

 下载:
下载:

-
Ticks are recognized as one of the most important vectors that cause diseases in animals and humans, and have significant public health impacts worldwide[1]. They have a wide range of hosts among terrestrial vertebrates, including mammals, birds, amphibians. The numerous zoonotic pathogens transmitted by ticks are the causative agents of Lyme disease, rickettsiosis, ehrlichioses, relapsing fever, Q fever, and tularemia. Cases of tick-borne diseases are increasing around the world and some emerging or re-emerging pathogens are being discovered, which pose great challenges for prevention and control.
Tick-borne Borrelia spp. are currently divided into two taxonomic groups, Lyme disease spirochetes and relapsing fever spirochetes. Lyme disease, caused by Borrelia burgdorferi sensu lato (B.b.s.l.), is widespread around the globe, having been reported in more than 70 countries. Phylogenetically, Borrelia miyamotoi is categorized as a relapsing fever spirochete, but it does not cause typical relapsing fever and is transmitted by hard-bodied ticks. This spirochete has a global distribution and often cocirculates with B.b.s.l., as they have overlapping vertebrate animal and tick vectors[2]. Most Rickettsiales described in the literature are well-known zoonotic emerging pathogens, some of which can cause serious disease in humans, including rickettsioses, anaplasmoses, and ehrlichioses. Spotted fever rickettsioses are caused by spotted fever group rickettsia (SFGR). Anaplasma phagocytophilum (AP) is the causative agent of human granulocytic anaplasmosis (HGA), with cases reported in the USA, Netherlands, Sweden, France, and other countries. Ehrlichia chaffeensis (EC) can lead to human monocytic ehrlichiosis (HME), and no clinical cases were reported in China until 2010[3].
Shanxi Province, located in the middle of China, features a rage of different landforms, including mountains, hills, and plains. Shrubs and grasslands are widely distributed in Shanxi and animal husbandry is well developed, which provides a suitable environment for tick survival. In this study, five pathogens was identified as being carried by hard ticks from Shanxi Province, and attempts are being made to control tick-borne pathogens in this area.
In May 2019, we used the flagging method to collect more than 600 ticks in the village of Liangping, Qi County, Shanxi Province. All ticks were identified based on their morphologies, and these identifications were verified based on their mitochondrial 16S rDNA sequences. After washing by 75% alcohol and distilled water, genomic DNA was individually extracted from 112 ticks using the DNeasy Blood & Tissue Kit (QIAGEN, Germany). Polymerase chain reaction (PCR) and real-time PCR assays were performed to identify the pathogens. Nested PCR was used to identify 1) B.burgdorferi based on 5S(rrfA)-23S(rrlB) rRNA intergenic spacer (IGS), and 2) B.miyamotoi based on glycerophosphodiester phosphodiesterase gene (glpQ). To detect SFGR, conventional PCR was performed by amplifying a fragment of the outer membrane protein A gene (ompA) of Rickettsia spp.. Amplification of the conserved 16S rRNA gene is an effective method for identifying AP. For EC, we designed primers and a Taqman MGB probe based on the 16S rRNA coding gene for real-time PCR. Table 1 lists the target genes and primers of five pathogens. PCR amplifications were performed on a Labcycler (Sensoquest, Germany), and the amplicon products were analyzed by electrophoresis in 1.5% agarose gel. The real-time PCR assays were performed on a LightCycler 480 (Roche, Switzerland). After purification and sequencing, the obtained nucleotide sequences were analyzed by the bioninformatics algrorithm BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi), and the results were compared with those of the sequences deposited at the US National Center for Biotechnology (NCBI). We also downloaded reference sequences from Genbank to enrich our data for phylogenetic analysis. The nucleotide sequences were aligned using the ClustalW method, phylogenetic trees were generated using MEGA 7.0 and the neighbor-joining algorithm, and bootstrap analysis of 1,000 replicates was performed to determine the reliability of the trees.
Table 1. Target genes and primers of tick identification and pathogen detection
Pathogens Target genes Primers Sequence (5′-3′) Size (bp) Reference Tick identification 16S rDNA mitochondrial gene 16s-F CTGCTCAATGATTTTTTAAATTGCTGTGG 460 [4] 16s-R CCGGTCTGAACTCAGATCAAGT B.burgdorferi 5S-23S rRNA IGS rrf-rrl(1f) CGACCTTCTTCGCCTTAAAGC 250 [5] rrf-rrl(1r) TAAGCTGACTAATACTAATTACCC rrf-rrl(2f) TCCTAGGCATTCACCATA rrf-rrl(2r) GAGTTCGCGGGAGA B.miyamotoi glpQ glpQ(1f) CACCATTGATCATAGCTCACAG 424 [6] glpQ(1r) CTGTTGGTGCTTCATTCCAGTC glpQ(2f) GCTAGTGGGTATCTTCCAGAAC glpQ(2r) CTTGTTGTTTATGCCAGAAGGGT SFGR ompA ompA-f ATGGCGAATATTTCTCCAAAA 632 [7] ompA-r GTCCGTTAATGGCAGCATCT AP 16S rRNA AP-f GTCGAACGGATTATTCTTTATAGCTTG 389 [8] AP-r TATAGGTACCGTCATTA CTTCCCTAC EC 16S rRNA EC-f AGCCTAACA CATGCAAGTCGAA 75 [9] EC-r CCCGTC TGCCACTAACAATTATT TaqMan-1 FAM-CAATTGCTTATAACCTTTTG-MGB Note. SFGR, Spotted fever group rickettsia; AP, Anaplasma phagocytophilum; EC, Ehrlichia chaffeensis. Based on their morphological characteristics, the ticks collected in this area were identified as either Haemaphysalis longicornis or Haemaphysalis japonica. Using a combination of morphological and conserved gene analyses improves the accuracy of tick identification and prevents subjective error. As such, the 112 ticks were subjected to mitochondrial 16s rRNA identification, and 103 H. longicornis and 9 H. japonica were identified.
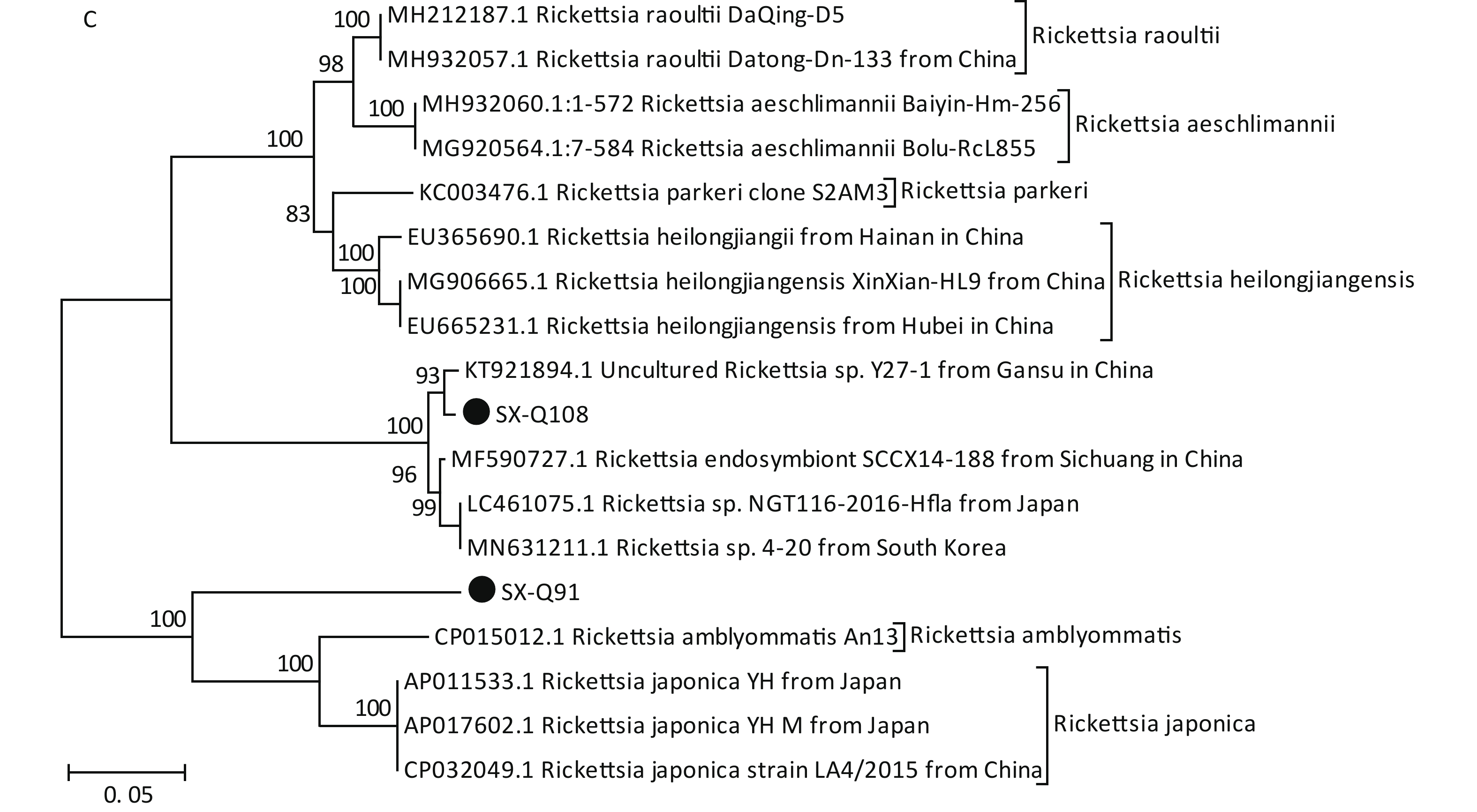
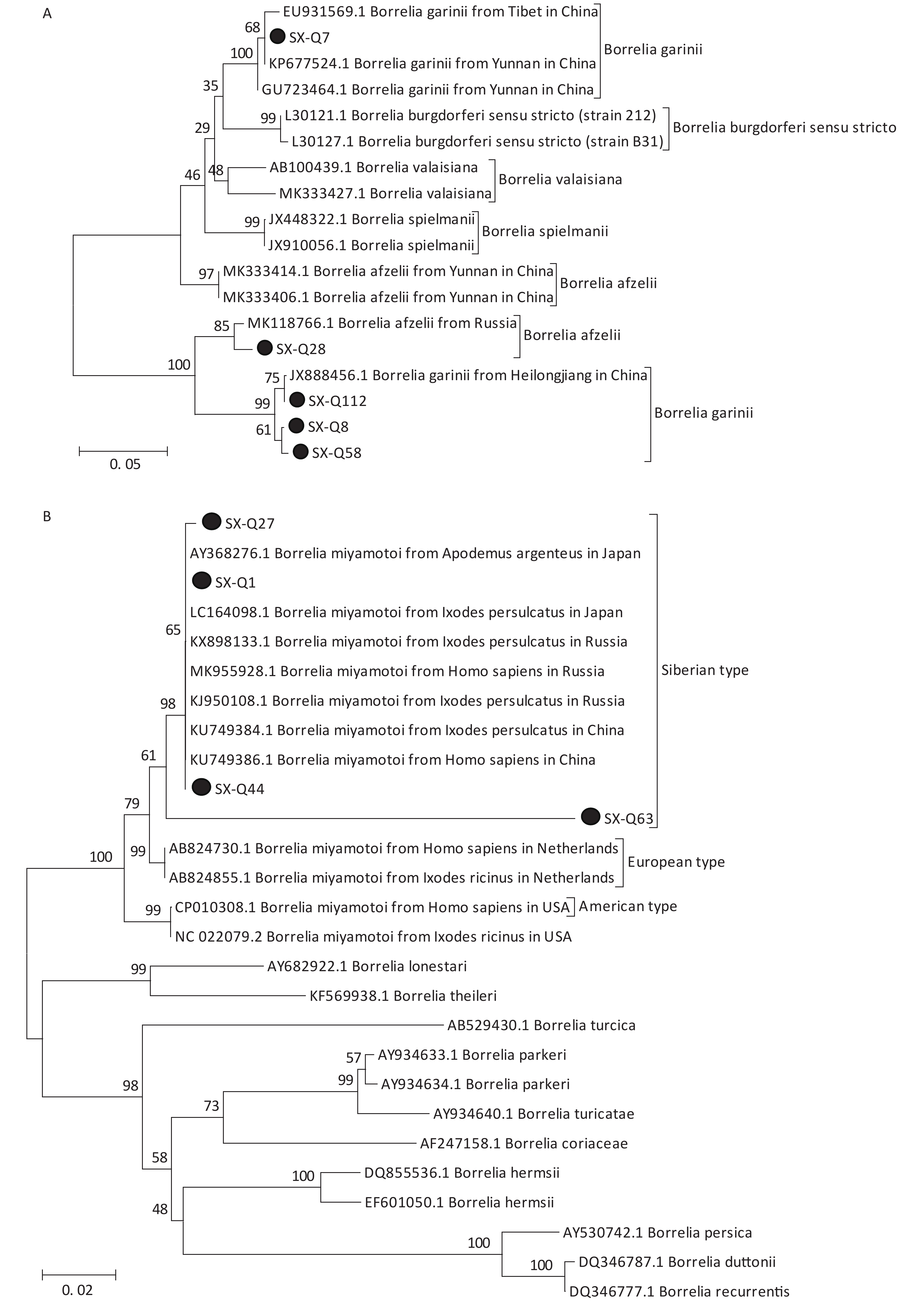
Based on the nested PCR assays, 11 ticks (9.82%) were identified as positive for B. burgdorferi (Table 2). All sequences from the tick samples matched by 99%–100% the registered sequences deposited at the NCBI. Four of the sequences were identical to each other (SX-Q58), with one sequence (SX-Q8) showing a 1-bp difference and another sequence (SX-Q112) showing a 3-bp difference. These six amplicons were closely related to Borrelia garinii from Heilongjiang in China (JX888456.1). Another three sequences were identical to each other (SX-Q7) and were closely related to Borrelia garinii from Yunnan in China (KP677524.1). In addition, two of the 11 sequences were identical to each other (SX-Q28) and were closely related to Borrelia afzelii from Russia (MK118766.1) (Figure 1A). At least two pathogenic genotypes (B. garinii and B. afzelii) occur in Qi County, Shanxi Province, and are also epidemic genotypes in China.
Table 2. Detection of five pathogens in 112 ticks of Qi County, Shanxi Province
Pathogens Detected gene Methods Positive Number Haemaphysalis longicornis
(n = 103)Haemaphysalis japonica
(n = 9)Total
(n = 112)B. burgdorferi 5S-23S rRNA IGS Nested PCR 9 2 11 (9.82%) B. miyamotoi glpQ Nested PCR 26 2 28 (25%) SFGR mopA Conventional PCR 6 6 12 (10.71%) AP 16S rRNA Conventional PCR 0 0 0 EC 16S rRNA RT-PCR 0 0 0 Note. SFGR, Spotted fever group rickettsia; AP, Anaplasma phagocytophilum; EC, Ehrlichia chaffeensis. 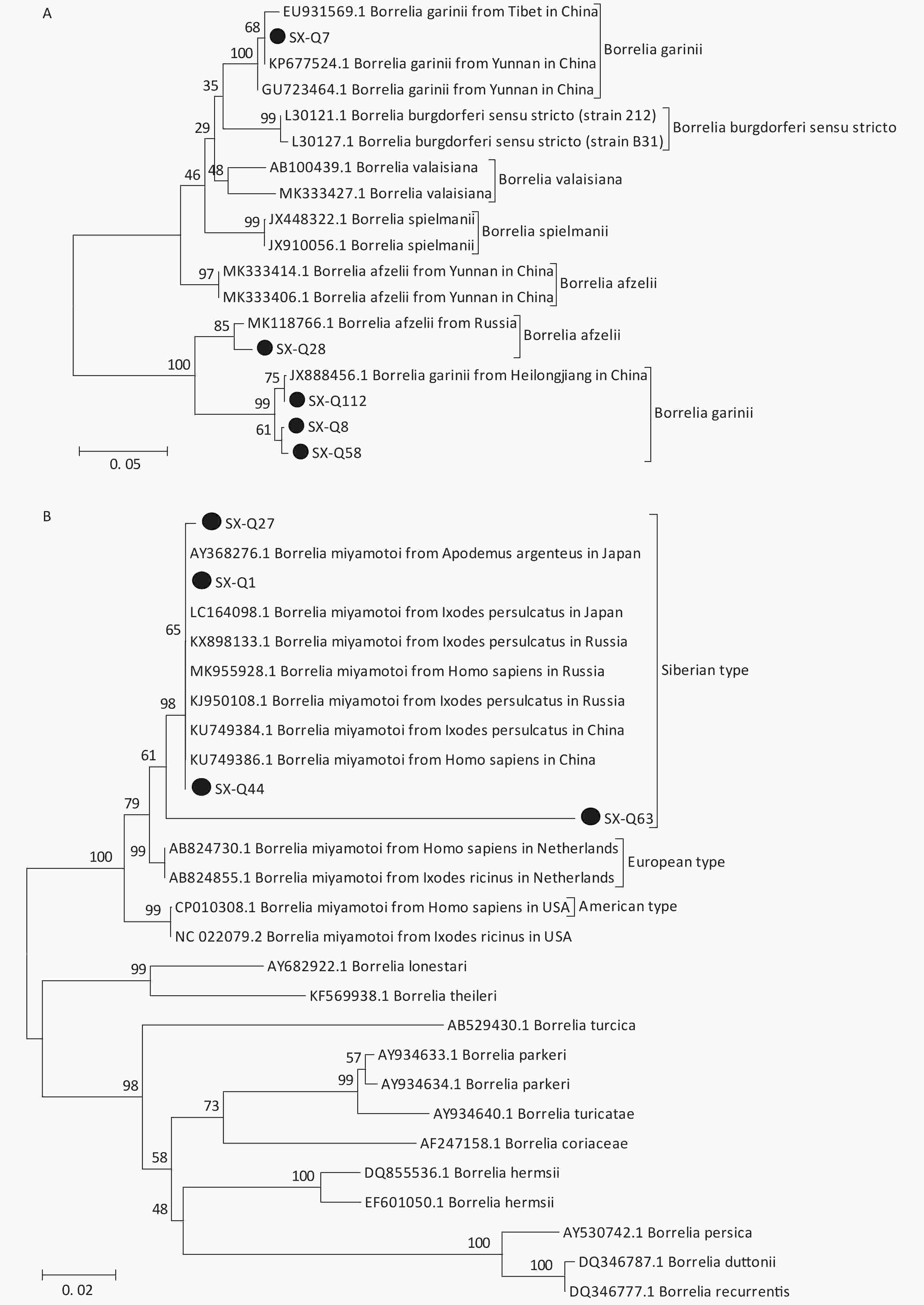
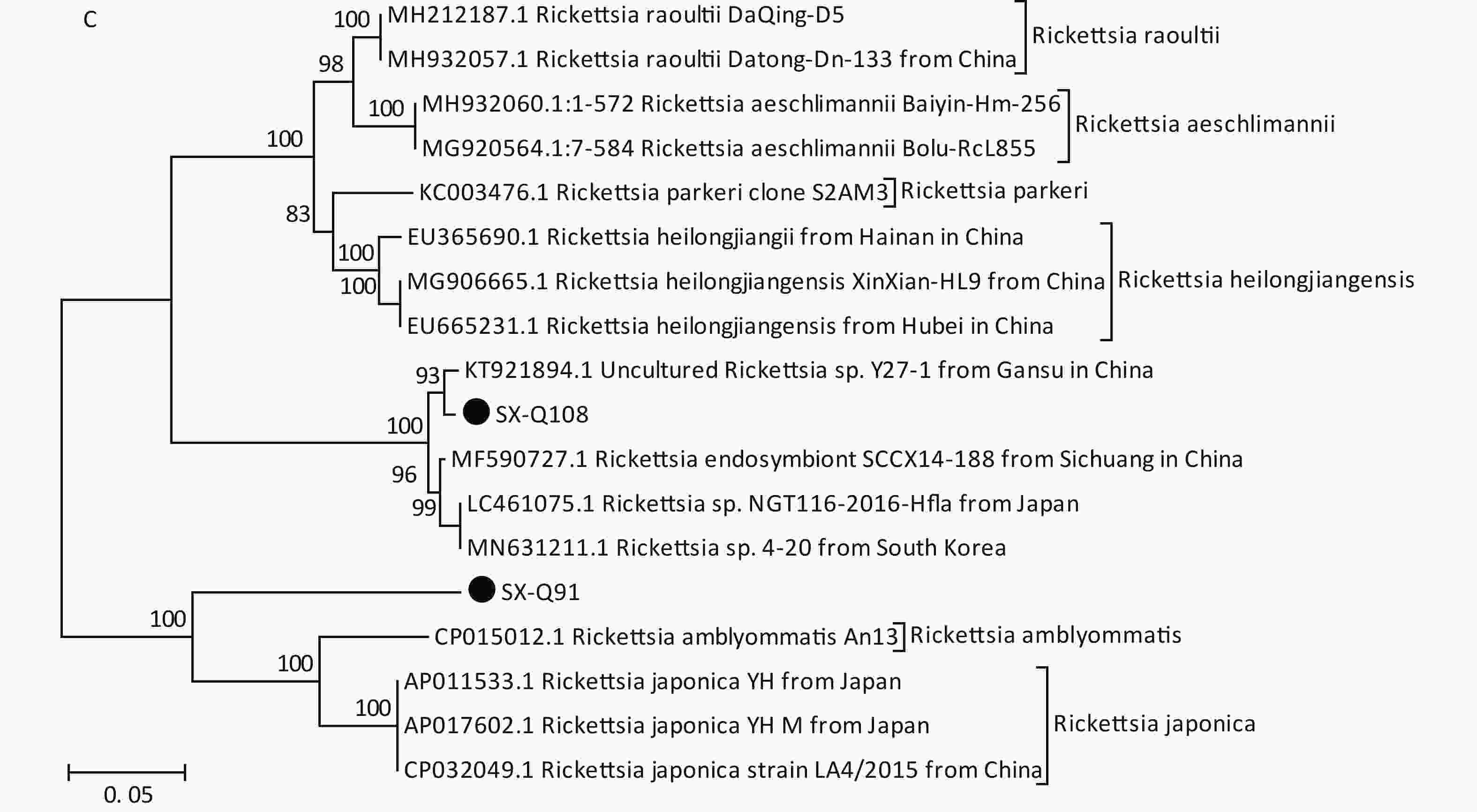
Figure 1. Phylogenetic analyses of target gene sequences of three pathogens from Shanxi province. The sequences obtained in this study are marked as “●”. (A) Sequence analysis for 5S-23S rRNA IGS of B. burgdorferi. (B) Sequence analysis for glpQ gene of B. miyamotoi. (C) Sequence analysis for ompA gene of SFGR.
We detected B. miyamotoi in 28 tick samples (25%) (Table 2). Of this total, 25 sequences were found to be identical to each other (SX-Q1), showing a 1-bp difference in one sequence (SX-Q27) and 1-bp deletion in another sequence (SX-Q44). B. miyamotoi can be divided into three geographically separate genotypes, i.e., Siberian (or Asian), European, and American. These 27 amplicons fall into the same branch as B. miyamotoi from China (KU749384.1, KU749386.1), Japan (AY368276.1, LC164098.1), and Russia (KX898133.1, MK955928.1, KJ950108.1), which belong to the Siberian type. However, one sequence (SX-Q63) showed an 87%–89% similarity with the sequences of B. miyamotoi deposited in the NCBI, with a relatively independent branch on the phylogenetic tree (Figure 1B).
Based on PCR amplification and sequence alignments for the ompA gene of SFGR, 12 ticks were positively identified (10.71%) (Table 2), nine of which were identical to each other (SX-Q108) and the other three had the same sequence (SX-Q91). SX-Q108 and SX-Q91 were distributed in different clusters in the phylogenetic tree (Figure 1C). These results suggest that there may be novel genotypes of SFGR in this area. Isolation efforts combined with the characterization of other genes (rrs, gltA, ompB, and geneD) are warranted. We found no presence of AP or EC in these tick samples.
Coinfection was observed in seven tick samples (6.25%), including one tick having triple coinfection (B. burgdorferi, B. miyamotoi, SFGR) and six with dual coinfection (two with B. Burgdorferi and B. miyamotoi, two with B. burgdorferi and SFGR, two with B. miyamotoi and SFGR). Ticks can become infected with multiple pathogens after a single blood meal from a coinfected host or by feeding on single infected hosts during sequential life stages. Researchers have found that ticks or host animals can be simultaneously infected with multiple pathogens and cases of human infection with multipathogen tick-borne disease also occur in some areas in China. Coinfection in humans induces nonspecific symptoms and makes diagnosis exceedingly difficult. Moreover, coinfection enhances the severity of clinical disease and poses treatment challenges. We recommend that practitioners apply a diagnostic panel rather than a single test when a patient presents with symptoms of tick-borne disease.
In Shanxi Province, clinically confirmed cases of Lyme disease and spotted fever have been reported, and serological examinations have also shown evidence of AP infection and coinfection with Lyme spirochetes. However, no other tick pathogens have been reported in Shanxi. In this study, we screened and successfully identified the presence of three pathogens. It is remarkable that our team identified B.miyatotoi and two pathogenic genospecies of B.b.s.l. (B. garinii, B. afzelii) in questing ticks, which can cause human tick-borne diseases and are associated with significant harm to human health in China. Coinfection of these pathogens was observed in seven samples. In China, B. miyamotoi was first reported in patients and ticks in 2018 in the city of Mudanjiang (Heilongjiang Province) and there have been no other related reports[10]. Thus, this represents the first detection of B.miyamotoi in ticks in central China and the discovery of coinfection of B. miyamotoi with other pathogens in China. It was also the first time that B.miyamotoi was detected from H. longicornis and H. japonica in China. The ticks collected during our study also exhibited an unexpectedly high infection rate (25%, 28/112) of B. miyamotoi. This finding implies that this area may be a natural epidemic focus for B. miyamotoi. Further investigations should be made regarding infections among humans and other hosts.
In China, the main vector of Lyme disease is Ixodes persulcatus; D. nuttalli is the main vector for spotted fever; A. testudinarium, H. yeni, and D. silvarum are vectors for HME; and Ixodes persulcatus, H. longicornis, and H. concinna are suspected to be the main vectors of HGA [3]. However, all of the questing ticks we collected belonged to Haemaphysalis spp., and there is little evidence that H. longicornis and H. japonica are vectors for the transmission of tick-borne spirochaetales and rickettsiales in humans. Many factors affect whether a tick can act as a vector for diseases, including the tick species, climate, the pathogens carried by the ticks, the pathogens carried by livestock and other hosts, the possibility of experiencing tick bites, and human immunity, to name a few. The DNA of potential pathogens in questing ticks is not an indicator of their prevalence, but only a marker of the community of tick-transmitted pathogens circulating in the target territory. However, from the presence of important pathogens in ticks, we can at least infer that some host animals are reservoirs in this area, which may have significant implications for the detection and transmission of tick-borne pathogens.
To clarify the role of these ticks, the 112 tick samples of this study may be insufficient for determining the overall prevalence of pathogens in ticks in this region. As such, the sample size must be increased to obtain more reliable results. Further studies are also needed to characterize tick infections in humans and animals. As the number of tick-borne diseases has continued to increase globally, the infectious microbes associated with these diseases pose a major healthcare threat. Details of the infectious microorganisms transmitted to humans by ticks are not yet fully elucidated, so further research is necessary to gain a better understanding of the complexity of these tick-transmitted enzootic agents on human health. Continuous monitoring of tick-borne pathogens and emerging tick-borne diseases is warranted.
Author Contribution YANG Xiao Na performed the experiments and wrote the manuscript. YANG Hui Jun, CAO Hong Bing, and HAO Rui E participated in the collection of ticks. ZHANG Lin, HOU Xue Xia, and MIAO Guang Qing participated in conducting the experiments. HAO Qin supervised the study and reviewed the manuscript.
doi: 10.3967/bes2021.055
-
&These authors contributed equally to this work.
注释: -
Figure 1. Phylogenetic analyses of target gene sequences of three pathogens from Shanxi province. The sequences obtained in this study are marked as “●”. (A) Sequence analysis for 5S-23S rRNA IGS of B. burgdorferi. (B) Sequence analysis for glpQ gene of B. miyamotoi. (C) Sequence analysis for ompA gene of SFGR.
Table 1. Target genes and primers of tick identification and pathogen detection
Pathogens Target genes Primers Sequence (5′-3′) Size (bp) Reference Tick identification 16S rDNA mitochondrial gene 16s-F CTGCTCAATGATTTTTTAAATTGCTGTGG 460 [4] 16s-R CCGGTCTGAACTCAGATCAAGT B.burgdorferi 5S-23S rRNA IGS rrf-rrl(1f) CGACCTTCTTCGCCTTAAAGC 250 [5] rrf-rrl(1r) TAAGCTGACTAATACTAATTACCC rrf-rrl(2f) TCCTAGGCATTCACCATA rrf-rrl(2r) GAGTTCGCGGGAGA B.miyamotoi glpQ glpQ(1f) CACCATTGATCATAGCTCACAG 424 [6] glpQ(1r) CTGTTGGTGCTTCATTCCAGTC glpQ(2f) GCTAGTGGGTATCTTCCAGAAC glpQ(2r) CTTGTTGTTTATGCCAGAAGGGT SFGR ompA ompA-f ATGGCGAATATTTCTCCAAAA 632 [7] ompA-r GTCCGTTAATGGCAGCATCT AP 16S rRNA AP-f GTCGAACGGATTATTCTTTATAGCTTG 389 [8] AP-r TATAGGTACCGTCATTA CTTCCCTAC EC 16S rRNA EC-f AGCCTAACA CATGCAAGTCGAA 75 [9] EC-r CCCGTC TGCCACTAACAATTATT TaqMan-1 FAM-CAATTGCTTATAACCTTTTG-MGB Note. SFGR, Spotted fever group rickettsia; AP, Anaplasma phagocytophilum; EC, Ehrlichia chaffeensis.  下载: 导出CSV
下载: 导出CSV
Table 2. Detection of five pathogens in 112 ticks of Qi County, Shanxi Province
Pathogens Detected gene Methods Positive Number Haemaphysalis longicornis
(n = 103)Haemaphysalis japonica
(n = 9)Total
(n = 112)B. burgdorferi 5S-23S rRNA IGS Nested PCR 9 2 11 (9.82%) B. miyamotoi glpQ Nested PCR 26 2 28 (25%) SFGR mopA Conventional PCR 6 6 12 (10.71%) AP 16S rRNA Conventional PCR 0 0 0 EC 16S rRNA RT-PCR 0 0 0 Note. SFGR, Spotted fever group rickettsia; AP, Anaplasma phagocytophilum; EC, Ehrlichia chaffeensis.
下载: 导出CSV
-
[1] Mrzljak A, Novak R, Pandak N, et al. Emerging and neglected zoonoses in transplant population. World J Transplant, 2020; 10, 47−63. doi: 10.5500/wjt.v10.i3.47 [2] Telford III SR, Goethert HK, Molloy PJ, et al. Borrelia miyamotoi disease: neither Lyme disease nor relapsing fever. Clin Lab Med, 2015; 35, 867−82. doi: 10.1016/j.cll.2015.08.002 [3] Wu XB, Na RH, Wei SS, et al. Distribution of tick-borne diseases in China. Parasit Vectors, 2013; 6, 119. doi: 10.1186/1756-3305-6-119 [4] Wang YZ, Mu LM, Zhang K, et al. A broad-range survey of ticks from livestock in Northern Xinjiang: changes in tick distribution and the isolation of Borrelia burgdorferi sensu stricto. Parasit Vectors, 2015; 8, 449. doi: 10.1186/s13071-015-1021-0 [5] Zhang L, Miao GQ, Hou XX, et al. Evaluation of nested PCR and real-time PCR in host surveillance of Lyme disease. Chin J Vector Biol Control, 2018; 29, 425−7. (In Chinese [6] Schwan TG, Schrumpf ME, Hinnebusch BJ, et al. GlpQ: an antigen for serological discrimination between relapsing fever and Lyme borreliosis. J Clin Microbiol, 1996; 34, 2483−92. doi: 10.1128/JCM.34.10.2483-2492.1996 [7] Roux V, Fournier PE, Raoult D. Differentiation of spotted fever group rickettsiae by sequencing and analysis of restriction fragment length polymorphism of PCR-amplified DNA of the gene encoding the protein rOmpA. J Clin Microbiol, 1996; 34, 2058−65. doi: 10.1128/JCM.34.9.2058-2065.1996 [8] Overzier E, Pfister K, Thiel C, et al. Anaplasma phagocytophilum in questing Ixodes ricinus ticks: comparison of prevalences and partial 16S rRNA gene variants in urban, pasture, and natural habitats. Appl Environ Microbiol, 2013; 79, 1730−4. doi: 10.1128/AEM.03300-12 [9] Zhang JB, Wen BH, Chen ML, et al. Development of a quantitative real-time polymerase chain reaction assay specific for Ehrlichiae chaffeensis. Acta Parasitol Med Entomol Sin, 2006; 13, 212−6. (In Chinese [10] Jiang BG, Jia N, Jiang JF, et al. Borrelia miyamotoi infections in humans and ticks, northeastern China. Emerg Infect Dis, 2018; 24, 236−41. doi: 10.3201/eid2402.160378 -

 点击查看大图
点击查看大图
图(2) / 表ll (2)
计量
- 文章访问数: 1401
- HTML全文浏览量: 622
- PDF下载量: 64
- 被引次数: 0

 Quick Links
Quick Links