

 下载:
下载:

-
Lyme disease (LD) is a multisystem disorder caused by infection with the spirochete Borrelia burgdorferi (sensu stricto; B.b.s.s), which is transmitted to humans by Ixodes ticks[1, 2]. B.b.s.s is phenotypically and genotypically heterogeneous, as confirmed by many studies[3-5]. Currently, at least 18 B.b.s.s genospecies have been described, among which five species are associated with disease in humans including Borrelia burgdorferi (sensu stricto), Borrelia garinii (B.g), Borrelia afzelii (B.a), Borrelia lusitaniae, and Borrelia spielmanii. Clinical manifestations of LD are diverse, which mainly includes erythema migrans skin lesions, acrodermatitis chronica atrophicans, and neurotropic and arthritogenic symptoms[6].
In a previous study, more than 100 Borrelia strains in China were isolated from ticks, animals, and patients[7]. A total of five species was reported: B.b.s.s, B.g, B.a, Borrelia sinica, and Borrelia yangtze sp. nov[8-10].
In various pathogenetic events during different stages of infection of B.b.s.s, outer surface proteins (OSPs) play an important role[11-13]. According to reports, these proteins can assist spirochetes’ adhesion to and invasion of the tissues in vitro, and promote pathogen infectivity in vivo. Moreover, by undergoing subtle antigenic changes, which give rise to immunoevasive mutants, they may also contribute to the development of chronic LD[14, 15].
The 66 kD protein (P66) is one of outer surface proteins of Borrelia burgdorferi (sensu lato; B.b.s.l), which is known as adhesin with pore-forming activity[16,17]. P66 plays a role in the process of adhesion to and invasion of host tissues by binding specifically to integrins[18,19]. According to previous reports, it has a surface-exposed domain, including a putative surface-exposed loop (residues 459 to 502) near the C terminus[20]. Sequence variation in the surface-exposed loop is greater than the remaining sequence of P66, which indicates that during mammalian infection, the loop may be under immune selection pressure[16, 21].
To understand P66 locus’ gene diversity of Chinese Borrelia strains, especially on the B-cell epitopes, the P66 gene of 59 Chinese strains from three genotypes in China were sequenced and analyzed.
-
Fifty nine B.b strains were selected, which were isolated in Beijing municipality and 12 provinces and autonomous regions in China. In a previous study, multilocus sequence analysis (MLSA) was used to genotype these strains[8]. They were identified to three pathogenic genotypes: B.b.s.s, B.g, and B.a. Because B.g strains are predominant in China, 1 B.b.s.s, 42 B.g and 16 B.a strains were selected in this study (Table 1).
Table 1. Distribution of 59 strains in different areas of China
Provinces Number of strains Genotypes Jilin 14 B.g Guangdong 1 B.g Inner Mongolia 8 B.g Shandong 1 B.a Liaoning 1 B.a Guizhou 3 B.a Sichuan 6 B.a Heilongjiang 5 B.g (3); B.a (2) Xinjiang 15 B.g Beijing 3 B.a Hebei 1 B.g Hunan 1 B.b.s.s Note. B.b.s.s, Borrelia burgdorferi (sensu stricto); B.g, Borrelia garinii; B.a, Borrelia afzelii. -
Strains were cultured in Barbour-Stoenner-Kelly medium, collected by centrifuging at 13,000 rpm/min (r = 15 cm), and then heat-inactivated at 100 ℃. DNA obtained by this method was used as a template for amplifying the gene of 66 kD protein. Primer 5 software was used to design the nucleotide sequences of the primers used in this study according to B31 genome sequence. These are as follows: 5′-GAGCTTGTTCCTGGGTTTGA-3′ and 5′-CTTCCGCTGTAGGCTATTTT-3′. Polymerase chain reaction (PCR) was performed in a total volume of 50 μL. The PCR mix contained 25 μL PCR buffer, 20 pmol/L of each primer, 2.5 mmol/L each of four dNTPs, and 1 U DNA Taq Polymerase (Takara). Amplification was performed for 10 min of an initial denaturation at 94 ℃; 35 cycles under the following conditions: denaturation at 94 ℃ for 45 s, annealing at 41 ℃ for 45 s, extension at 72 ℃ for 90 s, and final extension at 72 ℃ for 10 min. When PCR was performed, negative control (reagent only, no DNA) was included. The positive control was 300 ng DNA from the B.b.s.s strain B31, which is the standard strain in United States. The PCR products’ presence and size were determined by electrophoresis on 1.5% agarose gel in Tris-boric acid-EDTA buffer, followed by staining with GoldView. To validate reproducibility, PCRs were performed at least twice.
-
An ABI 3730xl DNA analysis was used to determine the sequences of all PCR products. Distances were calculated using the neighbor-joining method. Sequences that contained 235–1,674 bp of P66 were compared using MEGA 5.10 software[22].
-
To amplify the coding gene 235–1,674 bp (encoding 79−558 amino acids) of P66, PCR was used. Neighbor-joining method was used to cluster analysis all strains by MEGA 5.1 software based on the amino acid sequence of P66. The five strains of B.a (details in Result 2), namely LIP94-11, FP1, LB20, LB21, and SZ21, were not included in the cluster analysis because of special mutations in the strains.
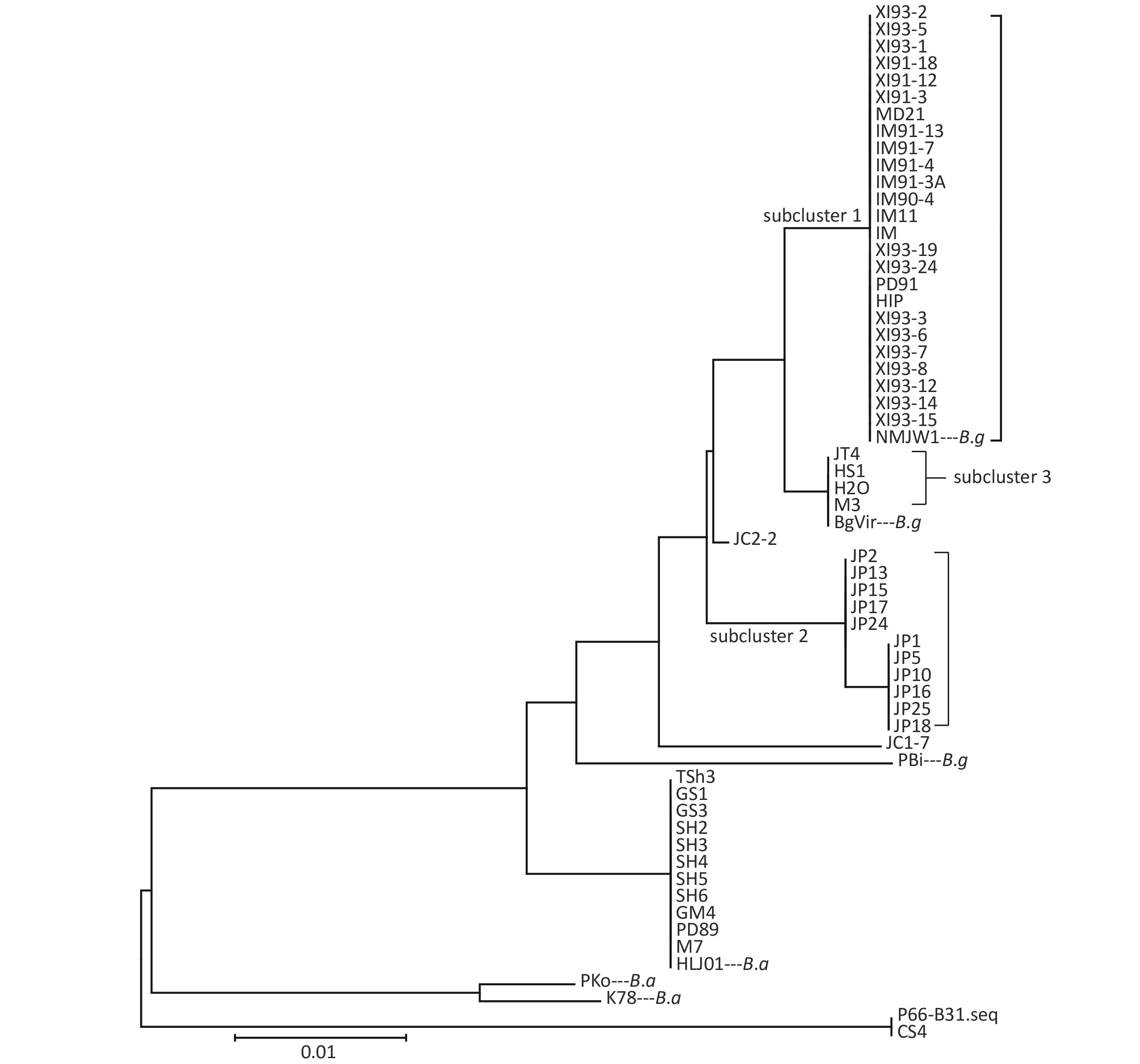
Clustering results of the 54 isolates were found to be in accordance with the seven locus sequence analysis[8]. In addition, the B.g strains were divided into three subclusters: subcluster 1, subcluster 2, and subcluster 3. Clustering results are shown in Figure 1.
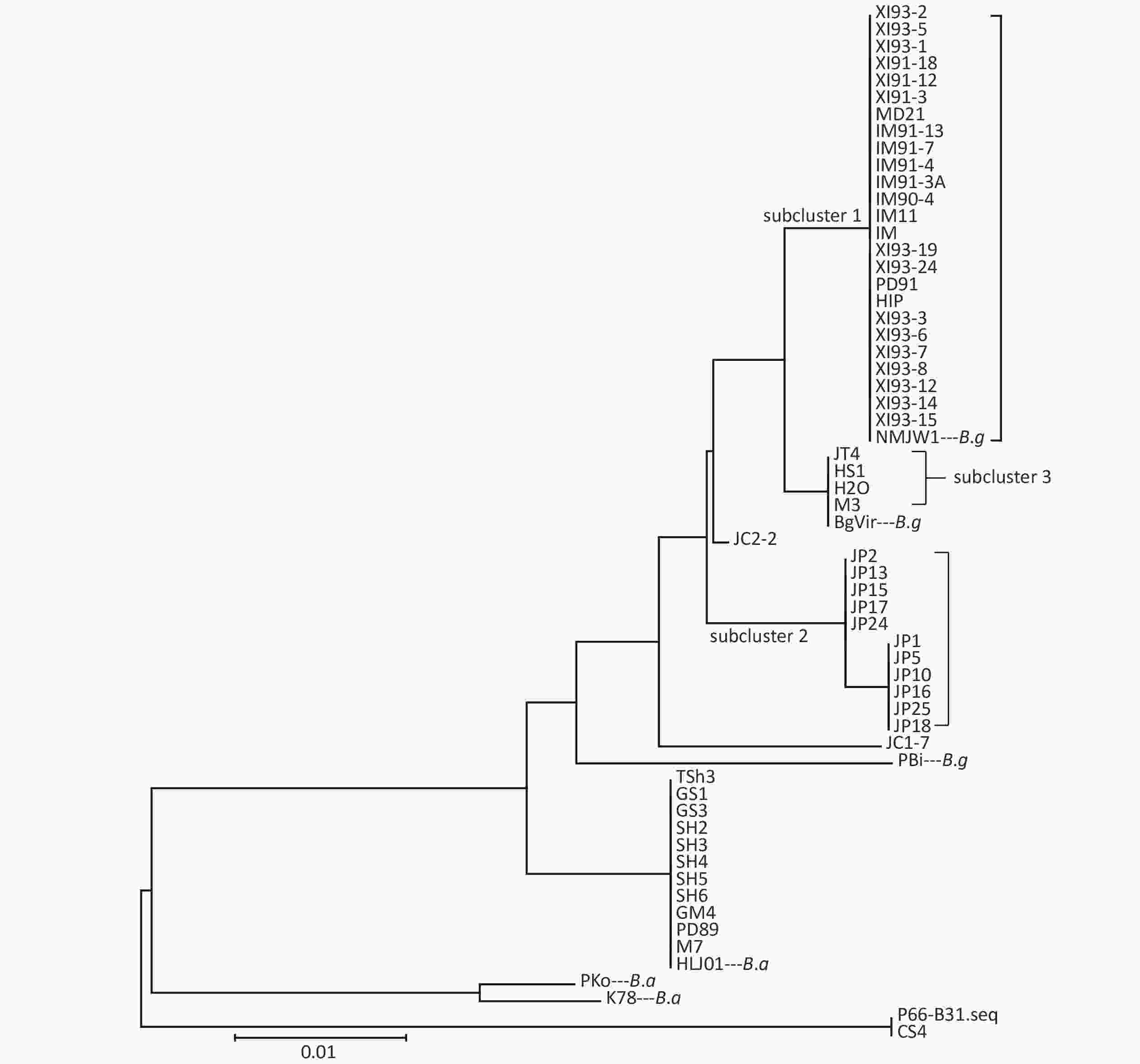
Figure 1. Clustering results of P66 amino acid sequences of 54 isolates.
-
The P66 gene of B.b.s.s strain CS4 was found to be exactly the same as the corresponding reference sequence of B31 strain and the 15 Xinjiang strains after the P66 gene sequences of all 59 isolates were combined, adjusted, and analyzed. It was represented by XI91-12 in the subcluster 1, which mutated from CAA to TAA codon in the 508 aa position and led to the premature termination of P66 amino acid sequences of these strains. The IM91-13 sequence in subcluster 1 was changed from TTA codon to TAA codon in 479 aa position, which resulted in the premature termination of the amino acid sequence of P66 protein and nonsynonymous mutations in subcluster 2, subcluster 3, JC1-7, and JC2-2 strains of B.g. Table 2shows the detailed information of the nonsynonymous mutations. In B.a strains, special mutations happened in five strains: a base G was inserted at 438 bp in the p66 gene of LIP94-11 strain, which caused termination of the amino sequence at the 172 aa position; bases A and C were inserted at 1 523 bp in the p66 gene of FP1, LB20, LB21, and SZ21 strains, leading to the early termination of the amino acid sequences.
Table 2. Nonsynonymous mutations in P66 gene of 42 B.g strains*
Genotype Strain AA
positionAA
mutationsGenotype Strain AA
positionAA
MutationsB. garinii Subcluster 2 118 M-I B. garinii JC1-7 109 I-V 153 G-R# 118 M-I 208 T-A 221 I-V 221 I-V 267 I-M 276 L-I 276 L-V 348 V-I 360 N-S 401 A-V 361 S-G 435 A-E 435 A-E 490 T-I 445 A-S 491 T-A 464 M-I 492 S-N 490 T-I 494 A-G 495 S-A 495 S-A 505 E-G 500 A-T 508 Q-K 505 E-G 510 I-T 509 A-T JC2-2 118 M-I 510 I-T 221 I-V Subcluster 3 118 M-I 276 L-V 435 A-E 435 A-E 440 T-A 495 S-A 495 S-A 501 G-E 510 I-A 505 E-G 511 T-I 510 I-T Note. *Reference strains: B.b.s.s-B31, B.g-NMJW1, B.a-HLJ01. #The mutation at 153 aa site occured in 6 strains of subcluster 2. -
According to the Immune Epitope Database (IEDB), there is one B-cell epitope (No. 63482) in P66 of B31 strain[23]. The corresponding amino acid sequence is located at 454–491 aa.
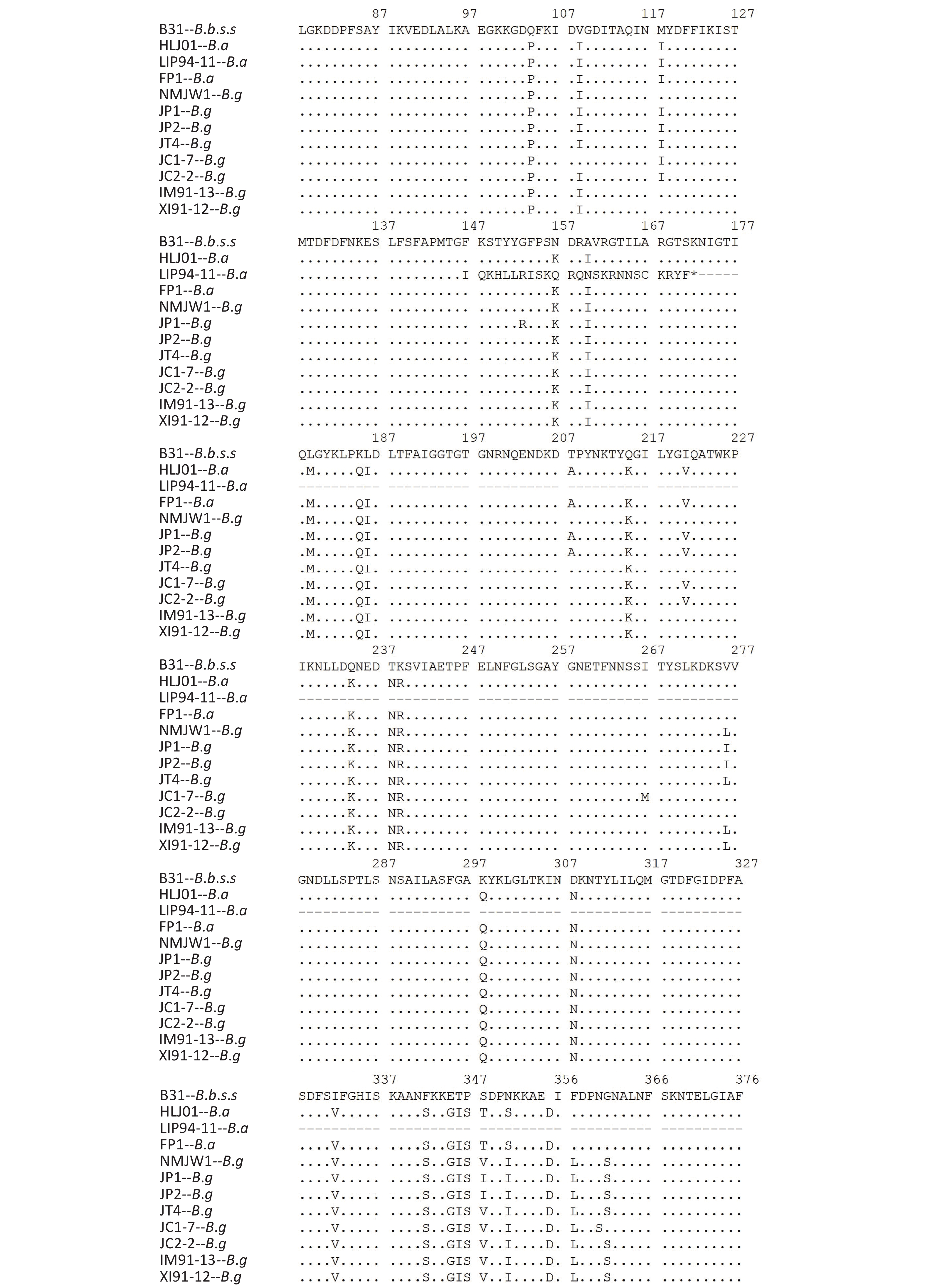
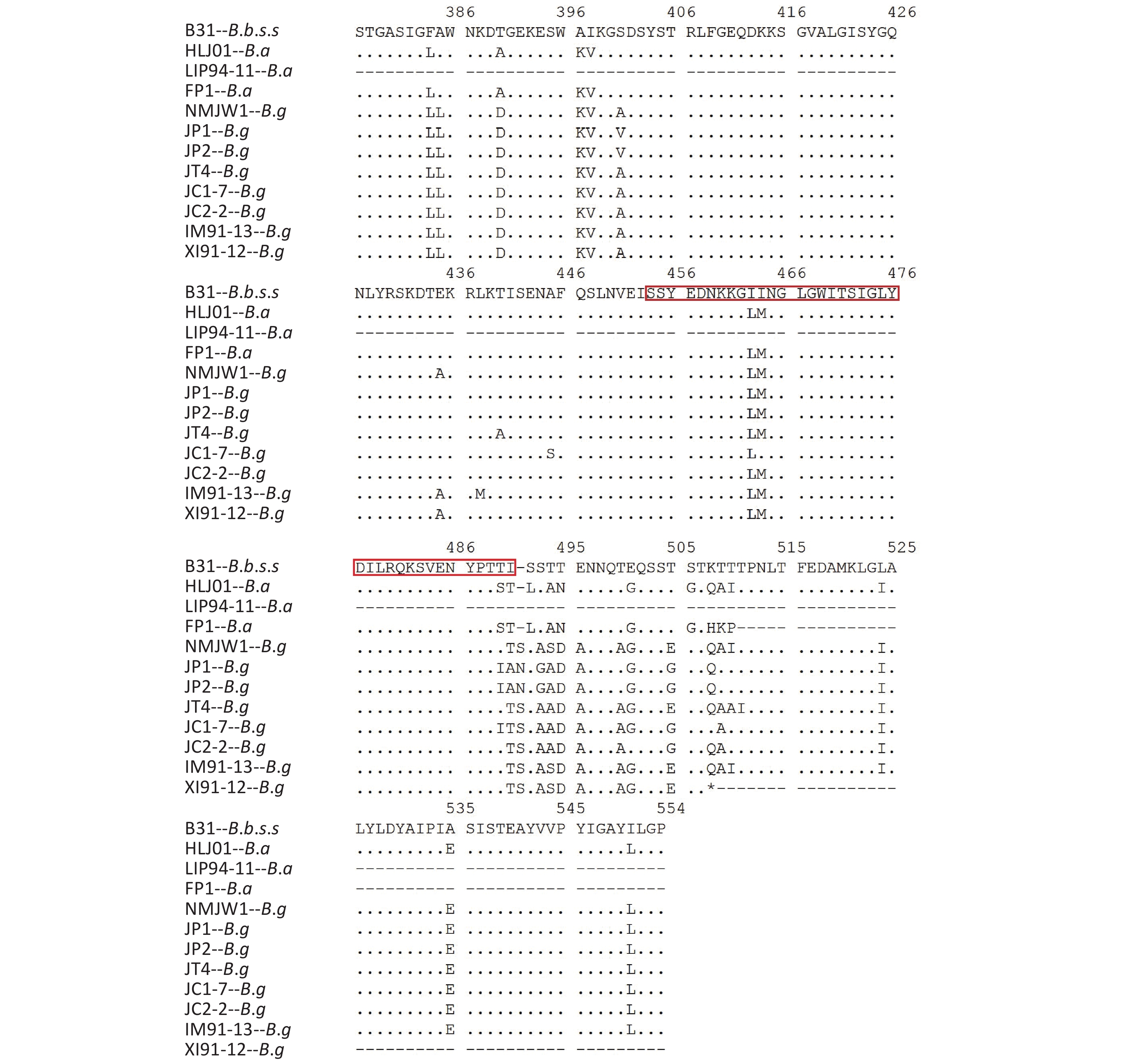
Isolates of different genotypes were analyzed according to reference strains of different genotypes. Figure 2 shows the specific mutation information in the different sequences. The amino acid sequence information of six isolates in subcluster 2 of the B.g genotype is represented by the JP1 strain and the other five isolates are represented by the JP2 strain. The detailed information is shown by the P66 amino acid cluster analysis in Figure 1. In subcluster 3 of the B.g genotype, JT4 strain represents the amino acid sequence information of four isolates; and XI91-12 represented amino acid sequence variation of 15 isolates. The variation of FP1 strain represented the four isolates (FP1, LB20, LB21, and SZ21) of the B.a genotype.
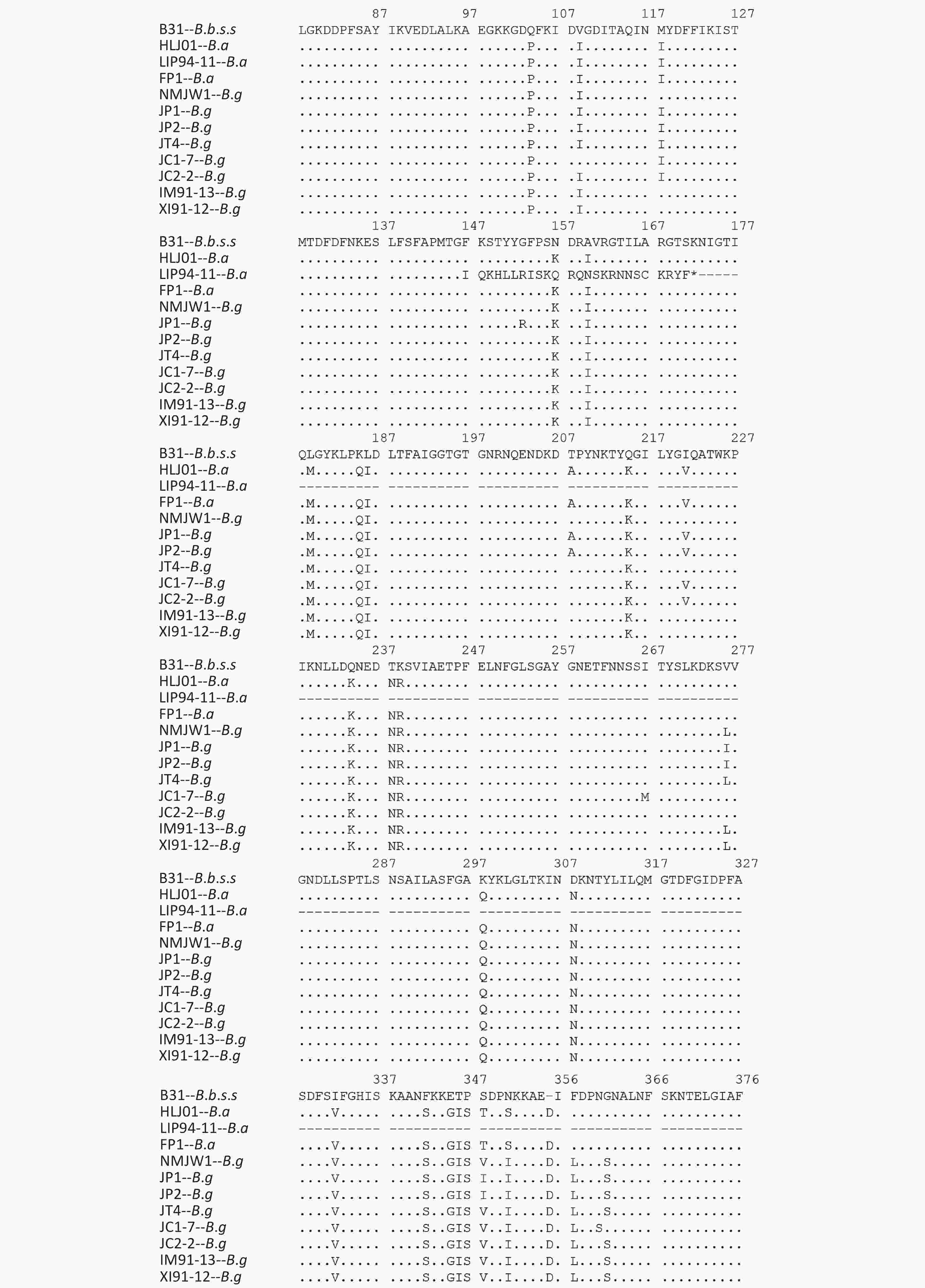

Figure 2. Variations of P66 amino acid sequences in 59 isolates.
The B.g and B.a genotype reference strains and isolates was found to insert aspartic acid (Asp, D) in the 356 aa position and alanine (Ala, A) at the 494 aa position
The 490 aa and 491 aa variations of the B.g subcluster 2 strains and the 464 aa and 490 aa variations of JC1-7 strain affect the only one B-cell epitope of the P66 protein antigen.
-
B.b has many kinds of OSPs and their heterogeneity in different strains is large. The study of polymorphism for OSPs can provide theoretical basis for LD pathogenesis, vaccine, and diagnostic reagents.
OspA, OspC, fla, etc. are more studied in the present. OspA is associated with the colonization of spirochetes in the tick’s midgut. The expression of OspA was downregulated when infected with mammalian, whereas OspC was upregulated. OspA and OspC can be taken as candidate antigens for vaccine because of their ability to produce protective antibodies by inducing the body. However, because OspC mutation is large and some strains do not express OspC protein, it is not used for the identification of strains. Gene sequence and amino acid cluster of 41 kD flagellin encoded by flaB gene was analyzed by Liu Huixin et al.[24] The results showed that the amino acid sequence of the 41 kD flagellin had variation among genotypes, but the variation within the genotype was small. Therefore, when 41 kD flagellin is used in the diagnosis of LD, the difference between genotypes should be considered. Both OspA and fla can be used for multilocus typing (e.g., MLSA).
Fifty nine isolates from 12 provinces of China were selected in this study, which covered the main epidemic areas of LD and the main pathogenic genotypes in China, so polymorphism of the P66 gene and B-cell epitopes of these strains were representative.
Cluster analysis of 59 strains based on P66 amino acids in China showed that the strains could be divided into three types (B.b.s.s, B.g, and B.a), which was in accordance with the MLSA genotyping results of the seven loci[8]. Among them, B.g strains were divided into three subclusters and two scattered strains JC1-7 and JC2-2, which indicated that P66 amino acid sequences of B.g strains had larger heterogeneity and that it has a certain regional character as shown on that clustering results. Xinjiang and Inner Mongolia strains were mainly located in Subcluster 1, Jilin strains in subcluster 2, and Heilongjiang and a few Jilin strains in subcluster 3.
The P66 amino acid sequence analysis of 59 Chinese isolates showed that large number of mutations happened not only among the three genotype strains but also in B.g and B.a strains, especially in the 480–523 aa region (Figure 2), which was consistent with the previous reports[20]. It was reported that at the carboxyl end of P66 protein located at 480–523 aa, there was a surface-exposed domain (surface-exposed ring structure). The region’s variation was larger than that of other P66 sequences, which indicates that surface-exposed structural rings may be involved in immune evasion in mammalian infection.
One of the main epidemic areas of LD in China is in Xinjiang Uyghur Autonomous Region[7,8]. A large number of Lyme-suspected patients is reported in some forest areas of Xinjiang, and the average infection rate of LD can reach 35.49%[25]. Etiology study showed that there were B.g and B.b.s.s pathogenic strains in Xinjiang[7, 8, 26]. This study found that 15 Xinjiang B.g strains terminate early at the 508aa position and it had a distinct regional character, which may be related to the B.g strains’ pathogenicity in this area.
Further analysis of B-cell epitopes (454–491 aa) of P66 revealed a variation of genotype strains existed in this region. The B-cell epitope region of B.g strains was more variable and that it is the immunoreactive region of P66 protein. Therefore, when P66 is used for immunoassay, the difference between genotypes should be taken into account.
doi: 10.3967/bes2021.048
-
Abstract:
Objective To study the polymorphism in P66 and its human B-cell epitopes of Borrelia burgdorferi strains in China. Methods Polymerase chain reaction (PCR) and sequencing were used to obtain the P66 sequences of 59 Chinese B. burgdorferi. Then the sequences were analyzed by MEGA 5.10 software and compared with the human B-cell epitope sequences from the Immune Epitope Database (IEDB) based on the reference strain of each genotype. Results Results showed that genetic and amino acid diversity presented in the 66 kD protein of all 59 Chinese strains, especially in Borrelia garinii (B.g) and Borrelia afzelii (B.a) strains. B.g strains were divided into three subclusters and two scattered strains JC1-7 and JC2-2 according to the amino acid sequences of P66. The P66 sequences of 15 Xinjiang strains represented by XI91-12 in the B.g subcluster 1, changed from CAA to TAA codon at 508aa position, resulting in early termination. Bases A and C were inserted at sequence position 1 523 bp of strains FP1, LB20, LB21, and SZ21 in the B.a genotype, which resulted to early termination at position 511 aa. G base was inserted at 438 bp of LIP94-11 strain, which led to early termination at position 172 aa. Conclusion In P66 of 59 Chinese strains, polymorphisms were widely distributed. More importantly, the P66 amino acid sequences of B.g strains had a certain regional character. One of the characteristics of Xinjiang B.g isolates might be the variation at the 508aa location in 15 Xinjiang B.g strains, which may be related to the strains’ pathogenicity in this area. -
Key words:
- Borrelia burgdoferi /
- P66 /
- B-cell epitope /
- Polymorphism
-
Figure 2. Variations of P66 amino acid sequences in 59 isolates.
The location of the human B-cell epitopes (in the red frame sequence) is labeled in the amino acid sequence of the B.b.s.s reference strain B31; *Represents the mutation position of the advanced terminating sequence.
Table 1. Distribution of 59 strains in different areas of China
Provinces Number of strains Genotypes Jilin 14 B.g Guangdong 1 B.g Inner Mongolia 8 B.g Shandong 1 B.a Liaoning 1 B.a Guizhou 3 B.a Sichuan 6 B.a Heilongjiang 5 B.g (3); B.a (2) Xinjiang 15 B.g Beijing 3 B.a Hebei 1 B.g Hunan 1 B.b.s.s Note. B.b.s.s, Borrelia burgdorferi (sensu stricto); B.g, Borrelia garinii; B.a, Borrelia afzelii.  下载: 导出CSV
下载: 导出CSV
Table 2. Nonsynonymous mutations in P66 gene of 42 B.g strains*
Genotype Strain AA
positionAA
mutationsGenotype Strain AA
positionAA
MutationsB. garinii Subcluster 2 118 M-I B. garinii JC1-7 109 I-V 153 G-R# 118 M-I 208 T-A 221 I-V 221 I-V 267 I-M 276 L-I 276 L-V 348 V-I 360 N-S 401 A-V 361 S-G 435 A-E 435 A-E 490 T-I 445 A-S 491 T-A 464 M-I 492 S-N 490 T-I 494 A-G 495 S-A 495 S-A 505 E-G 500 A-T 508 Q-K 505 E-G 510 I-T 509 A-T JC2-2 118 M-I 510 I-T 221 I-V Subcluster 3 118 M-I 276 L-V 435 A-E 435 A-E 440 T-A 495 S-A 495 S-A 501 G-E 510 I-A 505 E-G 511 T-I 510 I-T Note. *Reference strains: B.b.s.s-B31, B.g-NMJW1, B.a-HLJ01. #The mutation at 153 aa site occured in 6 strains of subcluster 2.
下载: 导出CSV
-
[1] Steere AC, Malawista SE, Hardin JA, et al. Erythema chronicum migrans and Lyme arthritis. The enlarging clinical spectrum. Ann Intern Med, 1977; 86, 685−98. doi: 10.7326/0003-4819-86-6-685 [2] Stanek G, Wormser GP, Gray J, et al. Lyme Borreliosis. Lancet, 2012. [3] Margos G, Vollmer SA, Cornet M, et al. A new Borrelia species defined by multilocus sequence analysis of housekeeping genes. Appl Environ Microbiol, 2009; 75, 5410−6. doi: 10.1128/AEM.00116-09 [4] Margos G, Vollmer SA, Ogden NH, et al. Population genetics, taxonomy, phylogeny and evolution of Borrelia burgdorferi sensu lato. Infect Genet Evol, 2011; 11, 1545−63. doi: 10.1016/j.meegid.2011.07.022 [5] Rudenko N, Golovchenko M, Grubhoffer L, et al. Updates on Borrelia burgdorferi sensu lato complex with respect to public health. Ticks Tick Borne Dis, 2011; 2, 123−8. doi: 10.1016/j.ttbdis.2011.04.002 [6] Strle F, Stanek G. Clinical manifestations and diagnosis of lyme borreliosis. Curr Probl Dermatol, 2009; 37, 51−110. doi: 10.1159/000213070 [7] Zhang ZF, Wan KL, Zhang JS. [Studies on epidemiology and etiology of Lyme disease in China]. Chin J Epidemiol, 1997; 18, 8−11. (In Chinese [8] Hao Q, Hou X, Geng Z, et al. Distribution of Borrelia burgdorferi sensu lato in China. J Clin Microbiol, 2011; 49, 647−50. doi: 10.1128/JCM.00725-10 [9] Chu CY, Liu W, Jiang BG, et al. Novel genospecies of Borrelia burgdorferi sensu lato from rodents and ticks in southwestern China. J Clin Microbiol, 2008; 46, 3130−3. doi: 10.1128/JCM.01195-08 [10] Masuzawa T, Takada N, Kudeken M, et al. Borrelia sinica sp. nov., a lyme disease-related Borrelia species isolated in China. Int J Syst Evol Microbiol, 2001; 51, 1817−24. doi: 10.1099/00207713-51-5-1817 [11] Yang XF, Pal U, Alani SM, et al. Essential role for OspA/B in the life cycle of the Lyme disease spirochete. J Exp Med, 2004; 199, 641−8. doi: 10.1084/jem.20031960 [12] Grimm D, Tilly K, Byram R, et al. Outer-surface protein C of the Lyme disease spirochete: a protein induced in ticks for infection of mammals. Proc Natl Acad Sci USA, 2004; 101, 3142−7. doi: 10.1073/pnas.0306845101 [13] Norris SJ, Coburn J, Leong JM, et al. Pathobiology of Lyme disease Borrelia. 2010. [14] Liang FT, Yan J, Mbow ML, et al. Borrelia burgdorferi changes its surface antigenic expression in response to host immune responses. Infect Immun, 2004; 72, 5759−67. doi: 10.1128/IAI.72.10.5759-5767.2004 [15] Liang FT, Brown EL, Wang T, et al. Protective niche for Borrelia burgdorferi to evade humoral immunity. Am J Pathol, 2004; 165, 977−85. doi: 10.1016/S0002-9440(10)63359-7 [16] Bunikis J, Noppa L, Ostberg Y, et al. Surface exposure and species specificity of an immunoreactive domain of a 66-kilodalton outer membrane protein (P66) of the Borrelia spp. that cause Lyme disease. Infect Immun, 1996; 64, 5111−6. doi: 10.1128/IAI.64.12.5111-5116.1996 [17] Skare JT, Mirzabekov TA, Shang ES, et al. The Oms66 (p66) protein is a Borrelia burgdorferi porin. Infect Immun, 1997; 65, 3654−61. doi: 10.1128/IAI.65.9.3654-3661.1997 [18] Coburn J, Chege W, Magoun L, et al. Characterization of a candidate Borrelia burgdorferi beta3-chain integrin ligand identified using a phage display library. Mol Microbiol, 1999; 34, 926−40. doi: 10.1046/j.1365-2958.1999.01654.x [19] Ristow LC, Bonde M, Lin YP, et al. Integrin binding by Borrelia burgdorferi P66 facilitates dissemination but is not required for infectivity. Cell Microbiol, 2015; 17, 1021−36. doi: 10.1111/cmi.12418 [20] Kenedy MR, Luthra A, Anand A, et al. Structural modeling and physicochemical characterization provide evidence that P66 forms a beta-barrel in the Borrelia burgdorferi outer membrane. J Bacteriol, 2014; 196, 859−872. doi: 10.1128/JB.01236-13 [21] Bunikis J, Luke CJ, Bunikiene E, et al. A surface-exposed region of a novel outer membrane protein (P66) of Borrelia spp. is variable in size and sequence. J Bacteriol, 1998; 180, 1618−23. doi: 10.1128/JB.180.7.1618-1623.1998 [22] Tamura K, Peterson D, Peterson N, et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol, 2011; 28, 2731−9. doi: 10.1093/molbev/msr121 [23] Vita R, Overton JA, Greenbaum JA, et al. The immune epitope database (IEDB) 3.0. Nucleic Acids Res, 2015; 43, D405−D412. doi: 10.1093/nar/gku938 [24] Liu HX, Liu W, Hou XX, et al. Polymorphism of 41 kD Flagellin Gene and Its Human B-Cell Epitope in Borrelia burgdorferi Strains of China. Bio Med Research International, 2016. doi: 10.1155/2016/1327320 [25] Yu-Hui T, Yong L, Huo S, et al. Surveillance of Lyme Disease in Xinjiang Uygur Autonomous Region During 2000-2004. Chin J Clin Neurosci, 2007; 15, 158−61. [26] Wang YZ, Mu LM, Zhang K, et al. A broad-range survey of ticks from livestock in Northern Xinjiang: changes in tick distribution and the isolation of Borrelia burgdorferi sensu stricto. Parasit Vectors, 2015; 8, 449. doi: 10.1186/s13071-015-1021-0 -

 点击查看大图
点击查看大图
图(3) / 表ll (2)
计量
- 文章访问数: 758
- HTML全文浏览量: 281
- PDF下载量: 49
- 被引次数: 0

 Quick Links
Quick Links