
-
Liver cancer accounts for 9.1% of all cancer deaths worldwide. Almost 83% of all liver cancer cases are diagnosed in underdeveloped nations [1], with 50% of global liver cancer diagnoses and deaths registered in China [2]. Hepatocellular carcinomas (HCCs) account for more than 75%–90% of all liver cancer cases, while the incidence rate of liver cancer increases fastest among all forms of cancer [3,4]. Many advanced therapies have positively impacted the diagnosis of HCC, but, owing to a high incidence of metastasis, the overall survival of HCC patients remains poor [5,6]. In addition, the continuously high rate of HCC recurrence results in poor survival [7]. Therefore, it is necessary to investigate urgently needed potentially potent targets for the early diagnosis and efficient treatment of HCC.
Programmed cell death 6 (PDCD6), also known as apoptosis-linked gene-2 (ALG-2), is a ubiquitously expressed calcium-binding protein. The PDCD6 gene encodes five serially repetitive EF-hand structures and an open reading frame of 191 amino acids [8]. Several studies have evaluated PDCD6 expression in clinical samples of tumor tissues or cell lines, reporting inconsistent impacts of PDCD6 in different tumors and species. PDCD6 is ubiquitously expressed in adult mouse tissues. In rats, PDCD6 is expressed at higher levels in hepatoma tissues than in normal liver tissues [9]. PDCD6 is highly expressed in human lung cancer tissue and metastatic ovarian cancer cells [9,10]. In addition, PDCD6 positively regulates the metastatic ability of ovarian cancer cells [10]. In contrast, low levels of PDCD6 expression were found in non-small cell lung cancer and gastric cancer [11,12]. Because PDCD6 has not yet been identified as an oncogene or tumor inhibitor in HCC, we investigated these potential roles of PDCD6 in human HCC tissue and cancer cell lines.
The high incidence of cell migration in HCC contributes to the poor prognosis of HCC patients. Epithelial-mesenchymal transition (EMT) was shown to play a crucial part in increasing the frequency of cancer metastasis [13,14]. Many signaling pathways, including Wnt/β-catenin and phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT), have been implicated in the regulation of EMT in cancer [15]. Furthermore, the same pathways are also critical for tumor proliferation [16,17]. AKT is a serine/threonine kinase activating many biological processes, including proliferation, cell death, metastasis, and angiogenesis [16,17]. Abnormally phosphorylated AKT is associated with malignancy and poor prognosis [18]. In HCC, the enhancement of AKT activity (phosphorylation status) promotes intracellular events, such as the inactivation (phosphorylation) of glycogen synthase kinase-3β (GSK3β) [19]. The phosphorylation at Ser9 blocks the catalytic activity of GSK3β, leading to increased accumulation of nuclear β-catenin that promotes the expression of carcinogenic factors, namely T-cell factor/lymphoid enhancer-binding factor (TCF/LEF), which in turn stimulate cell growth and metastasis [20].
The above evidence suggests that the exploration of the potential molecular mechanism may ultimately identify innovative therapeutic strategies for HCC. Our aim in this study was to explore the effect and mechanism of PDCD6 in HCC. We found that PDCD6 acted as a potent oncogene of HCC, promoting the proliferation and metastasis mediated by the AKT/GSK3β/β-catenin pathway. In conclusion, our results identify PDCD6 as a significant diagnostic and therapeutic target in HCC.
-
Human liver cancer cell lines HepG2 and Hep3B (American Type Culture Collection; MD, USA) and human normal hepatocyte cell line LO2 (Chinese Academy of Sciences, China) were cultured in Dulbecco’s modified eagle medium (DMEM) (Gibco, Thermo Fisher Scientific, USA) with 10% fetal bovine serum (Gibco, Thermo Fisher Scientific) and 1% penicillin/streptomycin (Gibco, Thermo Fisher Scientific) at 37 °C in 5% CO2. LY294002 (CAS# 154447-36-6; Sigma-Aldrich Co., MO, USA) was dissolved by DMSO as stock solution (20 mmol/L) and was diluted with culture medium prior to use (cells treated with 10 μmol/L LY294002).
-
We used a methylthiazol tetrazolium (MTT) assay, as implemented in the MTT Cell Proliferation Assay Kit (Beyotime, China), to assess cell metabolism (an indicator of cell viability and growth) and cytotoxicity of experimental drugs (LY294002) on HCC cells [21]. For this assay, HepG2 and Hep3B cells with stable expression or knockdown of PDCD6 were planted into 96-well plates (3,000 cells/well) and incubated with MTT for 4 h before they were placed in complete DMSO for 10 min. Metabolic activity, indicated by a change in optical density, was determined by the absorbance measured at 570 nm.
-
The cell’s migration and invasion ability were measured by using the transwell migration assay implemented in an HTS transwell-24 system with a transwell membrane of 8 μm pore size (Corning; NY, USA) [21]. In brief, 200 μL of cell suspension in serum-free DMEM medium was placed in the upper chamber, and 600 μL culture medium with 10% serum was placed in the lower chamber for the chemo-attraction of cells. After culturing for 48 h at 37 °C in 5% CO2, the cells sequestered in the upper chamber were deemed, by definition, non-metastatic, while the cells that passed through the membrane into the lower chamber were deemed invasive cells. The cells were fixed by methyl alcohol for 10 min and stained with 0.1% crystal violet for 10 min before the upper cells were removed with cotton swabs. Metastatic cells collected from five randomly selected areas of the upper chamber were manually counted under a light microscope. For the invasion assay, adhesion to Matrigel (a model of the basal layer of the epithelium) added to the upper chambers was monitored.
-
Western blotting assay was conducted according to the standard protocol [21]. All antibodies were diluted in a mixture (20:1) of Tris-buffered saline with Tween, as follows:
p-AKT (1:1,000), AKT (1:1,000), GSK3β (1:2,000), p-GSK3β (1:2,000), β-catenin (1:2,000), Vimentin (1:1,000), and E-cadherin (1:1,000), all purchased from Cell Signaling Technology (CST; MA, USA);
PDCD6 (1:2,000), β-actin (1:2,000), PCNA (1:2,000), β-tubulin (1:2,000), and the HRP-conjugated goat anti-rabbit/mouse secondary antibody (1:5,000), all purchased from Proteintech (China).
-
Total RNA was extracted from cells by using TRIzol reagents (TransGen, China). Reverse transcription of RNA into complementary DNA (cDNA) was performed using the PrimeScript RT reagent kit (TaKaRa, Japan) with the manufacturer’s protocol. RT-qPCR was performed by using the SYBR® Green Realtime PCR Master Mix (TaKaRa). Cycle threshold (Ct) was determined separately for each gene from the target fold-changes by the 2−ΔΔCT method after standardizing amplification to β-actin (the internal control). Each experiment was performed in triplicate. The following primers were used for RT-qPCR:
PDCD6
5′-ATGGCCGCCTACTCTTACC-3′ (Forward)
5′-TCCTGTCTTTATCGACCCTCTG-3′ (Reverse)
CCND1
5′-TGTCCTACTTCAAATGTGTGCA-3′ (Forward)
5′-ATTGGAAATGAACTTCACATCTGT-3′ (Reverse)
c-MYC
5′-TGAGGAGACACCGCCCAC-3′ (Forward)
5′-CAACATCGATTTCTTCCTCATCTTC-3′ (Reverse)
MMP-7
5′-CAGATGTGGAGTGCCAGATG-3′ (Forward)
5′-TGTCAGCAGTTCCCCATACA-3′ (Reverse)
MMP-9
5′-CTCTGGAGGTTCGACGTGAA-3′ (Forward)
5′-GGCTTTCTCTCGGTACTGGA-3′ (Reverse)
β-actin
5′-CATGTACGTTGCTATCCAGGC-3′ (Forward)
5′-CTCCTTAATGTCACGCACGAT-3′ (Reverse)
-
We used the recombinant lentivirus vector pLVX-IRES-GFP-puro loaded with cloned PDCD6 sequences, whole gene or fragments, to infect the human HCC cell lines and induce overexpression or knockdown of PDCD6, respectively. For overexpression, the whole PDCD6 gene sequence was cloned. For the knockdown, two custom-designed shRNA based on hairpin sequences of the PDCD6 gene were cloned into the pLKO.1 plasmid vector. The human PDCD6 shRNA sequences were as follows:
sh PDCD6 #1:
CCGGCAACTCCGGGATGATCGATAACTCGAGTTATCGATCATCCCGGAGTTGTTTTTG
sh PDCD6 #2:
CCGGGCAGAGGTTGACGGATATATTCTCGAGAATATATCCGTCAACCTCTGCTTTTTG
Recombinant lentiviral particles were made by transiently transfecting 293T cells. Briefly, using Lipofectamine 2,000, 4 μg of the PDCD6 overexpression or shRNA plasmid, 2 μg of the psPAX2 plasmid, and 1 μg of pMD2.G plasmid were simultaneously transfected into 293T cells seeded in 60-mm culture plates. After incubation for 48 h at 37 °C in 5% CO2, the supernatant of the cell culture containing the virus was collected and filtered. The harvested recombinant lentiviruses were used to infect HCC cells by incubating them together for 48 h after adding 3 μg/mL polybrene. After incubation, HCC cells with PDCD6 overexpression or knockdown were screened with 2 μg/mL puromycin before measuring the expression levels of PDCD6 in the cells by using Western blotting and RT-qPCR.
-
RNA-seq data obtained from Liver Hepatocellular Carcinoma samples were downloaded directly from The Cancer Genome Atlas (TCGA) data portal (https://portal.gdc.cancer.gov/projects/TCGA-LIHC). Of the 371 liver hepatocellular carcinoma cases represented in the extracted data, we selected for further analysis the 50 cases that had paired data available for the primary HCC and adjacent normal tissues. We quantified gene expression levels in terms of FPKM (fragments per kilobase million) based on the RNA-seq results.
-
The PDCD6 data extracted for the online liver hepatocellular carcinoma samples were analyzed by using Prism (version 8.0, GraphPad; CA, USA). The Kaplan–Meier survival analysis was performed to evaluate the prognostic value of PDCD6 using the Kaplan–Meier Plotter Online tool (
http://kmplot.com/analysis/ ). The two-tailed Student’s t-test implemented in Prism was used to identify the differentially expressed genes between the primary tumor and the adjacent normal samples. P < 0.05 was defined as statistically significant. -
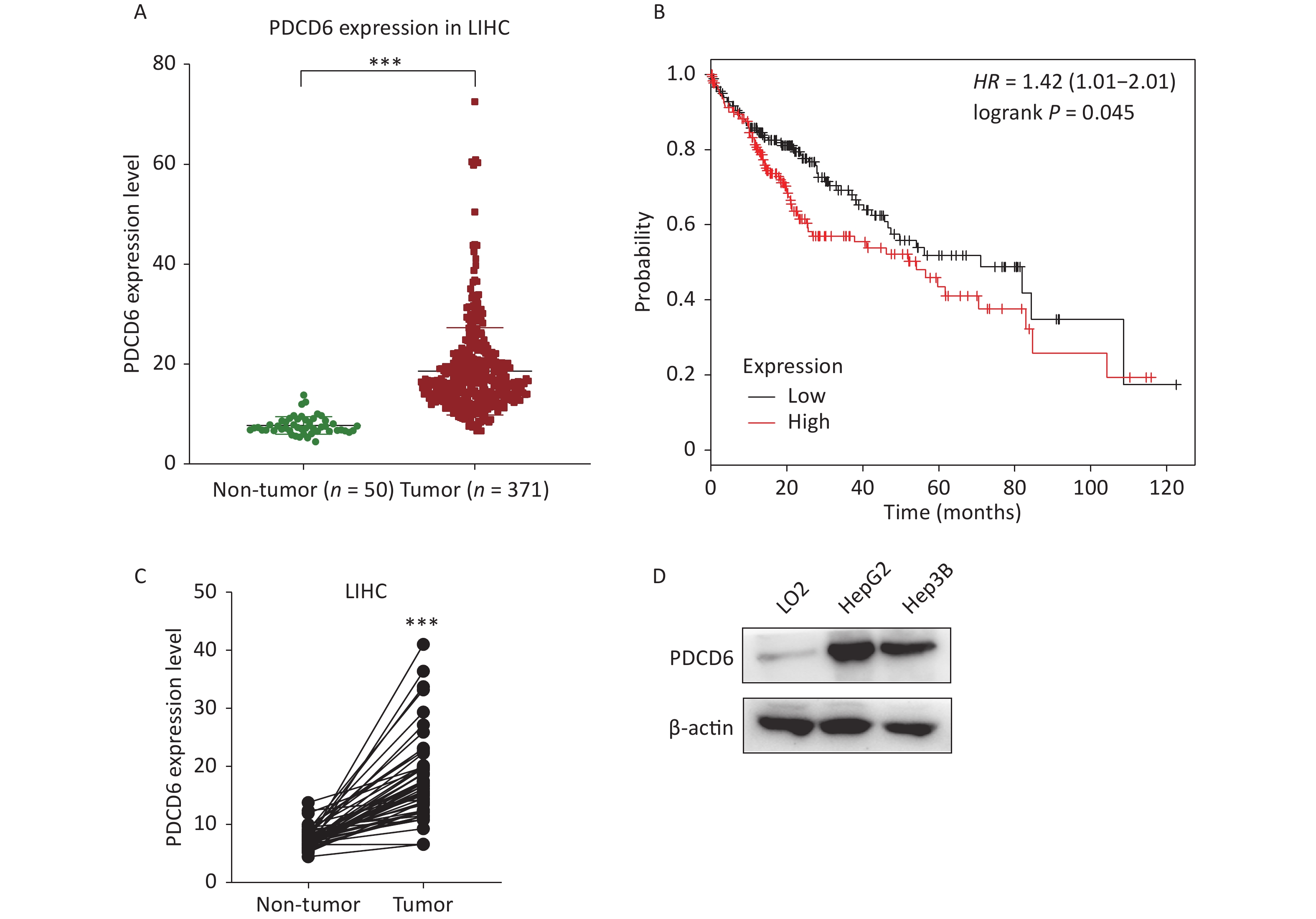
To examine the clinical significance of PDCD6 in HCC, we analyzed in silico the correlation between PDCD6 expression levels and HCC case outcomes using RNA-seq data from the TCGA project. The expression levels for PDCD6 were significantly upregulated in the 371 HCC patients compared with the 50 healthy individuals in the database (Figure 1A, P < 0.001). Moreover, the Kaplan–Meier survival analysis indicated a significantly shorter overall survival in patients with high levels of PDCD6 expression, as indicated by a hazard ratio of 1.4 (Figure 1B, log-rank test, P = 0.045). These findings suggest that high expression levels of PDCD6 are correlated with progressed stages of HCC. To further test the clinical significance of PDCD6 in HCC, the 50 paired data sets for primary tumor and adjacent non-tumor samples were selected from the TCGA data for further analysis. A two-tailed paired two-sample Student’s t-test revealed that PDCD6 levels were significantly higher in tumor tissues than in adjacent non-tumor tissues (Figure 1C, P < 0.001). We confirmed this result in a cell culture experiment, where we observed that PDCD6 expression was respectively increased by 8.171 and 6.321 folds in HepG2 and Hep3B cells relative to the levels measured for normal hepatocyte cell lines (Figure 1D). These findings strongly suggest that PDCD6 may play a crucial role in HCC.
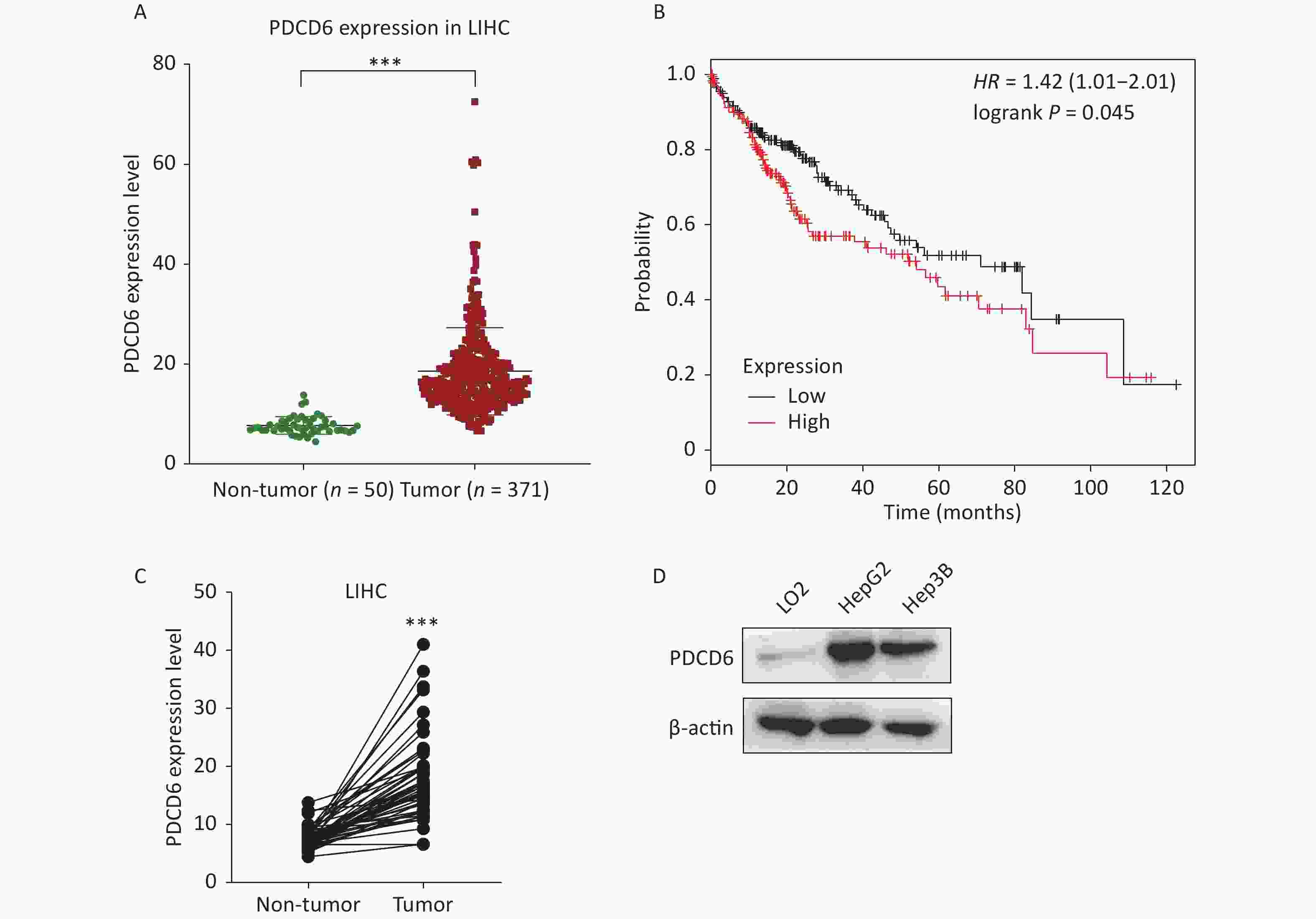
Figure 1. PDCD6 is significantly upregulated in human hepatocellular cancer tissues and cell lines. (A) PDCD6 expression levels extracted for 371 cases (median 18.57 unit) with tumors and 50 non-tumor cases (median 7.712 unit) from the TCGA database. (B) Kaplan-Meier survival analyses of patients with liver hepatocellular carcinoma (LIHC). High PDCD6 expression was associated with short survival (hazard ratio = 1.42). (C) PDCD6 levels in 50 LIHC tissues (median 17.95 unit) and 50 adjacent normal tissues (median 7.712 unit) from the TCGA database. (D) PDCD6 protein expression levels were detected by Western blotting in a normal hepatocyte cell line (LO2) (median 1.015 unit) and two HCC cell lines (HepG2 and Hep3B) cells (medians 8.171 unit and 6.321 unit, respectively). ***P < 0.001.
-
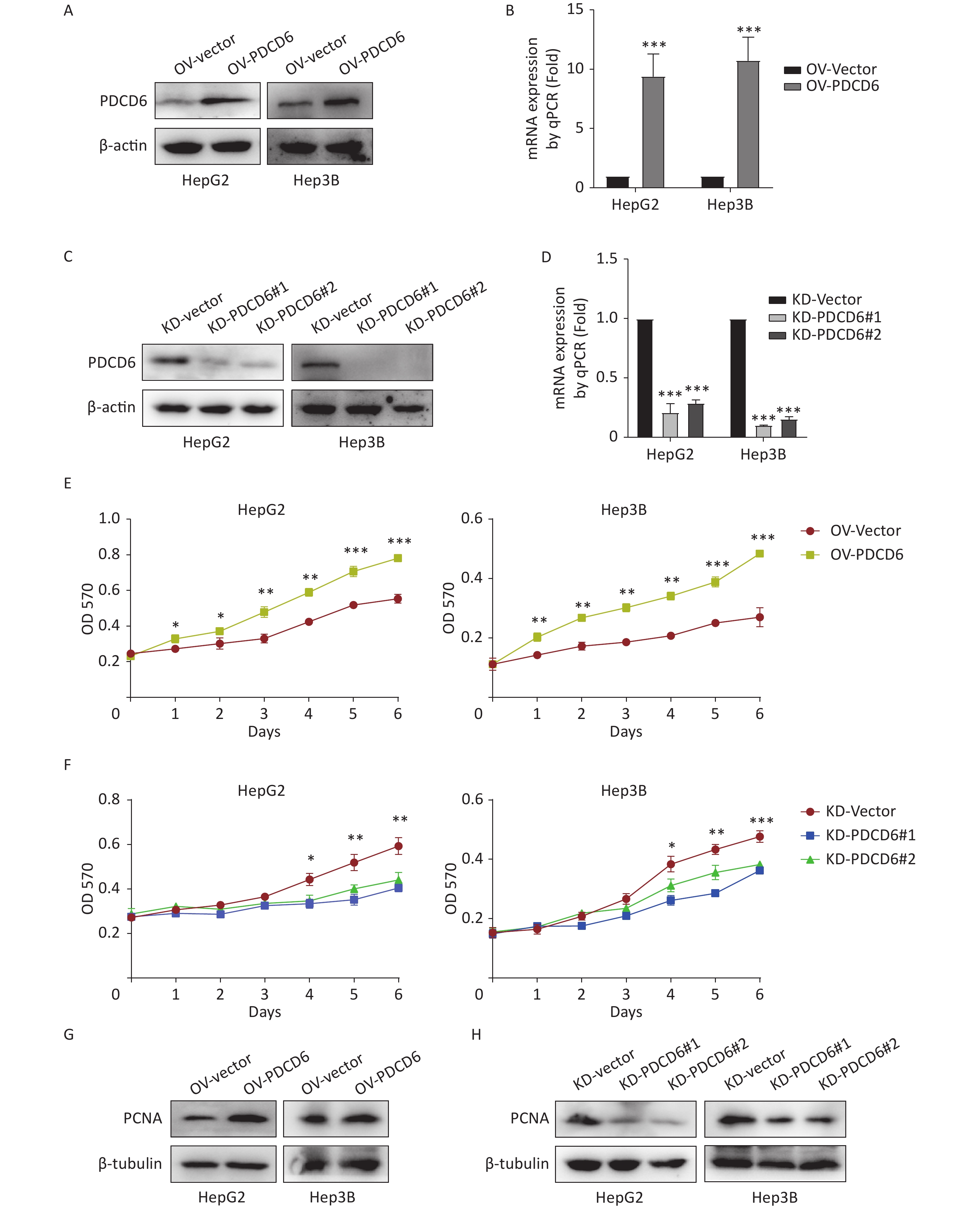
To ascertain the latent effect of PDCD6 in HCC progression, PDCD6 overexpression (OV) (Figure 2A–B) or knockdown (KD) (Figure 2C–D) in HepG2 and Hep3B cells were established using two alternative recombinant lentiviral systems and confirmed by Western blotting (Figure 2A, C) and RT-qPCR (Figure 2B, D). The MTT assay revealed that cell metabolic activity (a measure of cell viability) was significantly increased in HCC cells with PDCD6 overexpression (Figure 2E) and, in contrast, was significantly reduced in HCC cells with PDCD6 knockdown (Figure 2F).
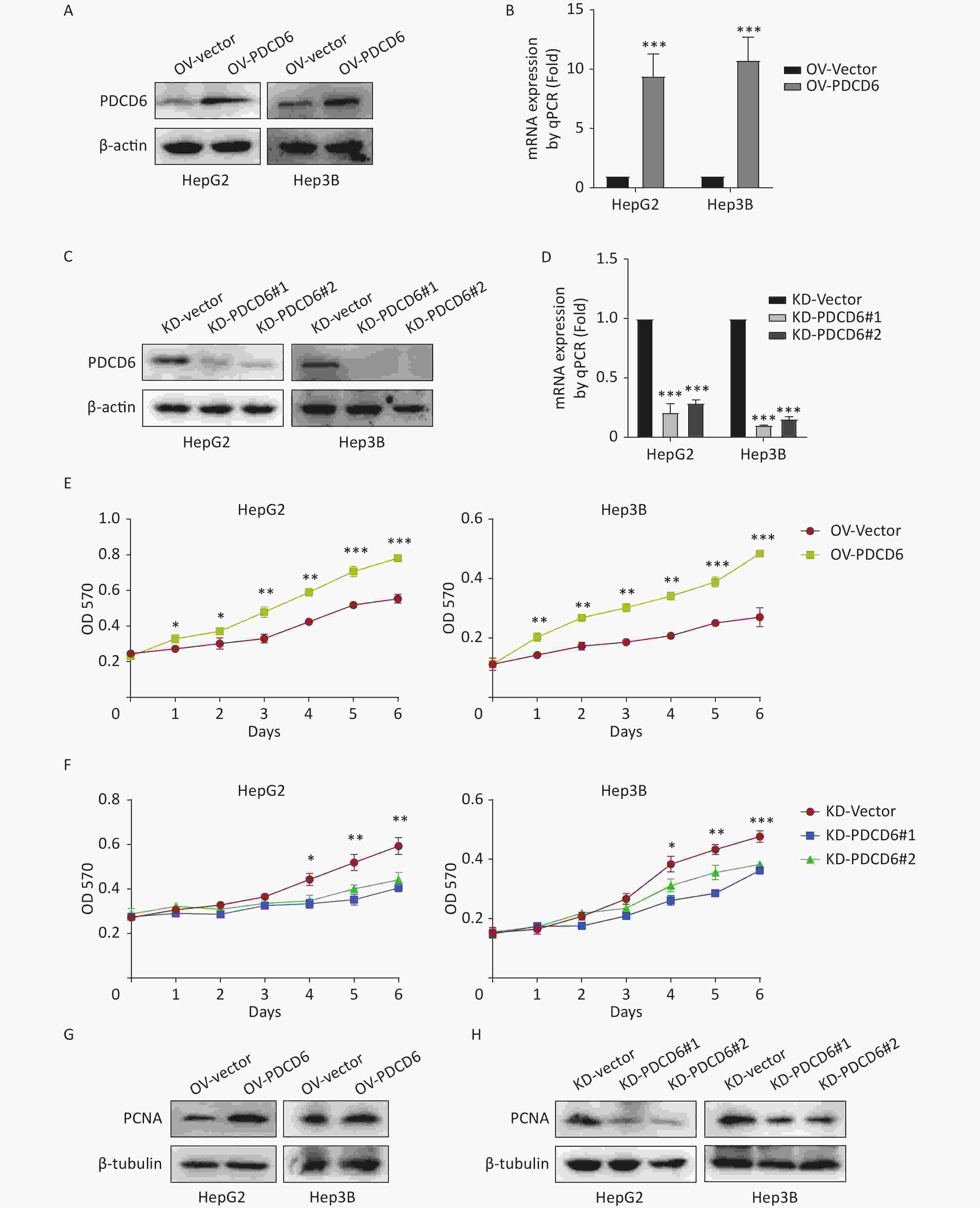
Figure 2. PDCD6 promotes cell proliferation in human HCC cell lines. Recombinant lentiviruses were used to establish (A, B) PDCD6 overexpression or (C, D) PDCD6 knockdown in HepG2 and Hep3B cells, which in turn were determined by (A, C) Western blotting and (B, D) RT-qPCR. MTT assay was performed to determine the effect on cell proliferation in HepG2 and Hep3B cells by (E) PDCD6 overexpression or (F) knockdown. The expression of proliferation-related protein PCNA was detected by western blotting analysis for HCC cells with (G) PDCD6-overexpression or (H) PDCD6-knockdown (H). Data shown in B, D, E, and F are the mean ± SD of three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
Proliferating cell nuclear antigen (PCNA), a key marker of cell proliferation, was increased when PDCD6 was overexpressed in HCC cells (Figure 2G) but decreased in HCC cells of PDCD6 knockdown (Figure 2H). These data indicated that PDCD6 was involved in the regulation of HCC cell proliferation.
-
Next, we investigated whether PDCD6 affected the migration and invasion of the HCC cells. We found that HepG2 and Hep3B cells stably overexpressing PDCD6 exhibited significantly heightened cell migration (Figure 3A and 3B) and significantly heightened ability to invade through the extracellular matrix coating (Figure 3C and 3D).
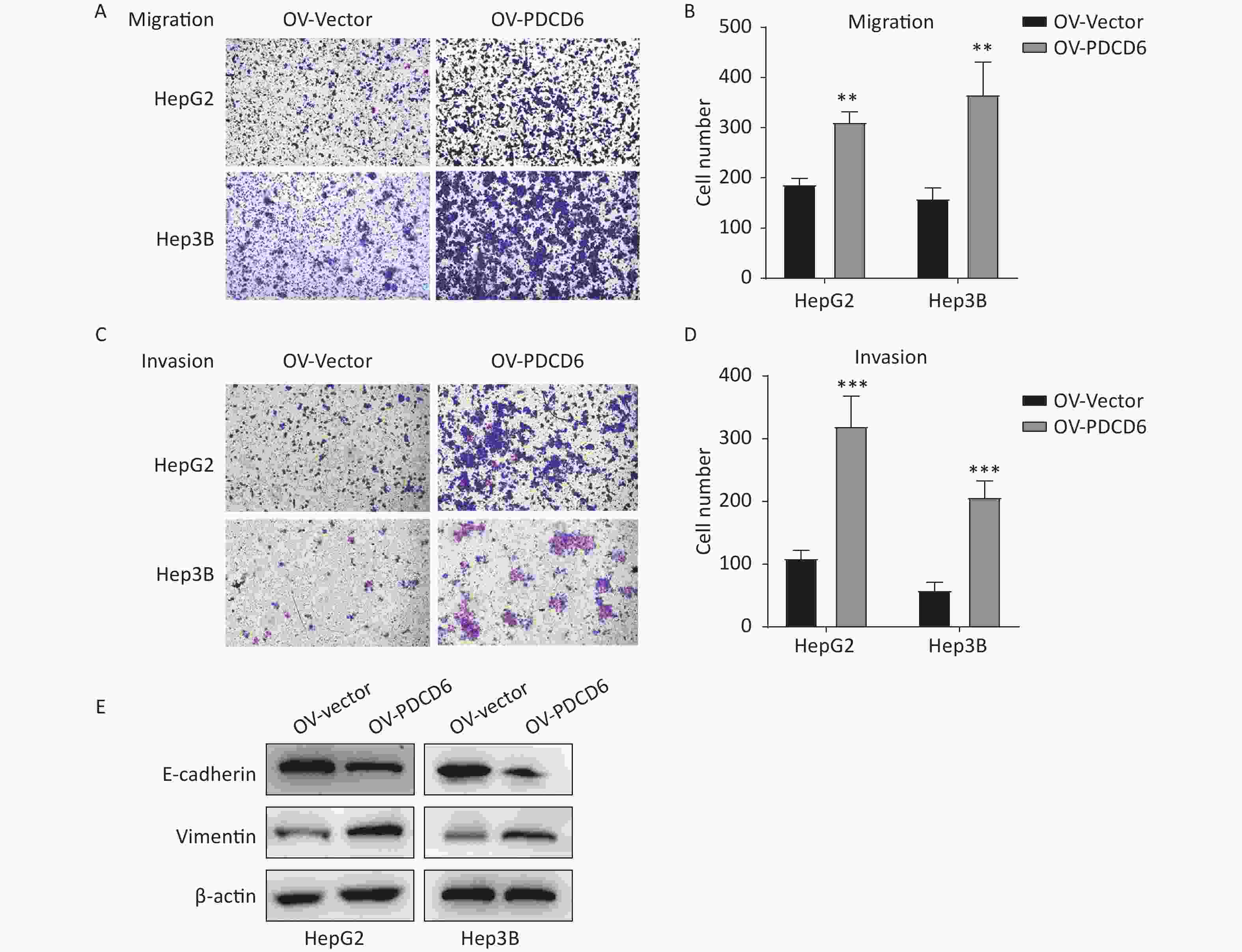
Figure 3. PDCD6 overexpression promotes migration and invasion in HCC cells. The transwell assays were used to determine the ability for (A, B) migration or (C, D) invasion in HepG2 and Hep3B cells with PDCD6 overexpression. (E) Expression levels of EMT marker proteins E-cadherin and vimentin were detected in the PDCD6-overexpressing HCC cell lines using Western blotting. The data shown are the mean ± SD of three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
In cancer cells, the progress of EMT is closely associated with cell metastasis. Thus, we examined the effect of PDCD6 on the expression level of EMT-related markers, such as E-cadherin and vimentin, in HepG2 and Hep3B cells. The results revealed that PDCD6 overexpression was associated with downregulated E-cadherin in combination with increased expression of vimentin, demonstrating that PDCD6 levels had a significant influence on the transformation from epithelial to mesenchymal phenotypes (Figure 3E).
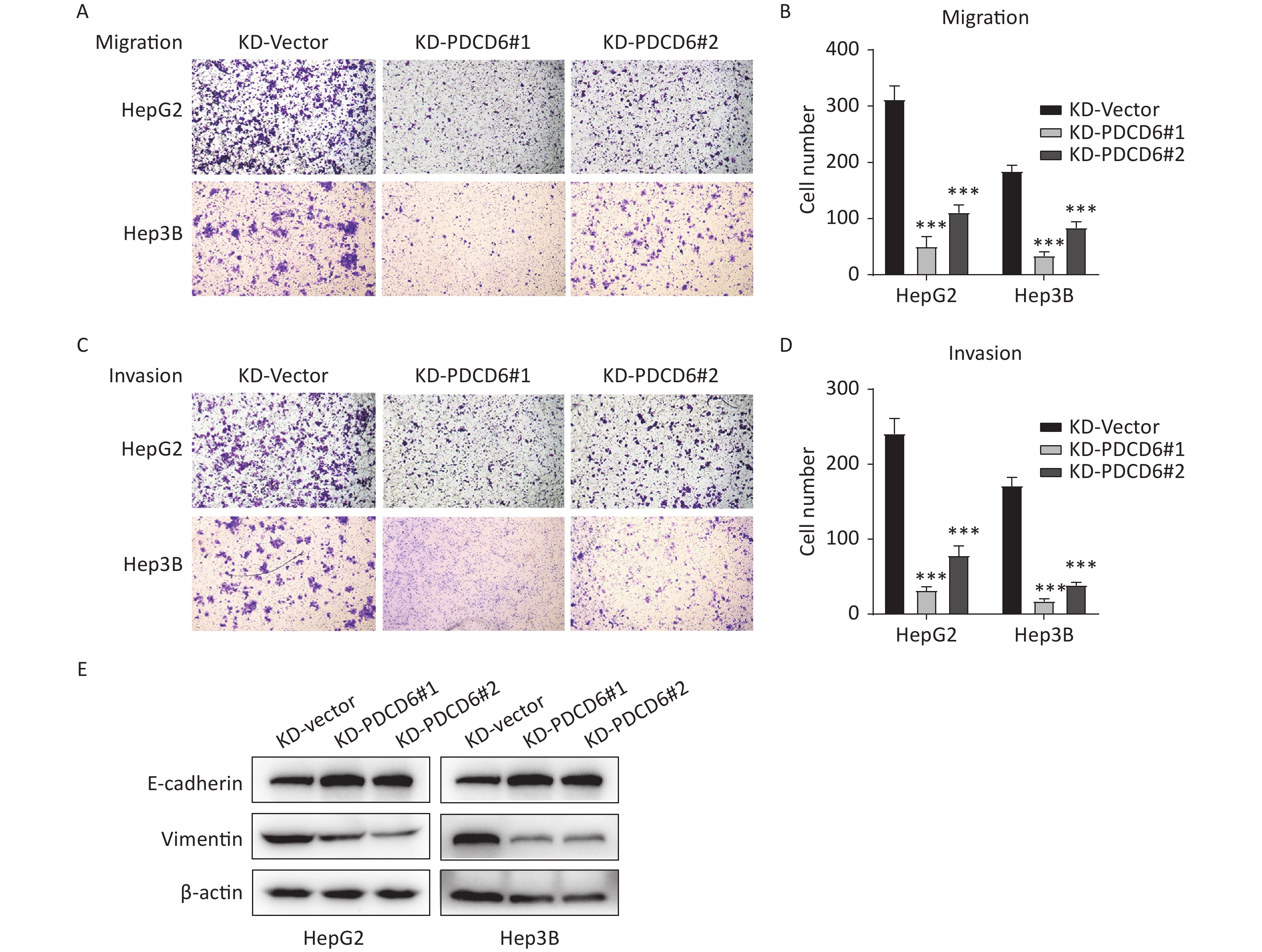
A similar analysis revealed the opposite results in the cells with PDCD6 knockdown. Compared with the control, the number of migrated cells was reduced (Figure 4A and 4B), and the ability for cell invasion was decreased (Figure 4C and 4D) in HCC cells with downregulated PDCD6 expression. PDCD6 knockdown was also associated with the inhibition of the EMT progress through the upregulation of E-cadherin and lowered levels of vimentin (Figure 4E), the opposite of the effects found for PDCD6 overexpression. In short, PDCD6 contributes to the augmented metastasis of HCC cells by inducing EMT.
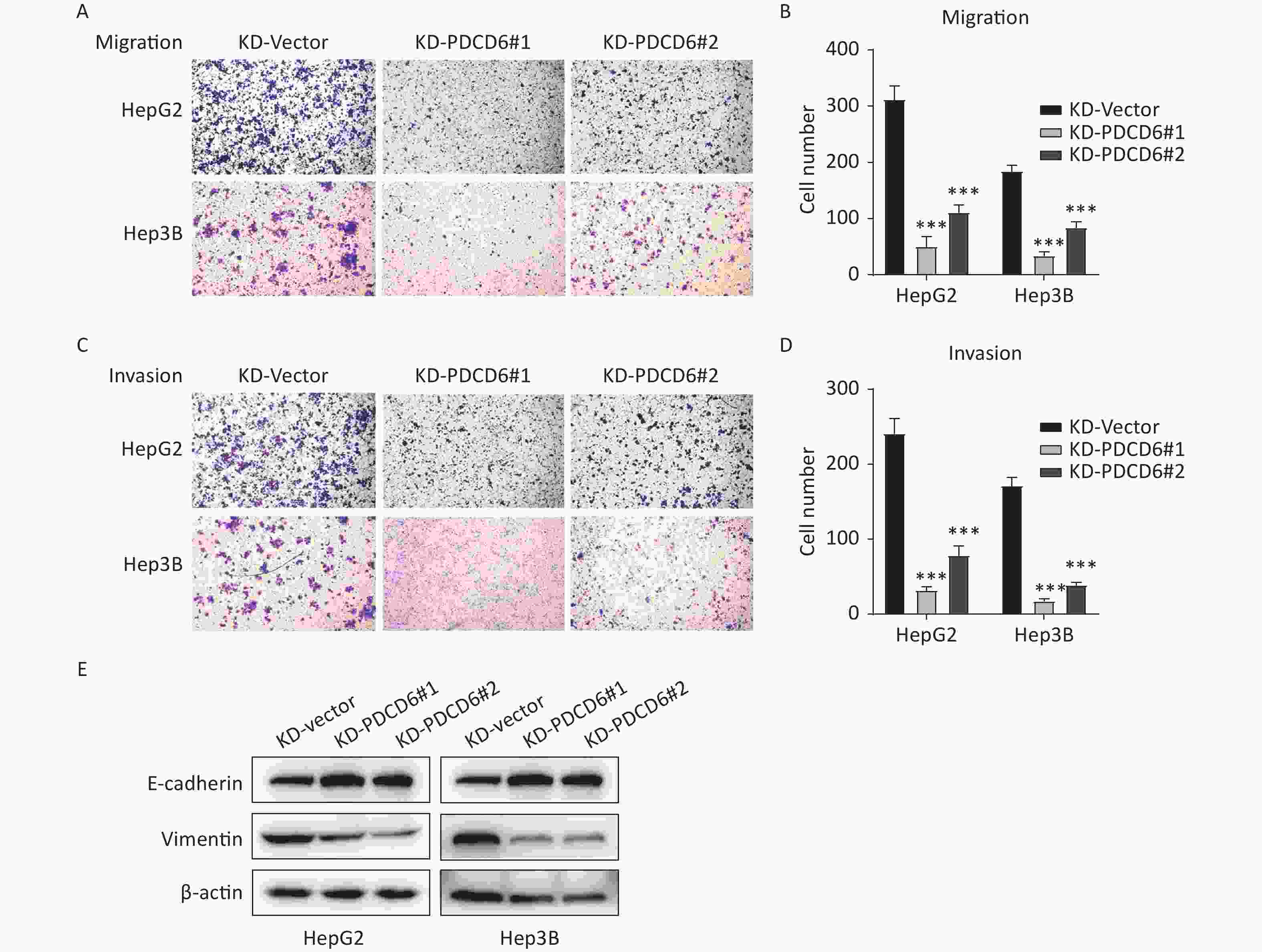
Figure 4. PDCD6 knockdown represses migration and invasion of HCC cells. The same experiments, with results similarly displayed, as in Figure 3, but for HepG2 and Hep3B cells with PDCD6 knockdown. The data shown are the mean ± SD of three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
-
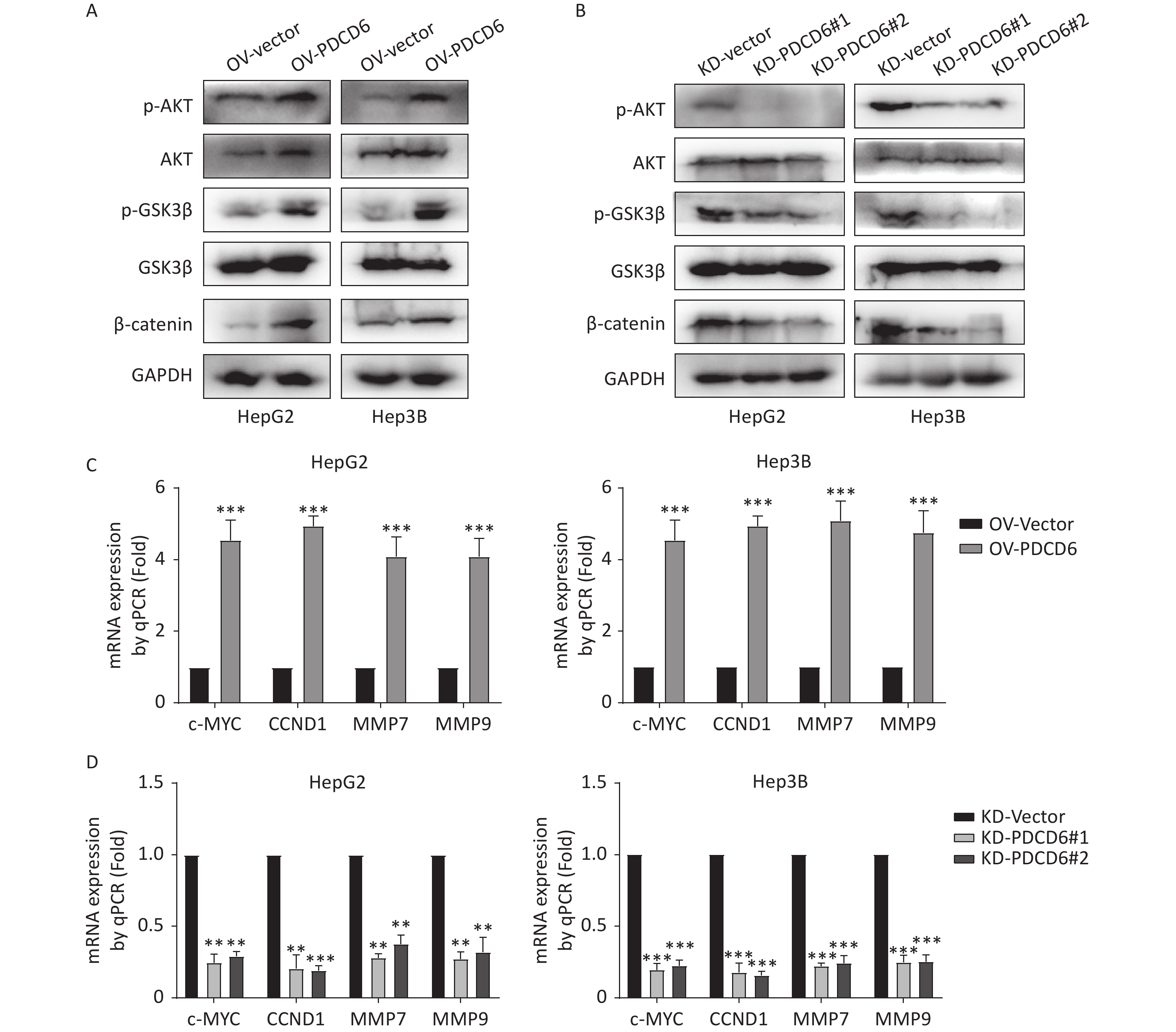
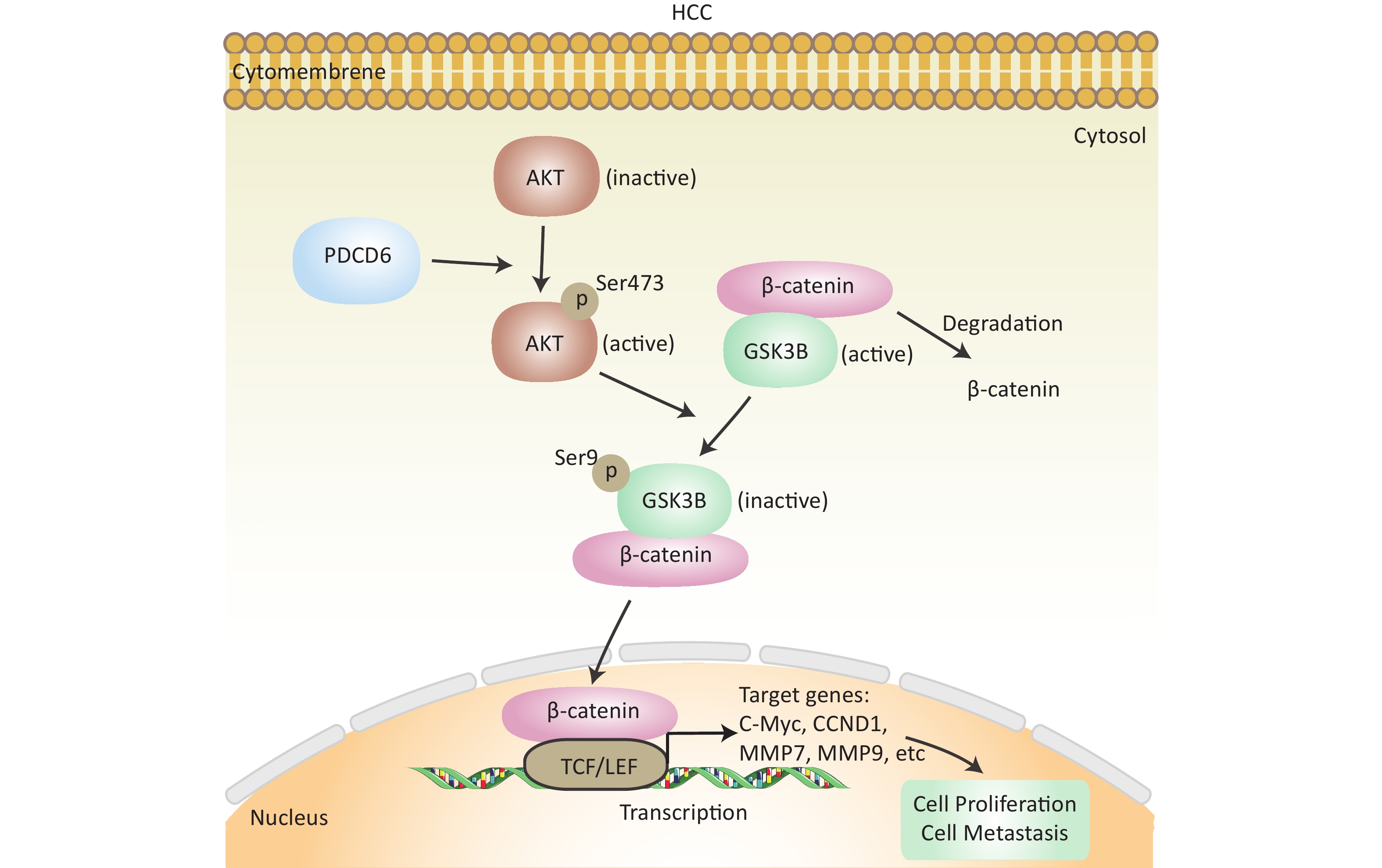
In cancer, phosphorylation activates AKT, that in turn inactivates GSK3β by phosphorylation at Ser9, thereby inhibiting GSK3β catalysis of β-catenin degradation that leads to increased cytoplasmic accumulation as well as the subsequent nuclear import of β-catenin. β-catenin is a transcriptional factor acting in the cell nucleus to regulate several downstream target genes, including cellular (c)-MYC, recombinant Cyclin D1 (CCND1), matrix metallopeptidase 7 (MMP7), and MMP9. Using the Western blotting assay, we confirmed that the level of phosphorylated (p)-AKT, p-GSK3β, and β-catenin increased when PDCD6 was overexpressed (Figure 5A), whereas the expression for each of the three proteins was inhibited when PDCD6 expression was silenced (Figure 5B). Furthermore, using RT-qPCR, we determined the expression levels for the downstream target genes of β-catenin. As shown in Figure 5C, PDCD6 overexpression enhanced the expression of c-MYC, CCND1, MMP7, and MMP9. Conversely, PDCD6 knockdown led to the downregulation of the same genes (Figure 5D). These findings suggest that PDCD6 activates the AKT/GSK3β/β-catenin signaling axis in HCC cells.
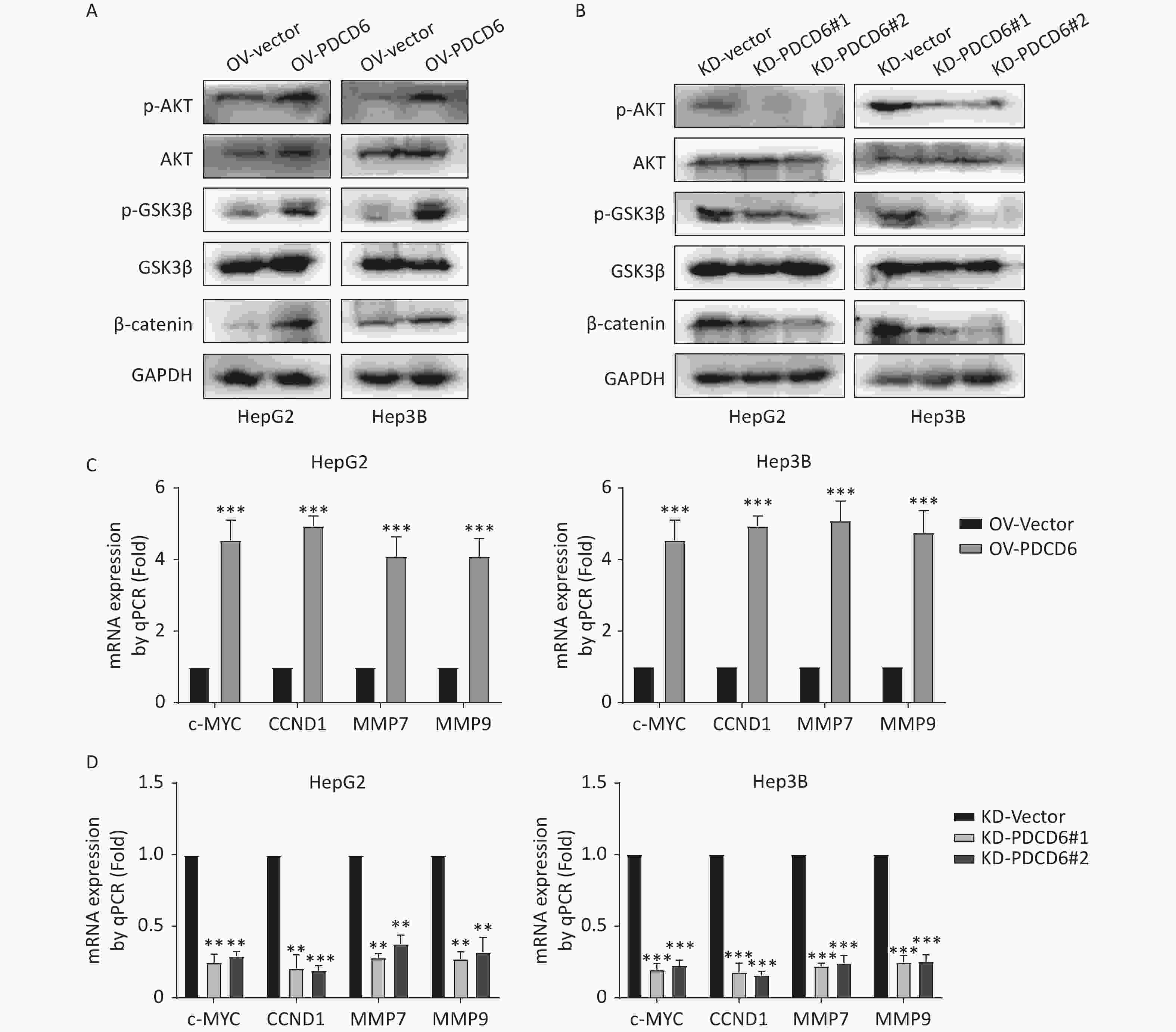
Figure 5. PDCD6 induces activation of the AKT/GSK3β/β-catenin signaling pathway. (A, B) Expressed AKT, p-AKT, GSK3β, p-GSK3β, and β-catenin were detected by Western blotting in HepG2 and Hep3B cells with (A) PDCD6 overexpression or (B) PDCD6 knockdown. (C, D) The mRNA levels for the downstream targets of β-catenin were quantified by using RT-qPCR from the total RNA extracted from the HepG2 and Hep3B cells with stable (C) PDCD6 overexpression or (D) PDCD6 knockdown. The data shown are the mean ± SD of three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
-
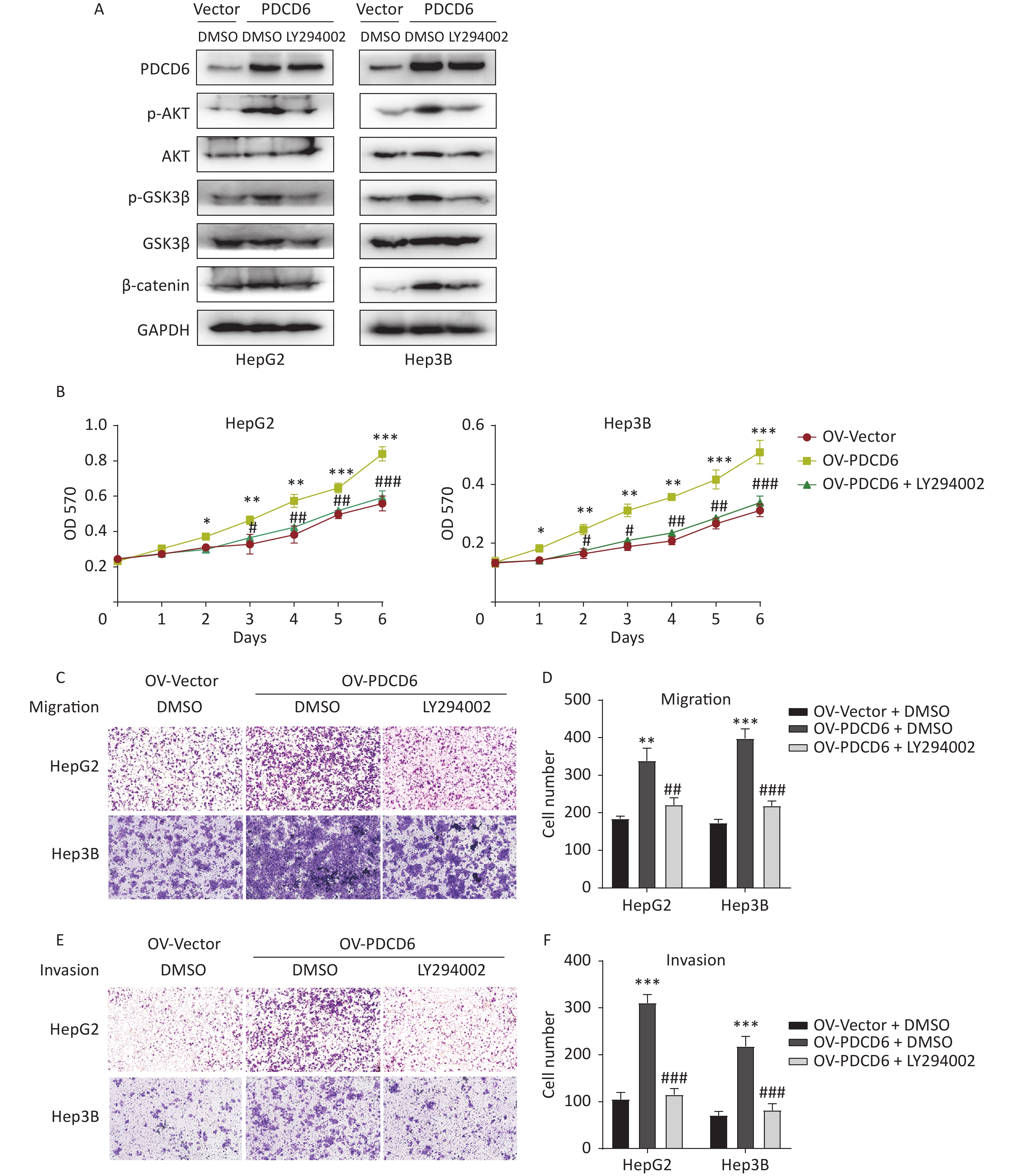
To determine whether the observed PDCD6 effects on proliferation and metastasis of HCC were mediated by the AKT/GSK3β/β-catenin signaling axis, we incubated the HepG2 and Hep3B cells with 10 μmol/L LY294002 (a PI3K inhibitor and an indirect AKT inhibitor through the PI3k/AKT pathway) for 48 h to limit the molecular activities in the AKT/GSK3β/β-catenin pathway. Next, we used Western blotting to determine how the expression of proteins known to be active in the AKT/GSK3β/β-catenin pathway changed in the AKT-inhibited cells. The analysis revealed that increased levels of p-AKT, p-GSK3β, and β-catenin caused by PDCD6 overexpression were reversed by the LY294002 treatment (Figure 6A). Moreover, LY294002 also fully reversed the enhanced proliferation viability of HCC cells caused by PDCD6 overexpression (Figure 6B). Similarly, LY294002 treatment of PDCD6-overexpressing HepG2 and Hep3B cells reversed the cells increased ability of migration (Figure 6C and 6D) and invasion (Figure 6E and 6F). These observations are taken as evidence that PDCD6 acted upstream of the AKT/GSK3β/β-catenin pathway to promote cell proliferation and metastasis in HCC.
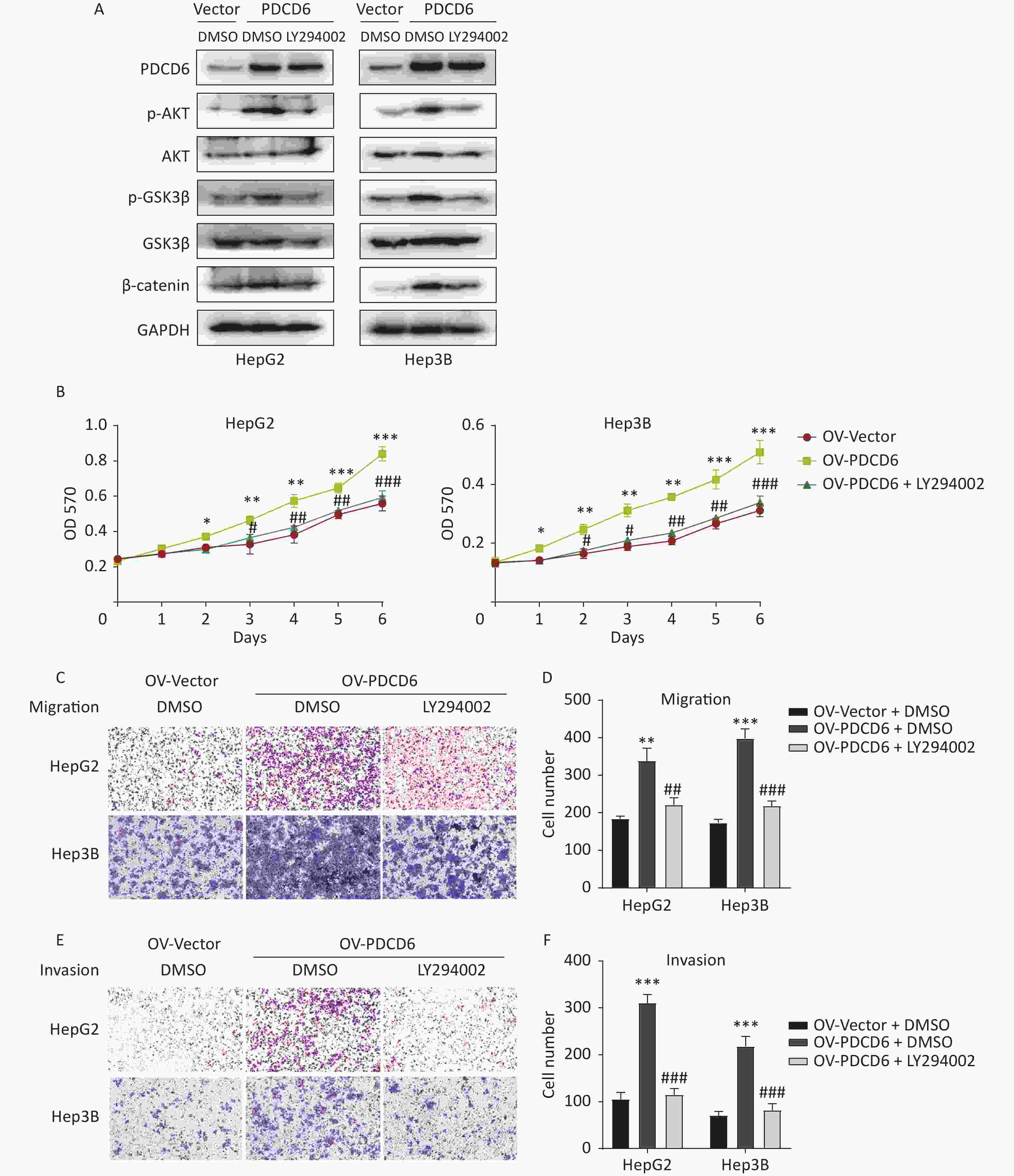
Figure 6. PDCD6 promotes tumor growth and metastasis through the AKT/GSK3β/β-catenin pathway. PDCD6-overexpressing HepG2 and Hep3B cells were pretreated with LY294002 (10 μmol/L) for 48 h. (A) Western blotting analysis indicated that LY294002 treatment led to the reversal of the PDCD6-overexpression induced enhancement of expression for AKT, p-AKT, GSK3β, p-GSK3β, and β-catenin. DMSO was used as a control. (B) MTT assay of cell metabolism (indicating viability and proliferation) indicated that LY294002 reversed the enhanced cell proliferation. Additionally, transwell assays demonstrated that LY294002 also reversed enhanced (C, D) cell migration and (E, F) cell invasion ability. The data shown are the mean ± SD of three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001, compared with the control group. #P < 0.05, ##P < 0.01, ###P < 0.001, compared with the PDCD6 overexpression group.
-
In this study, we investigated the role and molecular mechanisms of PDCD6 in HCC proliferation and metastasis. Based on accumulated evidence and the results presented here, we believe that PD may be an important oncogene in HCC. We showed, using bioinformatic analysis of the TCGA database, that high PDCD6 expression levels predicted a poor outcome for HCC patients, so PDCD6 may play an important role in the pathogenesis of HCC. We genetically manipulated HCC cells to overexpress or silence PDCD6 and found that PDCD6 overexpression enhanced HCC cell proliferation, migration, and invasion, while PDCD6 silencing did quite the opposite, weakening HCC cell viability and metastatic ability. Further, we found that, in HCC cells, PDCD6 overexpression activated AKT activity (by phosphorylation), which in turn phosphorylated GSK3β (at Ser9), inhibiting GSK3β catalytic activity. The inactivation of GSK3β led to increased accumulation of nuclear β-catenin, accelerating the transcription rates for downstream genes, including c-MYC, CCND1, MMP7, and MMP9, that stimulate HCC tumorigenesis and EMT (Figure 7). Overall, our study revealed that PDCD6 was upregulated in HCC, enhancing HCC cell proliferation and metastasis through the AKT/GSK3β/β-catenin pathway.
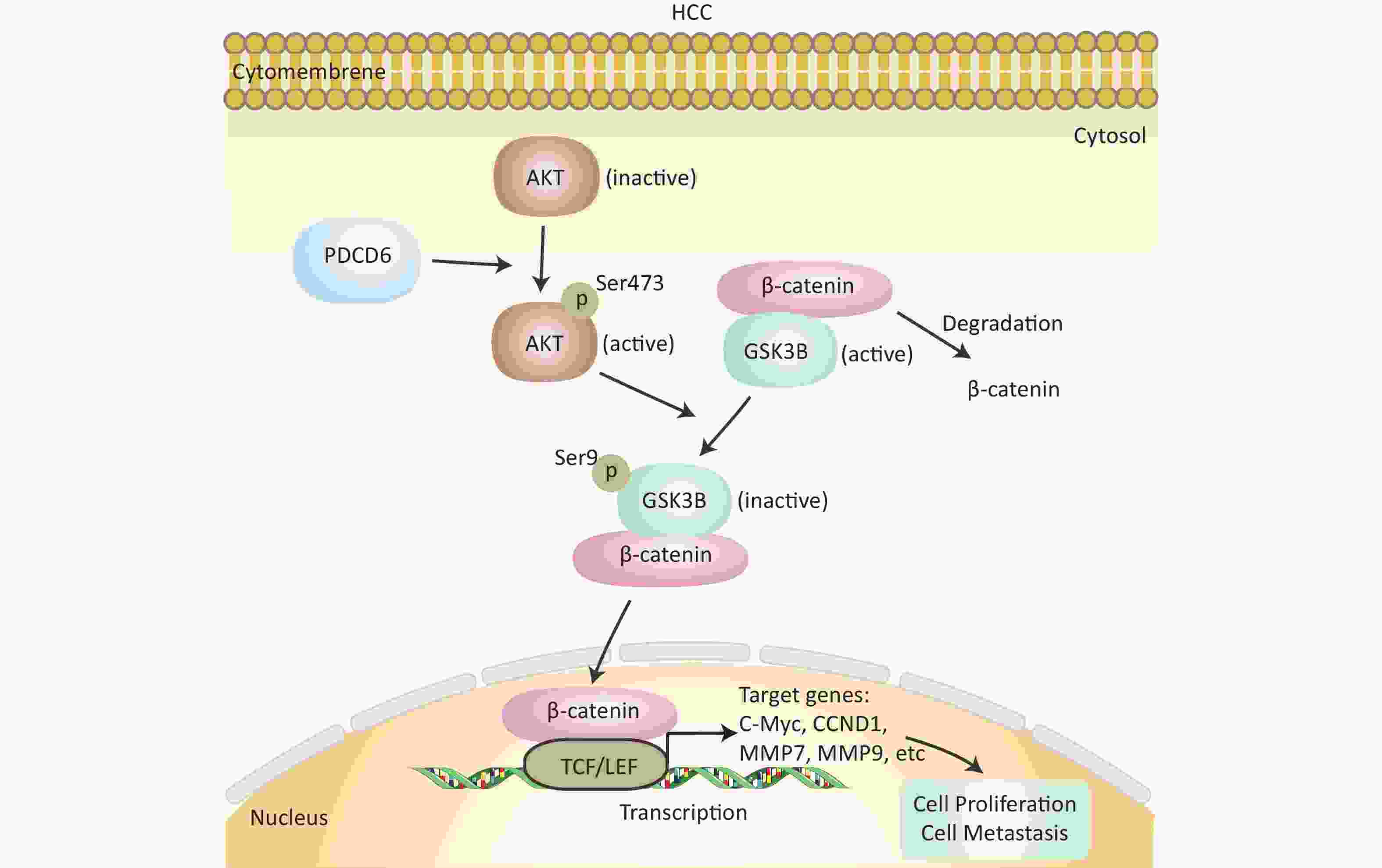
Figure 7. Schematics of the molecular regulatory pathways underlying PDCD6-promoted proliferation and metastasis in HCC. PDCD6 upregulates the expression of p-AKT and p-GSK3β. This, in turn, promotes β-catenin accumulation (reduced degradation by GSK3β) in the cytoplasm and β-catenin entry into the nucleus to activate gene transcription of downstream proteins in the TCF/LEF family for controlling cellular processes, such as proliferation and metastasis.
Studies have reported the opposite effects of PDCD6 in different tumors. Some results have reported that PDCD6 was a prognostic biomarker for advanced-stage gastric cancer and that PDCD6 was more highly expressed in metastatic ovarian cancer, facilitating cell migration and invasion [22,23]. Moreover, PDCD6 has also been considered a potential prognostic biomarker for early adenocarcinoma and advanced oral cancer [24,25]. Nevertheless, several reports revealed that PDCD6 could be regarded as an inhibitor gene in a variety of human tumors. In ovarian cells, PDCD6 positively regulated anti-tumor activity and apoptosis by targeting the PI3K/mTOR/p70S6K pathway [26]. Convincing evidence has been reported that PDCD6 was a novel p53-responsive gene, accumulating in the nucleus in response to apoptosis induced by DNA damage [27]. Our study suggests that PDCD6 is a crucial tumor oncogene regulating AKT/GSK3β/β-catenin pathway in HCC.
Patients with HCC are often diagnosed at an advanced stage due to the high metastatic rate in HCC that facilitates cancer recurrence. EMT is the process of changing cellular profile, in which epithelial cells lose polarized organization and gain the ability to migrate and invade the mesenchyma [28]. The EMT process is strongly linked to the development and progression of multifarious malignant tumors, including HCC [29,30]. Thus, it is noteworthy that about 80% of human malignancies arise from epithelial tissues such as the lung, liver and colon [31]. In the course of EMT, adhesion instability based on E-cadherin leads to the disruption of cell-cell adhesion and increases metastasis in tumors [32]. Several studies reported that aberrant PDCD6 expression induced the EMT pathway to drive invasion and metastasis in various cancers [13]. Nevertheless, further investigation is necessary to better understand the molecular mechanism by which PDCD6 regulates EMT.
There is accumulating evidence proving that several signaling pathways, including Wnt/β-catenin, PI3K/AKT, and NF-κB, are related to an aberrant EMT program in cancer [33,34]. AKT is positively associated with cell tumorigenesis and metastasis [35]. GSK3β improves the permeability of the mitochondrial membrane and drives cell apoptosis. However, AKT phosphorylates GSK3β, which inactivates and inhibits GSK3β to suppress cell apoptosis. GSK3β phosphorylation can cause the separation of β-catenin from the GSK3β complex and leads to its subsequent nucleus import. The increased levels of β-catenin in the nucleus activate TCF/LEF transcription factors, such as the MMP family, c-MYC, CCND1, CD44, Slug, and C-JUN, thereby inducing proliferation and EMT of cancer cells [36,37]. MMPs, a family of structurally related zinc- and calcium-dependent endopeptidases, degrade extracellular matrices and facilitate the EMT program [35]. Overexpression of c-MYC and CCND1 can drive proliferation and induce genes related to the cell cycle [38,39]. These studies demonstrated that the aberrantly activated AKT/GSK3β/β-catenin pathway plays an important role in malignant progression. Our findings strongly indicate that PDCD6 promotes cell proliferation and metastasis via AKT/GSK3β/β-catenin pathway in HCC.
In conclusion, our current findings revealed the effect and molecular mechanisms of PDCD6 in promoting HCC proliferation and metastasis ability. PDCD6 regulates the AKT/GSK3β/β-catenin signaling axis to promote these malignant features in HCC. Given the crucial role of PDCD6 in the aggressive progression of HCC, PDCD6 may be considered for further exploration as a potently diagnostic and therapeutic target for HCC.
HTML
Cell Culture and Chemicals
Cell Viability Assay
Transwell Assay
Western Blotting
RT-qPCR
Overexpression or shRNA Knockdown of PDCD6 in HCC Cell Lines
Bioinformatic Analysis of The Cancer Genome Atlas (TCGA) Data
Statistics
PDCD6 Expression was Significantly Promoted in HCC
PDCD6 is Involved in Cell Viability in HCC Cells
PDCD6 Improves HCC Cell Migration and Invasion by EMT
PDCD6 Activated AKT/GSK3β/β-catenin
PDCD6 Promotes the Proliferation and Metastasis of HCC Cells Through AKT/GSK3β/β-catenin Pathway
CONFLICTS OF INTEREST The authors declare no conflicts of interest.

 Quick Links
Quick Links
 DownLoad:
DownLoad: