-
The highly pathogenic avian influenza (HPAI) H5N1 virus has caused several outbreaks in domestic poultry. Despite great efforts to control the spread of this virus, it continues to evolve and poses a substantial threat to public health because of a high mortality rate. In this study, we sequenced whole genomes of eight H5N1 avian influenza viruses isolated from domestic poultry in eastern China and compared them with those of typical influenza virus strains. Phylogenetic analyses showed that all eight genomes belonged to clade 2.3.2.1 and clade 7.2, the two main circulating clades in China. Viruses that clustered in clade 2.3.2.1 shared a high degree of homology with H5N1 isolates located in eastern Asian. Isolates that clustered in clade 7.2 were found to circulate throughout China, with an east-to-west density gradient. Pathogenicity studies in mice showed that these isolates replicate in the lungs, and clade 2.3.2.1 viruses exhibit a notably higher degree of virulence compared to clade 7.2 viruses. Our results contribute to the elucidation of the biological characterization and pathogenicity of HPAI H5N1 viruses.
The highly pathogenic avian influenza (HPAI) H5N1 virus was first identified in 1996 in southern China, and it has already spread domestically via poultry reservoirs in numerous countries in the Middle East and rest of Asia. The H5N1 strain can cross the species barrier and infect different animal hosts. Previous studies have shown that H5N1 has infected the human population almost every year since the first confirmed human infection in Hong Kong in 1997, and the majority of infections resulted from direct or indirect contact with infected poultry. It is unlikely that the human population, both regionally and globally, harbors any immunity to this virus. Although there is no evidence of H5N1 transmission among humans, its continuous evolution and diversification add to the challenges of influenza prevention. Control of H5N1 circulation in poultry, the natural animal reservoir of this strain, is the first step in reducing the risk of a pandemic and benefitting global public health [1-2].
Asia, especially China, has the highest frequency of H5N1 outbreaks. In the past two decades, numerous outbreaks of H5N1 infections have occurred in poultry and humans in China. Despite sizeable efforts to control these outbreaks, new clades of H5N1 viruses, which tend to exhibit extreme differences in pathogenicity in infected poultry, are emerging in regions within Asia. New and different strains of localized, circulating H5N1 viruses, originating from phylogenetically distinct clades, greatly increase the possibility of further genetic reassortment and genetic drift/shift. An in-depth characterization of both the phylogenetics and pathogenicity of these geographically localized strains of H5N1 will significantly benefit monitoring of potential outbreaks by health agencies and allow for better contingency should a pandemic arise. There have been previous reports on sporadic outbreaks of H5N1 in Asia, including China (Qinghai province), Mongolia, and South Korea [3]. In our previous study, we found that 226 different subtypes of HPAI influenza virus that were isolated from domestic poultry in northern, southern, and eastern regions of China exhibited a wide range of pathogenicity and genetic characteristics both in vivo and in vitro [4].
To determine genetic features of the uncharacterized potential H5N1 isolates from the Shandong province, we first sequenced whole genomes of all eight viruses, namely A/CK/J01/2009, A/CK/J02/2009, A/CK/J03/2009, A/CK/J04/2009, A/CK/J05/2009, A/CK/J06/2009, A/DK/Y01/2009, and A/DK/Y02/2009 (abbreviated as CKJ01, CKJ02, CKJ03, CKJ04, CKJ05, CKJ06, DKY01, and DKY02, respectively) (Tables S1 and S2, see in the website www.besjournal.com), and compared them to those of 39 representative influenza viruses that have previously been isolated from Asia, especially China and south Asia. The 47 viruses (39 representative influenza viruses and 8 potential H5N1 isolates) were clustered into 11 different clades by phylogenetic analysis of the hemagglutinin (HA) gene according to the WHO influenza (H5N1) nomenclature system (WHO, 2015, Figure 1A). Of the eight newly isolated and sequenced viruses, the HA genes of four were clustered into clade 2.3.2.1, whereas those of the remaining four viruses were clustered into clade 7.2. Based on the phylogenetic tree for HA, the eleven clade 2.3.2.1 viruses, including CKJ01, CKJ06, DKY01, and DKY02 and which were all detected in eastern Asia between 2008-2012, shared high homology with clade 2.3.4 viruses, which were isolated in eastern China. The two clade 2.4 viruses were detected in China in 2004. Clade 2.2 comprised two viruses, which were isolated in China in 2006, and clade 2.5 included two viruses. Four clade 1 viruses were detected in Vietnam in 2008-2012, and two clade 9 viruses were isolated in eastern China in 2004. There is a higher degree of relatedness among viruses belonging to clades 1, 2, and 9, and it is very likely that some of these closely clustered clades of viruses share a common ancestor currently co-circulating in the environment. For example, viruses of clades 2.3.2.1 and 2.3.4 may co-circulate on account of high sequence homology and, in fact, may have all originally evolved from clade 9 viruses. Clade 0 comprised three viruses, two of which were detected in China and the other in Vietnam. The two clade 3 viruses were isolated in the south of China in 2001. Two clade 4 viruses were also detected in China. Clade 7.2 contained thirteen viruses, including CKJ02, CKJ03, CKJ04, and CKJ05. All thirteen of these viruses were distributed throughout China, with high concentrations detected in mainland China. Two of the clade 7.2 viruses were isolated as early as 2005 in the northern provinces of Xinjiang and Hebei. Viruses belonging to clades 0, 3, 4, and 7.2 possibly did not have any significant correlation with the current phylogenetic tree of the HA sequence.
Virus Isotype Province Sublineagea CKJ01 Chicken Shandong Clade 2.3.2.1 CKJ02 Chicken Shandong Clade 7.2 CKJ03 Chicken Shandong Clade 7.2 CKJ04 Chicken Shandong Clade 7.2 CKJ05 Chicken Shandong Clade 7.2 CKJ06 Chicken Shandong Clade 2.3.2.1 DKY01 Duck Shandong Clade 2.3.2.1 DKY02 Duck Shandong Clade 2.3.2.1 Note. aBased on the World Health Organization influenza (H5N1) nomenclature system. Table Table S1. Influenza Virus Isolated from Poultry in China
Virus Isolate ID GISAID Accession No. for Segment PB2 PB1 PA HA NP NA MP NS CKJ01 EPI_ISL_169392 EPI553218 EPI553216 EPI553215 EPI553219 EPI553221 EPI553217 EPI553222 EPI553220 CKJ02 EPI_ISL_169393 EPI553230 EPI553229 EPI553228 EPI553223 EPI553225 EPI553231 EPI553224 EPI553227 CKJ03 EPI_ISL_169416 EPI553282 EPI553281 EPI553280 EPI553276 EPI553278 EPI553283 EPI553277 EPI553279 CKJ04 EPI_ISL_169417 EPI553290 EPI553289 EPI553288 EPI553284 EPI553286 EPI553291 EPI553285 EPI553287 CKJ05 EPI_ISL_169418 EPI553298 EPI553297 EPI553296 EPI553292 EPI553294 EPI553299 EPI553293 EPI553295 CKJ06 EPI_ISL_169419 EPI553306 EPI553305 EPI553304 EPI553300 EPI553302 EPI553307 EPI553301 EPI553303 DKY01 EPI_ISL_169420 EPI553314 EPI553313 EPI553312 EPI553308 EPI553310 EPI553315 EPI553309 EPI553311 DKY02 EPI_ISL_169421 EPI553322 EPI553321 EPI553320 EPI553316 EPI553318 EPI553323 EPI553317 EPI553319 Table Table S2. The GISAID Accession Numbers of H5N1 Viruses Isolated from the Poultry in China

Figure 1. phylogenetic trees of HA(A), NA(B), NP(C), NS(D), PB2(E), PB1(F), and PA(H) genes in the H5N1 infiuenza A viruses. The trees were generated using the distance-based neighbor-joining method (Bootstrap test: 1000 replicates) by the MEGA5.2 software. Horizontal distances are proportional to the genetic distance.
Phylogenetic analyses of the neuraminidase (NA) gene of the eight viruses (Figure 1B) isolated in our study showed two clusters. The four clade 7.2 viruses and three viruses isolated in Shandong, Ningxia, and Jiangsu provinces in 2008-2011 belonged to group 1, indicating that the NA gene segment was still circulating in poultry in China at that time. Group 2 contained viruses belonging to clade 2.3.2.1. We also found that the NA gene segment was circulating in the Asian mainland, especially in eastern Asia. Then, the PB2, PB1, M, PA, NS, and NP segments of the eight viruses were compared and found to cluster into two or three groups. The four viruses categorized as part of clade 7.2 were still tightly clustered, while the three viruses grouped as clade 2.3.2.1 were also part of a highly homologous clade, with the exception of the relatively outlying CKJ01 sequence.
The concentration of clades of viruses may be region-specific. Previous studies showed that the clade 2.2 viruses, which caused large outbreaks in Qinghai Lake in China in 2005, spread from west to central and southern Asia and even to Europe and Africa. In Vietnam, the dominant circulating viruses have been shown to cluster to clade 1 and the clade 2.3.4 viruses [5], whereas the clade 2.3.4 viruses have been shown to circulate in numerous south-east Asian countries, such as Lao PDR, China, and Myanmar. In this study, we found that almost all of the clade viruses were still circulating in China, especially clade 2.3.2.1 and clade 7.2 viruses. The clade 2.3.2.1 viruses were found to circulate mainly in eastern Asia, and clade 7.2 viruses circulated in China from the west to the east. Other clade viruses were also shown to circulate mainly in eastern Asia. There are two main avian migratory flyways across mainland China providing opportunities for contact between different clade viruses [6-7].
The multiple gene products and amino acid sites could explain the virulence of certain influenza viruses in mice. In this study, the deduced amino acid sequences of each segment of the eight viruses were analyzed, and results showed that all viruses in our study were characteristic of HPAI H5N1 variants, based on the presence of multiple basic amino acids at the HA cleavage site (-RRRKR-) (Table S3, see in the website www.besjournal.com) [4]. Amino acids at positions 226 and 228 of the HA segment are Gln(Q) and Gly(G), respectively, confirming the functional receptor binding site of HA protein. The sequence at this site also indicated that these viruses may retain high-affinity binding to α2, 3-NeuAcGal linkages, the avian receptor. Interestingly, a deletion at amino acid positions 49-68 in the NA stalk was detected in CKJ01, CKJ06, DKY01, and DKY02, while a deletion at positions 48-67 was detected in the other four viruses clustering in clade 7.2. The amino acids at positions 119, 274, and 292 in NA in all eight viruses are Gln(E), His(H), and Arg(R), respectively, indicating that these viruses would be sensitive to treatment with the commercially available neuraminidase inhibitors, oseltamivir and zanamivir. Presence of Gln(E) and Asp(D) at positions 627 and 701 in polymerase basic (PB) 2 in all viruses sequenced in this study also confirms a typical characteristic of avian influenza. Substitutions of N30D and T215A at matrix 1 positions could increase the virulence in mice. Both these mutations were detected in all eight viruses, although the mutations associated with the resistance to adamantine drugs were not detected in matrix 2. Finally, all eight viruses were confirmed to harbor a 15-nucleotide deletion from positions 612 to 626 in the NS gene, resulting in a 5-amino acid deletion, which could affect the virulence of influenza virus [8-9].
Virus HA NA PB2 M1 M2 226 228 Cleavage ite 119 274 292 Stalk Deletion (positions) 627 701 30 215 26 31 CKJ01 Q G -RRRKR- E H R 49-68 E D D A L S CKJ02 Q G -RRRKR- E H R 48-67 E D D A L S CKJ03 Q G -RRRKR- E H R 48-67 E D D A L S CKJ04 Q G -RRRKR- E H R 48-67 E D D A L S CKJ05 Q G -RRRKR- E H R 48-67 E D D A L S CKJ06 Q G -RRRKR- E H R 48-67 E D D A L S DKY01 Q G -RRRKR- E H R 49-68 E D D A L S DKY02 Q G -RRRKR- E H R 49-68 E D D A L S Table Table S3. Important Amino acid characteristics in the HA, NA, PB2, M1, M2, and NS1 proteins associated with interspecies transmission and drug resistance of H5N1 virus
To assess the lethality of H5N1 in infected mice, female BALB/c mice were placed under mild anesthesia followed by inoculation with 105 TCID50 of viruses and observed for mortality and changes in body weight for 14 d post inoculation. The experiment was approved by the ethics committee of Beijing Institute of Microbiology and Epidemiology (ID: SYXK2012-005). All live virus experiments were performed in Biosafety Level 3 facilities in accordance with institutional guidelines. As shown in Figure 2, CKJ01, CKJ06, DKY01, and DKY02, clustered in clade 2.3.2.1, were markedly more pathogenic in mice. After 2 weeks post inoculation, mice individually infected with these four isolates lost over 30% of their body weight, and most mice died within the 14-day observation period (Figure 2A, B). Upon infection with clade 7.2 viruses including CKJ02, CKJ03, CKJ04, and CKJ05, the mice were observed to have lost less than 10% of their body weight, and all the mice were alive during the observation period (Figure 2A, B). To determine the minimal dosage of viruses that is lethal to 50% of mice (MLD50), we formed groups of 4-6-week-old female BALB/c mice, inoculated them with 10-fold serial dilutions of the viruses ranging from 100-106 TCID50, and monitored them daily for 2 weeks. The MLD50 values of the four clade 2.3.2.1 viruses ranged from 2.55-2.33 log10TCID50, with the exception of that of DKY01. This particular virus was lethal to all the inoculated mice, even at a TCID50 of 1 (Table 1). No mortality was observed in mice challenged with both clade 7.2 viruses following a 14-day incubation period. We also assessed the kinetics of replication of the H5N1 isolated in mice. The mice were euthanized and their organs, including nasal turbinate, lung, spleen, and brain, were harvested for virus titration in embryonated eggs 5 days post- inoculation. As summarized in Table 1, three viruses, CKJ01, DKY01, and DKY02, replicated to higher titers and could be detected in the lungs, nasal tissue, brain, and spleen of sacrificed mice post inoculation. CKJ06, although detected in lungs, nasal tissue, and spleen of mice, was only detected in undiluted samples obtained from the brain of sacrificed mice post inoculation. CKJ05 was detected in the lungs and nasal tissue of infected mice, while CKJ02, CKJ03, and CKJ04 were only detected in the lungs of infected mice. Neither of these viruses was detected in other organs. During the 5-day observation period, mice infected with CKJ01, CKJ03, DKY01, and DKY02 were more lethargic and developed severe neurological dysfunctions compared to those infected with CKJ02, CKJ03, CKJ04, or CKJ05. These results indicated that avian influenza viruses, isolated from birds, could indeed infect mice. Viruses belonging to clade 2.3.2.1 displayed a high degree of lethality in mice, whereas viruses belonging to clade 7.2 were comparatively mild in their pathogenicity in infected mice. Moreover, these new isolates of H5N1, found circulating in poultry, can replicate in mouse models. Importantly, viruses grouped in clade 2.3.2.1 replicated more effectively in most of the examined organs compared to viruses grouped in clade 7.2.
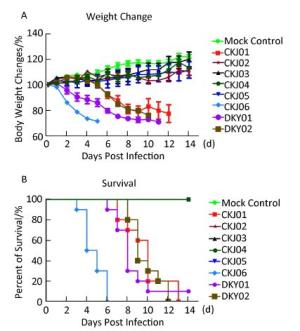
Figure 2. Pathogenicity of H5N1 influenza A viruses in mice. Body weight changes (A) and survival rates (B) of mice incubated with different H5N1 viruses. Groups of five mice each were inoculated intranasally with 105 TCID50 (50 μL) or with the same volume of allantoic fluid as the mock control and monitored daily for 14 d post infection.
Virus Virus Replication in Organs (log10EID50/mL ± SDb) MLD50 Lung Brain Spleen Nasal CKJ01 3.83 ± 0.47 1.55 ± 0.08 3.50 ± 0.00 6.39 ± 0.08 2.75 CKJ02 1.98 ± 0.05 NDc ND ND > 6 CKJ03 1.33 ± 0.00 ND ND ND > 6 CKJ04 1.67 ± 0.21 ND ND ND > 6 CKJ05 2.44 ± 0.16 ND ND 1.94 ± 0.40 > 6 CKJ06 5.16 ± 0.36 Dd 0.89 ± 0.55 2.94 ± 0.91 2.55 DKY01 5.44 ± 0.16 4.33 ± 0.00 2.39 ± 0.08 5.55 ± 0.08 < 1.00 DKY02 5.78 ± 0.32 2.39 ± 0.80 3.50 ± 0.00 5.39 ± 0.08 3.23 Note. aSix-week-old BALB/c mice were used for this study. bStandard-deviation. cThe data were not detected. dThe data were only detected in undiluted samples. Table 1. Replication of H5N1 Viruses in Micea
Influenza viruses are still considered to be more significant than other viruses, such as Zika virus [10]. Even though these viruses were isolated and identified years ago, the elucidation of the origin, reasons for persistence, and process of continuous evolution of these viruses is essential for disease prevention. In addition, whether the isolated virus function as an intersection of the ‘gene pool’ circulating in Asia warrants further investigation. The phylogenetic and biological diversity of eight HPAI H5N1 viruses isolated from domestic poultry in eastern China were investigated, and the replication and pathogenicity of these viruses were assessed in mice. Our data suggest thatAsiamight still be the main source of H5N1 virusses, resulting in epidemics. The current co-existence of multiple clades of these viruses as well as opportunities to co-infect avian reservoirs will lead to the emergence of new isolates and potentially make these viruses highly virulent in mammalian hosts. It is quite feasible that high frequencies of genetic reassortment and virus co-infections may lead to an increase in the transmissibility of these strains to humans. Further studies will benefit government monitoring of H5N1 viruses and the planning for potential future pandemics.
Conflict of interest The authors declare no financial or commercial conflicts of interest.
HTML
and the National High Technology Research and Development Program of China 2015AA020924 and 2013ZX10004003
This work was supported in part by the funding from the National Natural Scientific Foundation 81370518
YANG Penghui was supported by a grant from the Beijing Nova Program No.Z141107001814054

 Quick Links
Quick Links
 DownLoad:
DownLoad: