL were used to evaluate the sensitivity of the M-RAP method for BSI. DEPC water was used as the negative control for each test, and each concentration was tested eight times. In addition, other BSI-related pathogens were used to evaluate the specificity of M-RAP methods, including SA, PA, AB, Klebsiella pneumoniae, Escherichia coli, Staphylococcus epidermidis, Enterococcus faecium, Enterococcus faecalis, Enterobacter cloacae, Neisseria meningitidis, Pseudomonas maltophilia, Proteus mirabilis, Listeria monocytogenes, Candida albicans, Candida tropicalis, Candida parapsilosis, Candida glabrata, and Candida krusei.
-
To compare the detection sensitivity of M-RAP with that of qPCR alone, the recombinant plasmids SA, PA, and AB and the nucleic acid of the standard strain were used for qPCR detection. The reaction was performed based on the guidelines outlined in the Entrans qPCR Probe Set V2 user manual. The reaction procedure is described in Table 1. The SA amplification process was performed as follows: 95 °C 10 s, 60 °C 10 s, 70 °C 15 s, 40 cycles; PA amplification program: 95 °C 15 min, 95 °C 15 s, and 60 °C 1 min, in 40 cycles. Alternatively, the AB amplification was performed as follows: 95 °C 10 min, 95 °C 30 s, and 62 °C 1 min, in 40 cycles.
-
The SA, PA, and AB standard strains pre-frozen at −80 °C were inoculated in Columbia blood plate (Hopebiol, Qingdao, China), respectively. The plates were then placed in the CO2 incubator (Thermo Fisher Scientific, United States) and incubated at 37 °C for 24 h. Single colonies were selected and inoculated in Brian Heart Infusion culture medium (Hopebiol, Qingdao, China) and grown overnight at 37 °C in an incubator shaking at a speed of 220 rpm (JΜLABO, Germany). Subsequently, 1 mL of the bacterial solution was withdrawn and centrifuged. The supernatant was discarded, and 1 mL of PBS (GIBCO, United States) was added for resuspension. After two repetitions of this process, the OD600 values of the three bacterial fluids were adjusted to 0.45–0.5 after adding the PBS (concentration: 108 CFU/mL). Then, bacterial fluids of < 2, 4, 6, 8, 10, 50, 100, 200, 300, 500, and 1,000 CFU were added to 1 mL of sterilized human plasma to prepare the simulated plasma samples (final concentrations: < 2, 4, 6, 8, 10, 50, 100, 200, 300, 500, and 1,000 CFU/mL). The bacterial solution was mixed with 1 mL of PBS in a similar manner to prepare the simulated PBS samples for the calculation of the number of colonies on the plate. The actual number of colonies on the plate was counted after an overnight incubation.
To prepare the M1 beads, 1 mL of PBS was added to 1 mg of protein A magnetic beads (Biocanal, Wuxi, China). The mixture was separated in a magnetic rack for 3 minutes, and the supernatant was discarded. This process was repeated twice. The beads were then resuspended in 1 mL of PBS. Next, 232 µg of M1 protein was added, and the mixture was blended in a mixer for 30 minutes. Afterward, it was separated in a magnetic rack for 3 minutes, the supernatant was discarded, and the M1 beads were prepared by resuspending them with 100 μL PBS.
-
Simulated plasma samples (2, 4, 6, 8, and 10 CFU) were introduced to a 14-mL round-bottomed tube, and M1 beads (1 mg) were added. Subsequently, the system was replenished to 10 mL with PBS. A final concentration of 4 mmol/L Cacl2 (Yuanye, Shanghai, China) was added, and the mixture was thoroughly mixed for 1 h. After placing it on a magnetic rack to separate magnetically, the supernatant was discarded. The system was then washed three times using PBS to remove the plasma components and pathogenic bacteria that had not been adsorbed onto the M1 beads. Finally, the solution was resuspended in 400 μL of PBS. The PBS-simulated samples (2, 4, 6, 8, and 10 CFU) were also enriched using the same method; after overnight cultivation on the plate, the number of colonies was counted. A Nucleic Acid Extraction Kit (Vazyme Biotech, Nanjing, China) was used to extract DNA from both unenriched simulated plasma specimens and enriched M1 bead suspensions as templates for M-RAP and qPCR. The preparation and enrichment processes for the M1 beads are shown in Figure 1A.
-
After nucleic acid extraction from the simulated plasma samples with different concentrations of SA, PA, and AB bacteria before and after enrichment, M-RAP testing and qPCR testing were performed separately, and the results were compared. These experiments were performed in triplicate. For clinical sample testing, 30 heparin anticoagulant plasma samples were collected from patients with positive results on blood culture tests, including 10 with SA, 10 with PA, and 10 with AB, as well as 30 blood culture-negative samples. After enrichment with M1 beads, M-RAP and qPCR assays were performed. The consistency of the two detection methods was compared to evaluate the clinical performance of the M-RAP detection method.
-
All statistical analyses were performed using the IBM SPSS Statistics version 25 (IBM Corporation, Armonk, NY, USA). Kappa analysis was used to evaluate the consistency of these two methods, and a P value of < 0.05 was considered significant.
-
The upper RAA reaction time was adjusted from 10 to 16 min to determine the optimal reaction time for the first stage of M-RAP. The sensitivity of the RAA reaction gradually decreased until the optimal range for the reaction was achieved. The experimental results (Figure 2A–C) indicated that in the M-RAPs, the detection results were better when the first-stage RAA reaction times for SA, PA, and AB were set at 14, 14, and 16 min, respectively. Due to the comparable cycle threshold (Ct) values of AB at 14 and 16 min of RAA reaction, a 14-minute enrichment time was subsequently adopted.
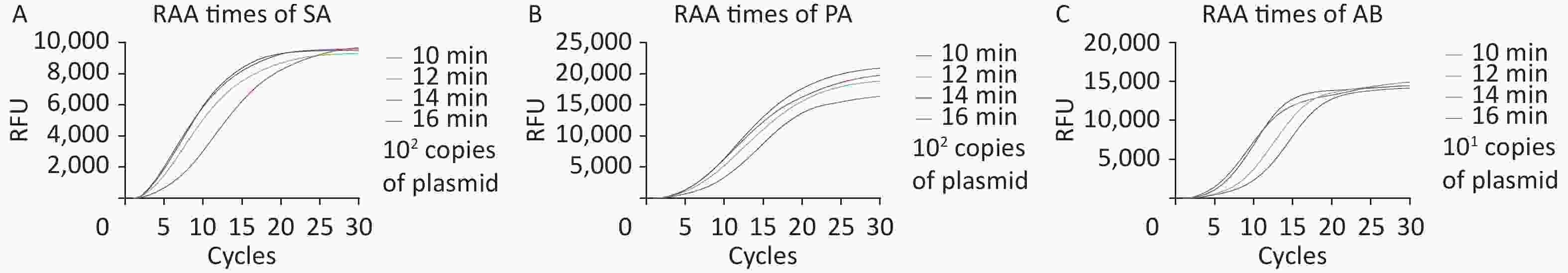
Figure 2. Relative fluorescence curves of the SA, PA, and AB RAP assays with different RAA incubation durations (10, 12, 14, and 16 min). RFU, relative fluorescence units; SA, Staphylococcus aureus; PA, Pseudomonas aeruginosa; AB, Acinetobacter baumannii; RAA, recombinase aided amplification.
-
Recombinant plasmids for SA, PA, and AB and diluted standard nucleic acids were used to evaluate the sensitivity of the M-RAP approach. The recombinant plasmid sequence was short and contained sequences that complemented the sequences of primers and probes tested. By contrast, the nucleic acids of the standard strains contained complete genetic information of the pathogens. Therefore, both recombinant plasmids and nucleic acids should be used to comprehensively evaluate the detection methods. In eight repeated experiments (
Supplementary Table S1 , available inwww.besjournal.com ), M-RAP detected 1, 10, and 1 copies/μL of SA, PA, and AB recombinant plasmids, respectively (Figure 3A–C). M-RAP successfully detected nucleic acid concentrations of 10−6, 10−5, and 10−6 ng/μL from SA, PA, and AB (Figure 3D–F). No amplification curve was observed in the negative control, and repeated tests showed consistent results.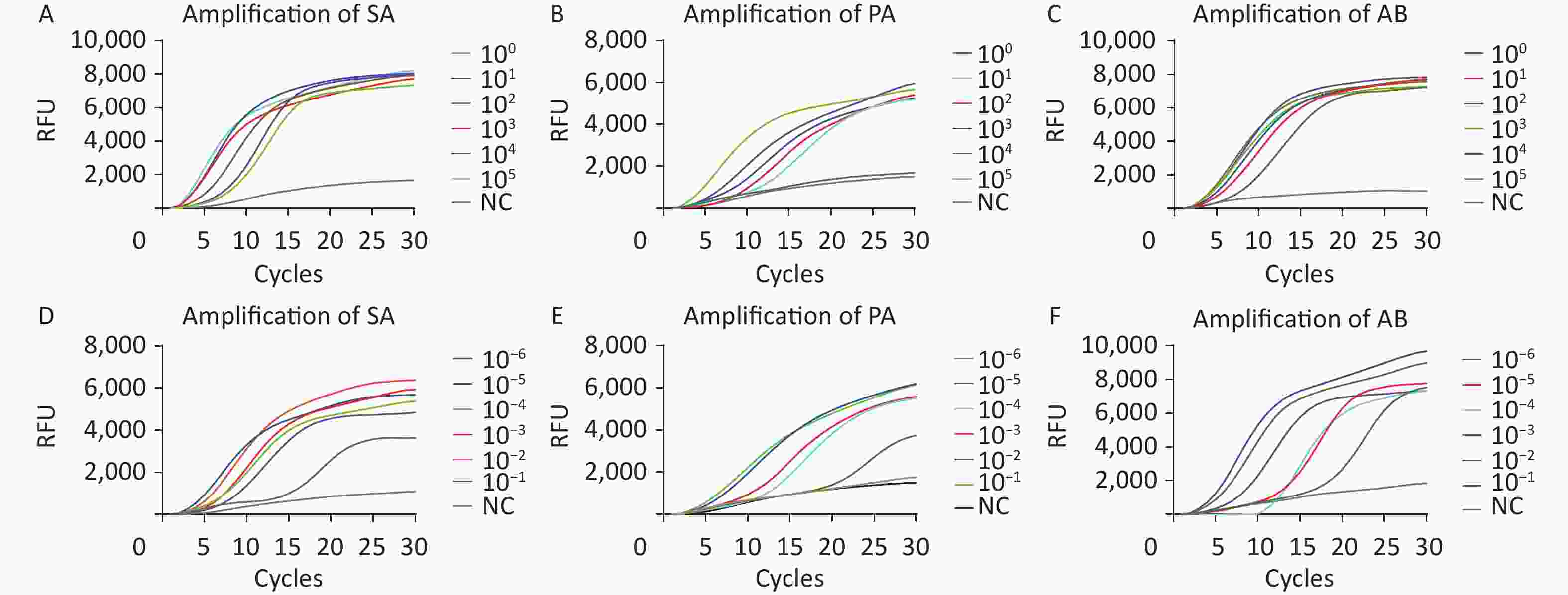
Figure 3. M-RAP sensitivity evaluation by copy number determination of the plasmids and nucleic acids of standard strains. The curves with different colors correspond to the amplification efficiency of the qPCR stage of M-RAP with different template concentrations (plasmids or international standard strain). (A–F) Sensitivity analysis of the SA, PA, and AB M-RAP assay results. Relative fluorescence curves of SA (A), PA (B), and AB (C) with different concentrations (1–105 copies/μL) using M-RAP. Relative fluorescence curves of SA (D), PA (E), and AB (F) with standard strain nucleic acids from 10−6 to 10−1 ng/μL using M-RAP. RFU, relative fluorescence units; NC, negative control; SA, Staphylococcus aureus; PA, Pseudomonas aeruginosa; AB, Acinetobacter baumannii.
-
The M-RAP assay specifically detected the fluorescence signals of SA, PA, and AB in the three channels and had no cross-reactivity with the other 18 common pathogens associated with BSI (Table 2).
Strains Origin M-RAP (SA) M-RAP (PA) M-RAP (AB) Staphylococcus aureus ATCC 29213 Pos (8/8a) Neg (0/8) Neg (0/8) Pseudomonas aeruginosa ATCC 27853 Neg (0/8) Pos (8/8) Neg (0/8) Acinetobacter baumannii ATCC 19606 Neg (0/8) Neg (0/8) Pos (8/8) Klebsiella pneumoniae ATCC 11296 Neg (0/8) Neg (0/8) Neg (0/8) Escherichia coli Isolated strains Neg (0/8) Neg (0/8) Neg (0/8) Staphylococcus epidermidis Isolated strains Neg (0/8) Neg (0/8) Neg (0/8) Enterococcus faecium Isolated strains Neg (0/8) Neg (0/8) Neg (0/8) Enterococcus faecalis Isolated strains Neg (0/8) Neg (0/8) Neg (0/8) Enterobacter cloacae Isolated strains Neg (0/8) Neg (0/8) Neg (0/8) Neisseria meningitidis Isolated strains Neg (0/8) Neg (0/8) Neg (0/8) Pseudomonas maltophilia Isolated strains Neg (0/8) Neg (0/8) Neg (0/8) Proteus mirabilis Isolated strains Neg (0/8) Neg (0/8) Neg (0/8) Listeria monocytogenes Isolated strains Neg (0/8) Neg (0/8) Neg (0/8) Candida albicans ATCC 753 Neg (0/8) Neg (0/8) Neg (0/8) Candida tropicalis ATCC 750 Neg (0/8) Neg (0/8) Neg (0/8) Candida parapsilosis ATCC 22019 Neg (0/8) Neg (0/8) Neg (0/8) Candida glabrata ATCC 2001 Neg (0/8) Neg (0/8) Neg (0/8) Candida krusei ATCC 6258 Neg (0/8) Neg (0/8) Neg (0/8) Note. aThe first number indicates the number of times a positive result was obtained. The second number indicates the number of times a negative result was obtained. Neg, negative; Pos, positive; SA, Staphylococcus aureus; PA, Pseudomonas aeruginosa; AB, Acinetobacter baumannii; M-RAP, multiple recombinase-aided PCR. Table 2. Bacterial strains used in the specificity test
-
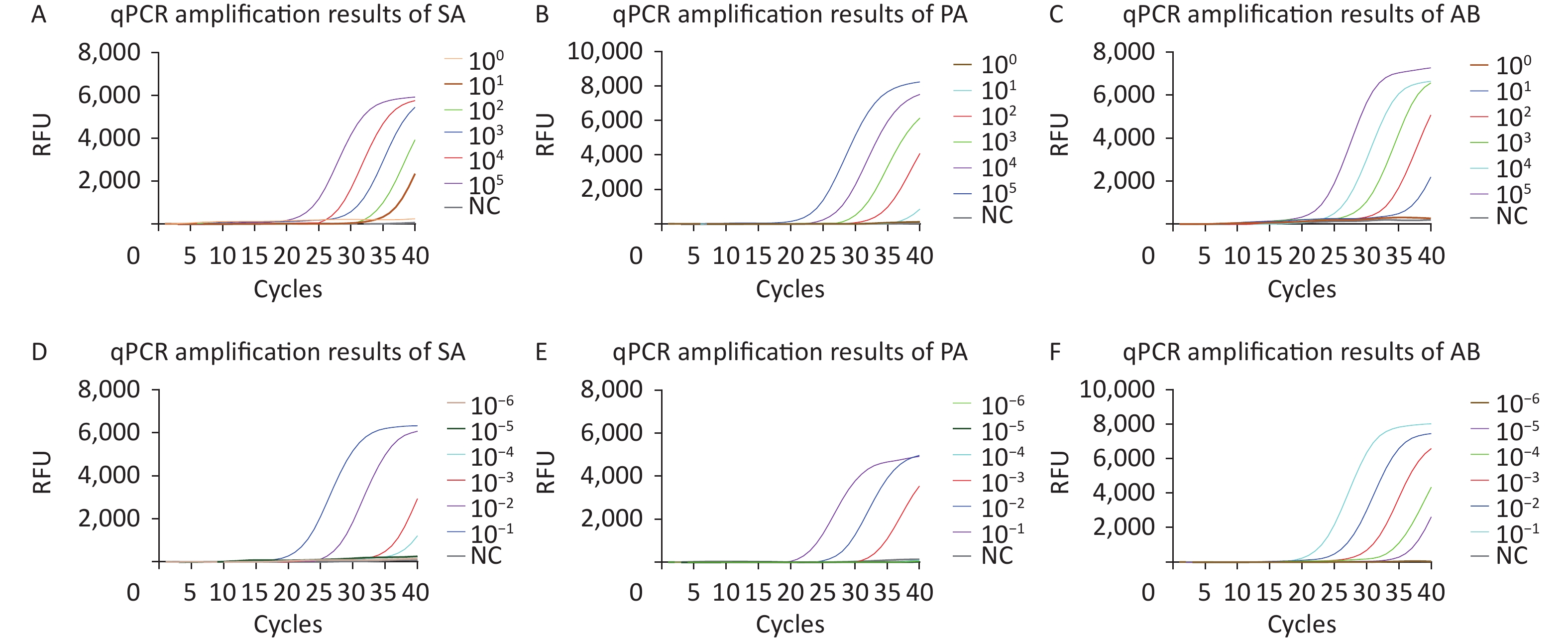
Recombinant plasmids for SA, PA, and AB as well as diluted standard strain nucleic acids were used to evaluate the sensitivity of the qPCR methods. In eight repeated experiments, qPCR successfully detected 10, 102, and 10 copies/μL of SA, PA, and AB recombinant plasmids, respectively (Figure 4A–C). qPCR successfully detected nucleic acid concentrations of 10−4, 10−3, and 10−5 ng/μL from SA, PA, and AB (Figure 4D–F). No amplification curve was observed in the negative control, and repeated tests showed consistent results.
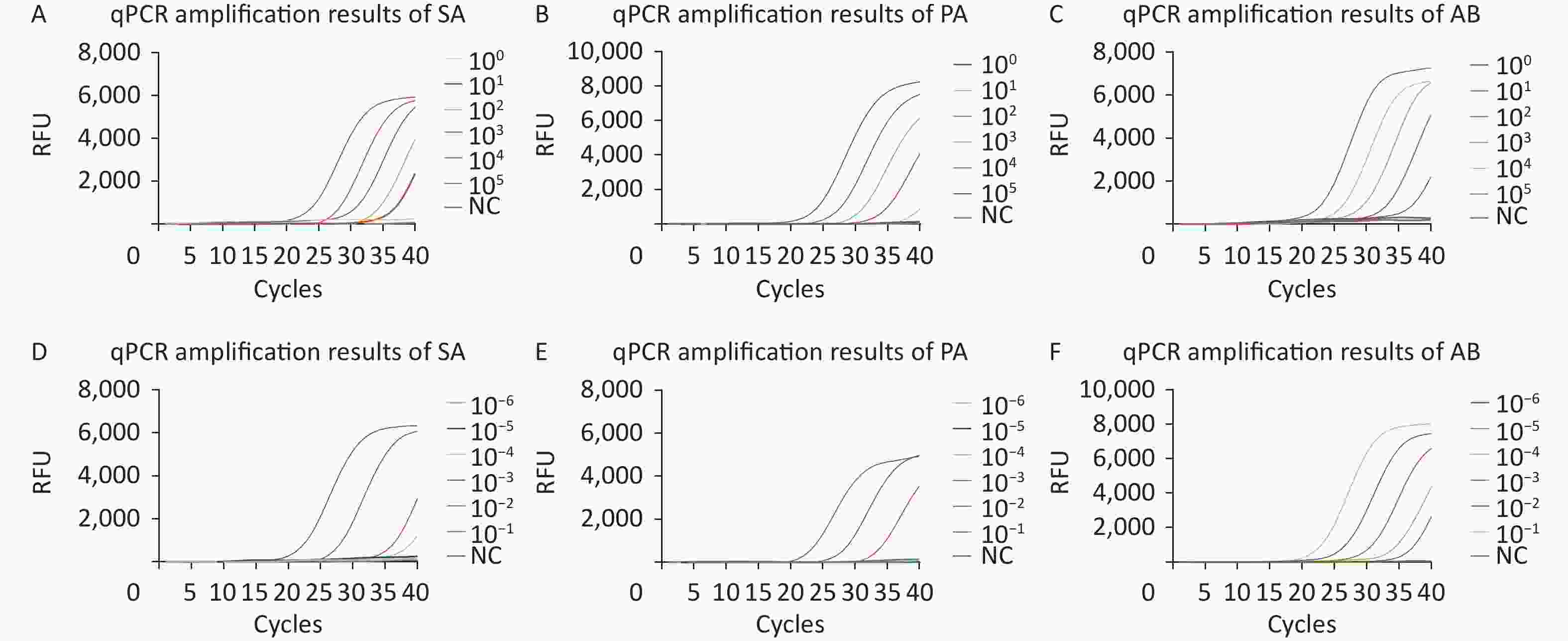
Figure 4. qPCR sensitivity evaluation by copy number determination of the plasmids and the standard strain nucleic acids. The curves with different colors correspond to the amplification efficiency at optimal reaction conditions (plasmids or standard strain). (A–F) Sensitivity analysis of the SA, PA, and AB qPCR assay results. Relative fluorescence curve of SA (A), PA (B), and AB (C) at different concentrations (1–105 copies/μL) using qPCR. Relative fluorescence curve of SA (D), PA (E), and AB (F) standard strain nucleic acid from 10−6 to 10−1 ng/μL using qPCR. RFU, relative fluorescence units; NC, negative control; SA, Staphylococcus aureus; PA, Pseudomonas aeruginosa; AB, Acinetobacter baumannii; qPCR, quantitative polymerase chain reaction.
-
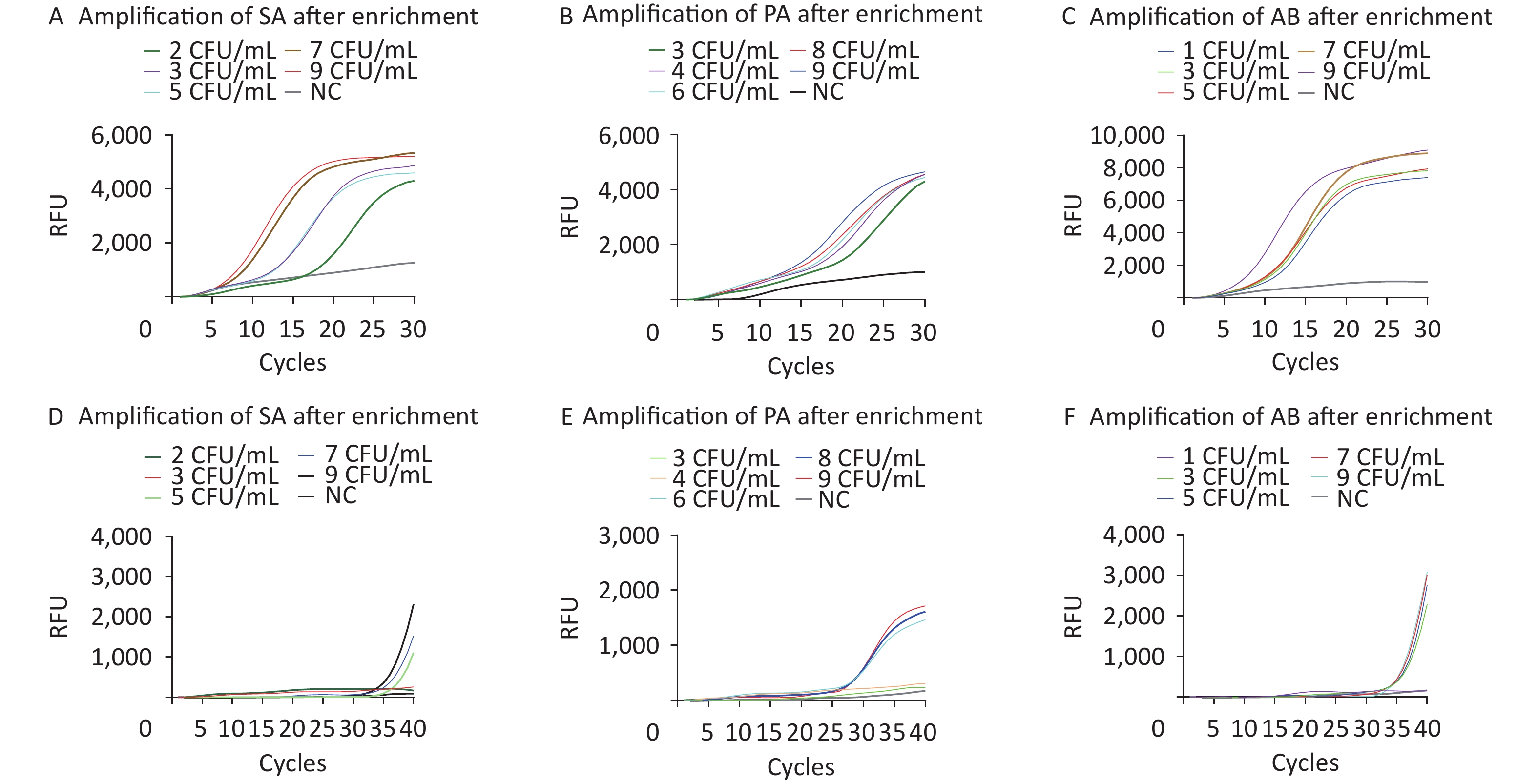
The limits of detection (LOD) of M-RAP for SA, PA, and AB simulated plasma samples were approximately 100, 300, and 50 CFU/mL, respectively, before enrichment with M1 beads (Figure 5A–C). The LOD of qPCR for SA-, PA-, and AB-simulated plasma samples were approximately 300, 500, and 200 CFU/mL, respectively, before enrichment with the M1 beads (Figure 5D–F). After enrichment with M1 beads, the LODs of M-RAP for SA, PA, and AB simulated plasma samples were determined to be 2, 3, and 1 CFU/mL (Figure 6A–C), respectively, corresponding to the number of spread plate colonies (
Supplementary Figure S1A –C , available inwww.besjournal.com ). Meanwhile, the LODs of qPCR for SA-, PA-, and AB-simulated plasma samples were 5, 6, and 3 CFU/mL (Figure 6D–F), corresponding to the number of spread plate colonies (Supplementary Figure S1A –C ).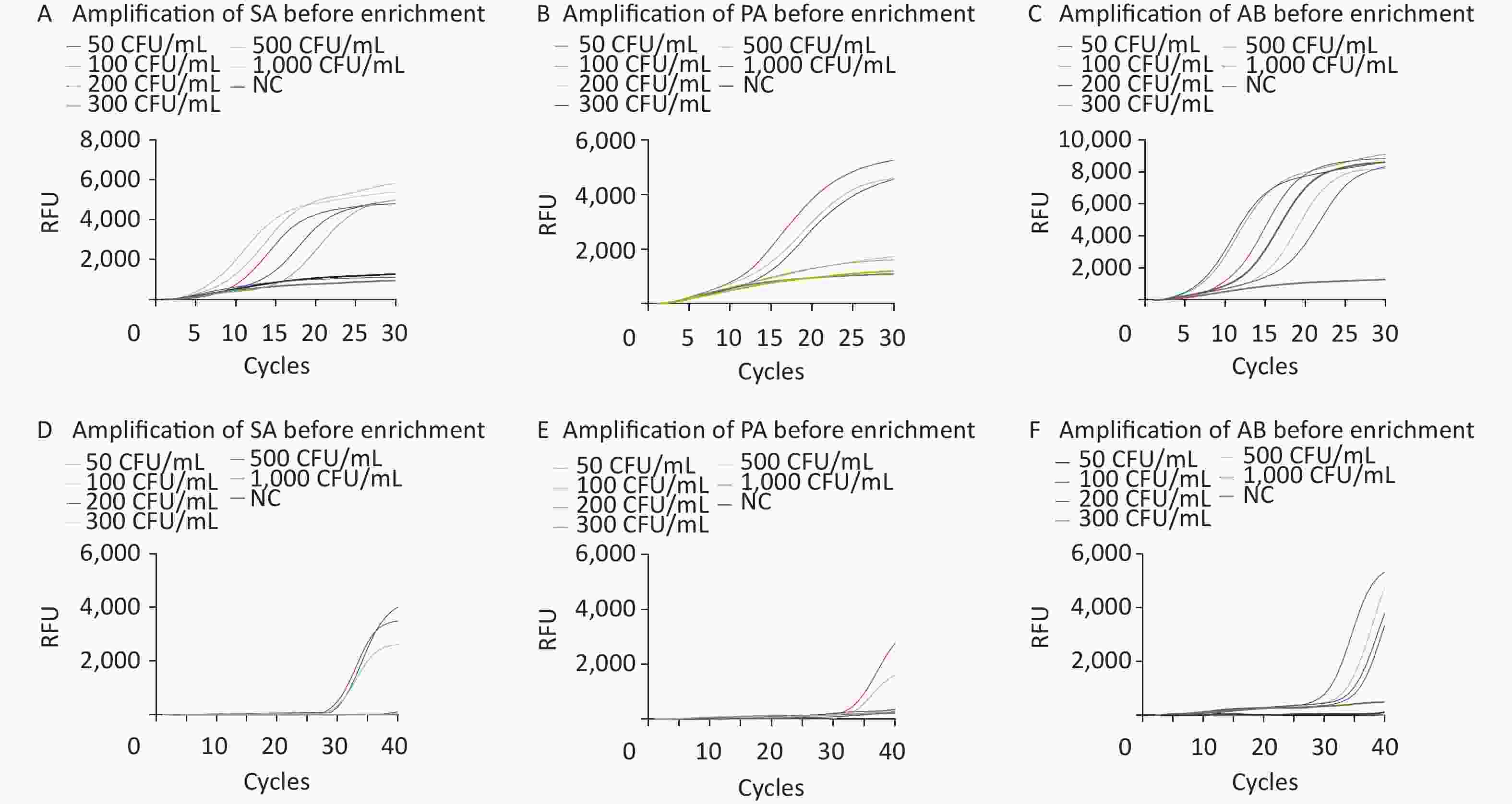
Figure 5. Evaluation of the LODs of simulated plasma samples prior to enrichment (A–F). The SA, PA, and AB M-RAP assays (A–C). The SA, PA, and AB qPCR assays (D–F). RFU, relative fluorescence units; NC, negative control; CFU, colony forming unit; SA, Staphylococcus aureus; PA, Pseudomonas aeruginosa; AB, Acinetobacter baumannii.
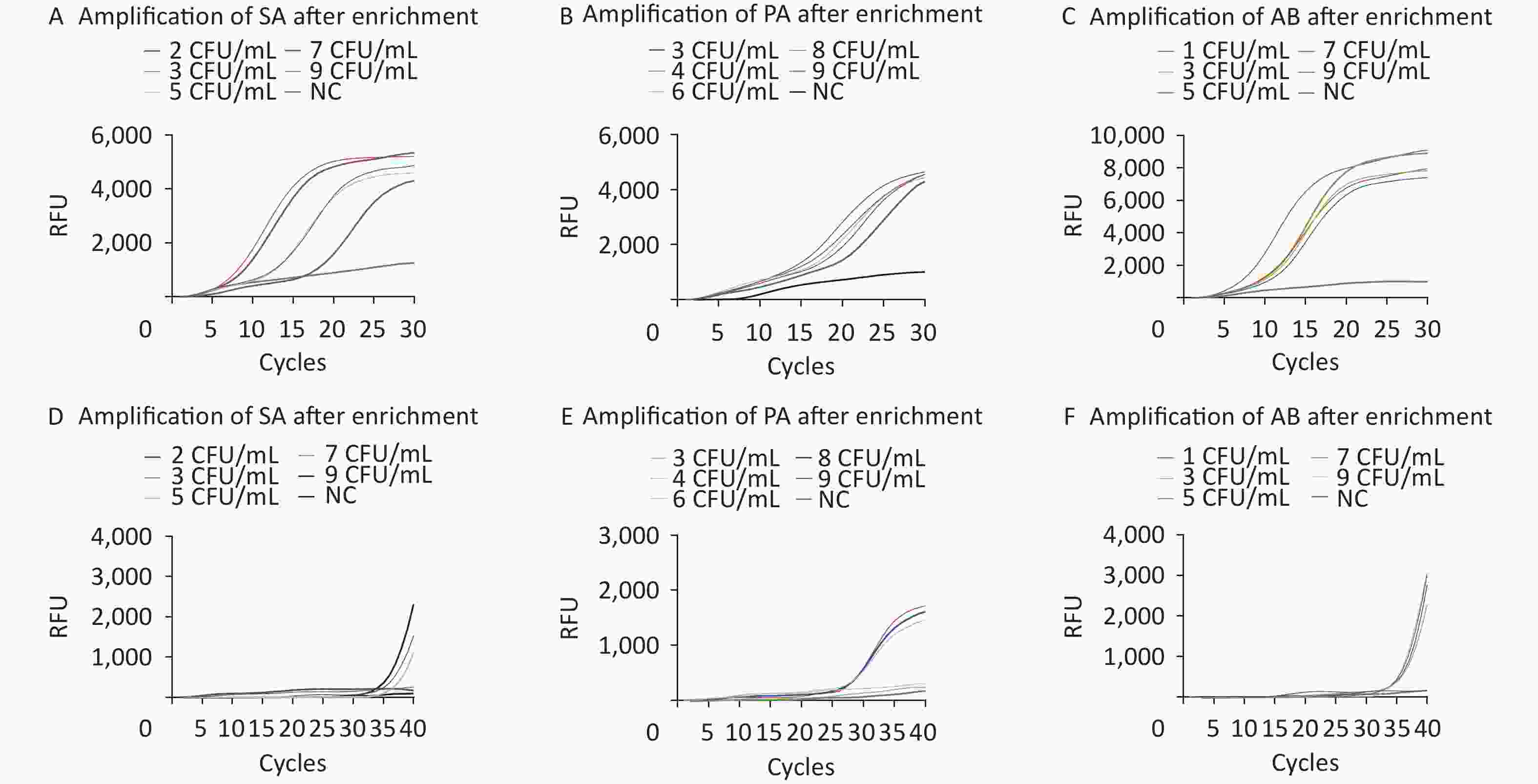
Figure 6. Evaluation of the LODs of the simulated plasma samples after enrichment (A–F). The SA, PA, and AB M-RAP assays (A–C). The SA, PA, and AB qPCR assays (D–F). RFU, relative fluorescence units; NC, negative control; CFU, colony forming unit; SA, Staphylococcus aureus; PA, Pseudomonas aeruginosa; AB, Acinetobacter baumannii.
-
Blood culture remains the gold standard for detecting BSI; however, factors such as insufficient blood collection, low number of pathogenic bacteria, incorrect timing of blood collection, use of antibiotics before blood collection, improper transportation, and failure to detect bacteria under strict nutritional conditions can lead to a low positive blood culture rate[27]. In addition, different sample types were used for blood culture and PCR. Whole blood samples were employed for blood culture, while plasma samples were utilized for PCR. The absence of components such as white blood cells in the plasma may have resulted in the loss of some bacteria, leading to a lower PCR positivity rate compared with that of the blood culture. A total of 60 plasma samples, of which 30 plasma specimens from blood culture-positive patients and 30 plasma specimens from blood culture-negative patients were enriched by M1 beads to assess the clinical manifestations of M-RAP. qPCR assays of both sets of specimens were performed, and the results were compared (Table 3). The Kappa values of 0.839, 0.815, and 0.856 were obtained, all exceeding 0.75, with a P value of < 0.05 indicating a high level of consistency in the detection results of the two methods.
Species M-RAP qPCR Kappa value Positive Negative Positive accuracy rate (%) Positive Negative Positive accuracy rate (%) SA
PA
AB8
7
952
53
5180
70
906
5
754
55
5360
50
700.839
0.815
0.856Note. SA, Staphylococcus aureus; PA, Pseudomonas aeruginosa; AB, Acinetobacter baumannii; M-RAP, multiple recombinase-aided PCR; qPCR, quantitative polymerase chain reaction. Table 3. Detection of SA, PA, and AB in clinical samples
-
In recent years, the incidence and mortality rates of BSI caused by bacteria have increased with the increase in invasive surgery and long-term use of broad-spectrum antibiotics and corticosteroids[28-30]. Among these, SA is the most common cause of gram-positive BSI in developed countries, with an annual incidence rate of 10–30/100,000, and the incidence of methicillin-resistant SA (MRSA) is increasing each year[31-32]. PA and AB are common bloodstream infectious bacteria that can invade a patient’s blood to form disseminated BSI, especially in patients with a compromised immune system. These bacteria exhibit a high mortality rate and are prone to developing resistance to multiple drugs[33-34]. Therefore, the prompt diagnosis of BSI holds great significance for the effective treatment of patients and contributes to enhancing their prognosis.
The M-RAP detection method established in this study combines the advantages of RAA rapidity and qPCR universality, with a shorter reaction time and higher sensitivity compared with qPCR alone. The M-RAP system includes a pair of RAA primers (outer primers), qPCR primers (inner primers), and probes. In the first stage, RAA amplification enriched the DNA template; then, the temperature was increased to inactivate the enzymes in the RAA system and dissolve the docosane. The amplified products were then introduced into the qPCR system; in the second stage, amplification and detection were carried out using a pair of qPCR primers and the corresponding qPCR probes. Due to the presence of both inner and outer sets of primers in the RAP system, it also has high specificity.
The entire process was completed in a closed PCR tube without opening the cover in the middle to avoid contamination of the sample and environment[35]. RAP combines the fast and efficient performance of RAA with the flexibility of TaqMan short probes in qPCR, eliminating the need to design intricate RAA probes. In addition, RAP does not rely on isothermal detection equipment and can be amplified using conventional qPCR instruments. Hence, most laboratories use this technology. The polysaccharide residues recognized by the M1 protein exist widely on the surface of various pathogens, including bacteria, fungi, viruses, etc.[36]. M1 beads have good enrichment effects on common pathogenic bacteria in BSI[37]. Owing to the low concentration of pathogenic bacteria in the patient’s blood, conventional molecular biology methods have limited sensitivity and cannot effectively detect them[38-39]. Therefore, M1 beads should be used for the pretreatment of the plasma sample to capture pathogenic bacteria. After enrichment, the relative concentration of pathogenic bacteria can be increased, and other interfering components in the plasma can be removed, thereby enhancing amplification efficiency and sensitivity[40]. Enriched live, intact bacteria can also be used for drug sensitivity testing. Since M1 beads can be stored for 14 d at 4 °C, they need to be specially prepared before each test, which increases the complexity of the experiment. Therefore, M1 beads can be prepared in advance, eliminating the preparation process and simplifying the experimental steps. During the experiment, some individual plasma samples might have exhibited agglutination around the M1 beads after enrichment. This could be attributed to inadequate heparin anticoagulation and the presence of residual fibrinogen in the plasma, which formed fibrin on the surface of the magnetic beads and wrapped around the magnetic beads, thus affecting the enrichment efficiency of the magnetic beads[41].
Many studies have reported the sensitivity of isothermal amplification or qPCR to detect pathogens; among them, the LOD for loop-mediated isothermal amplification (LAMP) in detecting MRSA was 102 copies/μL, while the LODs for recombinase polymerase amplification (RPA) in detecting Vibrio Cholerae and Vibrio Parahaemolyticus were 37 and 99 CFU/μL, respectively. The LOD for reverse transcription recombinase-aided amplification (RT-RAA) in detecting Wuxiang virus was 10 pfu/reaction viral RNA, while the LOD for qPCR in detecting Clostridioides difficile was 88.56 copies/reaction[42-45]. These detection methods cannot achieve the accuracy of blood culture, while the LOD of M-RAP was 1–10 copies/μL for SA, PA, and AB in this study. We observed a substantial reduction in the LODs of SA, PA, and AB following the addition of M1 beads to enhance pathogen enrichment. This finding suggests the significant role played by M1 beads in the process. In addition, the commonly used qPCR technology is unable to directly detect blood specimens containing fewer than < 10 CFU/mL of bacteria owing to its low sensitivity without the enrichment culture of the patient’s blood specimen. Obtaining positive blood cultures after preprocessing the blood specimen is a time-consuming process, which is not conducive to the early treatment of patients with bacteremia. In clinical plasma specimens from 30 blood culture-positive patients, even in the presence of M1 bead enrichment, the qPCR assay showed two more false-negative results for SA, PA, and AB, which could easily lead to missed detection in clinical settings. Inaccurate results could interfere with clinicians’ early judgment. Although we were unable to collect clinical blood samples that were infected with multiple pathogenic bacteria, M-RAP successfully established a multiple reaction system, demonstrating the feasibility of the detection method. Our established M-RAP assay revealed higher sensitivity and accuracy. It could directly detect plasma from patients with < 10 CFU/mL bacteremia without the need for blood specimen culture and could obtain valid results within 4 h. This presents a novel approach for detecting bacteremia in patients.
However, the M-RAP assay has some limitations. Previous experiments in our laboratory suggested that this may be due to the by-products produced by the RAA reaction, which inhibit the amplification efficiency of qPCR[15]. Due to the extremely high amplification efficiency of RAA, some samples already reached the threshold of qPCR detection during the RAA amplification period; hence, no CT value was generated in the second stage. Consequently, M-RAP cannot accurately quantify the copy number in samples; it serves as a qualitative detection method[15]. Additionally, although the M-RAP was performed in a single tube, the two reaction stages were separated by physical barriers, increasing the complexity of the experiment.
-
In summary, the integration of M-RAP with magnetic bead separation technology enables the direct detection of clinical plasma samples. This approach facilitates the rapid detection of trace amounts (< 10 CFU/mL) of common bacteria in the blood, eliminates the time-consuming microbiological culture process, shortens the detection time, and provides a new method for the early detection of trace pathogens in the blood of patients with BSI, which is of great significance and advances the rapid diagnosis of BSI in the early stages.
Although the M-RAP assay has some limitations, it is a stable, fast, highly sensitive, and specific detection method. M-RAP technology can not only be used to detect bacteria, fungi, viruses, and other pathogens but can also be combined with other technologies, such as microfluidics technology, to achieve high-throughput detection in different scenarios with broad application potential in clinical practice.
-
Standard DNA (copies/μL) SA PA AB Standard DNA (ng/μL) SA PA AB 105 8/8a 8/8 8/8 10-1 8/8 8/8 8/8 104 8/8 8/8 8/8 10-2 8/8 8/8 8/8 103 8/8 8/8 8/8 10-3 8/8 8/8 8/8 102 8/8 8/8 8/8 10-4 8/8 8/8 8/8 101 8/8 7/8 8/8 10-5 7/8 6/8 8/8 100 5/8 0/8 6/8 10-6 4/8 0/8 5/8 Note. aThe first number indicates the number of times a positive result occurred. The second number indicates that the experiment was repeated eight times. SA, Staphylococcus aureus; PA, Pseudomonas aeruginosa; AB, Acinetobacter baumannii; M-RAP, Multiple-recombinase-aided PCR. Table S1. The reproducibility of M-RAP
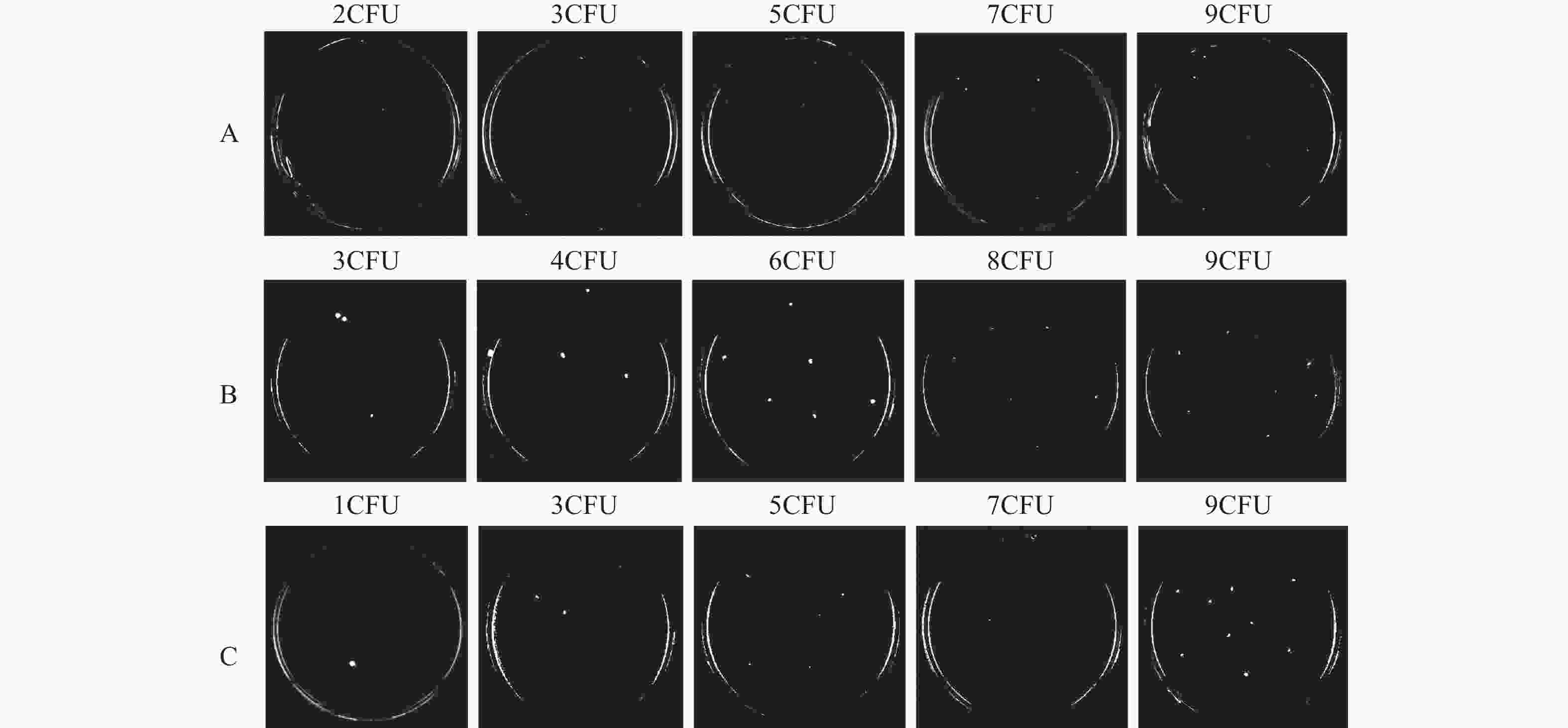
Figure S1. After enrichment with M1 beads, simulated PBS samples of SA (A), PA (B), and AB (C) were used to grow on plate culture corresponding to the bacterial colonies.
HTML
Sample Collection and Nucleic Acid Extraction
Design of M-RAP Primers and Probes and Construction of Plasmids
Establishment and Optimization of M-RAP Assay
Sensitivity, Repeatability, and Specificity of M-RAP Assay
Detection Sensitivity of Ordinary qPCR
Preparation of Simulated Plasma Samples and M1 beads
M1 Beads Capture Bacteria and Preparation of Nucleic Acid
Evaluation and Comparison of Simulated Samples and Clinical Samples
Statistical Analysis
Optimization of M-RAP Assay
Sensitivity and Repeatability of M-RAP Assay
Specificity of M-RAP Assay
Sensitivity of Ordinary qPCR Detection
Comparison of Results Between M-RAP and qPCR before and after Enrichment of Simulated Samples
Evaluation and Comparison of Clinical Sample Detection Results Using M-RAP and qPCR
 23278+Supplementary Materials.pdf
23278+Supplementary Materials.pdf
|

|
 Quick Links
Quick Links
 DownLoad:
DownLoad: