
-
Hematophagous arthropods, such as ticks and midges, are vector organisms that can transmit disease-causing pathogens to humans, livestock or wildlife through blood-sucking or stabbing behavior, causing serious diseases[1]. Vector organisms harbor a broad spectrum of bacterial microbes with crucial physiological functions and ecological importance[2]. Evidence has suggested that some bacteria affect the transmission capacity of arthropods by increasing or reducing pathogen replication and dissemination in the hosts[3], together with various aspects of their physiology and metabolism. Bacterial communities can play a factor affecting the reproductive fitness and survivability of parasites in vector hosts[4], and therefore they are likely to play a pivotal role in vector transmission, evolution and ecology[2,3,5].
Vector-borne diseases have emerged frequently over a decade[6-8]. With the application of high-throughput sequencing technology in the field of bacterial research, studies of vector microbiota have gradually become a research focus. Historically, most studies regarding arthropod communities have focused on describing the bacterial component and taxonomical profiles of specific genera and demonstrating how they are affected by habitat or geographical factors[9,10]; however, few have been studied at the species level. Our understanding of the bacterial diversity of arthropod vectors remains incomplete.
There are great varieties of vector organisms, including mosquitoes, ticks, midges, and sand flies. Epidemiologically, vector-borne diseases, accounting for more than 17% of infectious diseases worldwide based on WHO data (World Health Organization (2017) Global Vector Control Response (2017–2030), WHO), are related to vector species, pathogen dynamics, and changes in geographic and host ranges[11-13]. Among these, biting midges (Diptera: Ceratopogonidae) are both biting pests and important vectors of various pathogens, especially viruses, but also parasites and protozoa, such as avian hamosporidians[14]. To date, more than 50 viruses have been isolated from biting midges worldwide, several causing international epidemic diseases. Animal diseases caused by viruses of international significance including African horse sickness virus and bluetongue virus are transmitted by biting midges[15]. Midges are important vectors strongly linked to human and animal diseases, yet their core microbiota and potential pathogens are widely unknown. Recently Sarkar et al.[16] isolated and identified several hemolytic bacteria in Culicoides peregrinus Kieffer, those culturable bacteria with hemolytic activities may be involved in blood digestion in the gut of midges. Ticks are the second most broadly recognized human disease vector worldwide[5], followed by mosquitoes[17]. As the most versatile vector and blood-sucking ectoparasites, ticks carry a variety of pathogens, composed of viruses, bacteria, fungi, and protozoa, capable of spreading to humans and wildlife to cause diseases such as spotted fever, Ehrlichiosis, anaplasmosis, and Lyme disease. Ticks are of medical and veterinary importance and pose a serious threat to human health and public health security.
The purpose of this study was to investigate the bacterial community structure diversity of two arthropod vectors (midges and ticks) collected from Poyang Lake area, and whether vectors carry any bacterial pathogens that may cause diseases to humans. Specifically, we used a metataxonomic strategy to analyze the taxonomic composition, dominant groups, along with distribution of common pathogens that are likely to cause diseases in humans and discussed their possible biological significance. We hope to gain an understanding of the microorganisms carried by the vectors in Poyang Lake, to provide scientific basis for prospective pathogen discovery and disease prevention and control.
-
In June 2019, ticks and midges were collected from three sites in the Poyang Lake area, Jiangxi Province, China, namely, Qunlu Practice Base (Pengze County, 29°48’ N, 116°39’ E), Peach Blossom Garden (Pengze County, 29°53’ N, 116°41’ E), and Huangtong Animal Husbandry (De’an County, 29°24’ N, 115°43’ E) (Supplementary Table S1, available in www.besjournal.com). The temperature in the sampling area was about 20–30 °C, and the humidity was approximately 70%–90%. A total of 3,650 biting midges were gathered by UV (24 W, 220 V) light traps at dusk from animal sheds (sheep and chickens inside). There were 140 free ticks sampled by flagging grass and shrubs with a white cotton flag, and 95 ticks parasitizing on deer and sheep were collected. All ticks parasitizing on animals were in the engorgement stages. Depending on morphological features and detection of mitochondrial molecular genes, all arthropods were identified as adults and classified (Supplementary Table S1). Various molecular markers have been used to identify vectors by amplification and sequencing the mitochondrial molecular genes, including cytochrome c oxidase subunit 2 (cox2)[18], cytochrome b (cytb)[18] and cytochrome c oxidase subunit I (COI)[19]. The primers were shown in Supplementary Table S2, available in www.besjournal.com. As previously reported[20], the most widespread species of midges was Culicoides arakawae Arakawa (found at all sites), followed by Culicoides nipponensis Tokunaga and Culicoides punctatus Meigen. All ticks were identified as Haemaphysalis longicornis Neumann. The collected arthropod samples were stored in 2 mL sterile tubes, subsequently transferred to our laboratory in Beijing at 4 °C and kept in the refrigerator at −80 °C until DNA extraction.
Species Sample types Sample No. ACE Chao1 Shannon Simpson goods_Coverage Midges Culicoides spp. M01 167.79 165.20 1.17 0.40 1.00 M02 148.68 136.20 1.79 0.58 0.99 M03 185.32 189.14 1.22 0.28 0.99 M04 266.44 254.00 2.67 0.77 0.99 M05 30.10 26.33 0.90 0.34 1.00 M06 124.80 133.13 1.65 0.52 1.00 M07 125.99 107.38 1.56 0.47 1.00 M08 77.95 81.00 1.85 0.60 0.99 M09 176.82 141.93 3.03 0.78 1.00 M10 181.30 168.05 3.44 0.80 0.99 M11 198.50 246.86 2.86 0.73 0.99 M12 170.08 144.17 3.42 0.77 0.99 M13 96.45 78.00 1.84 0.62 0.99 M14 52.07 49.25 2.37 0.74 1.00 M15 121.29 107.17 1.56 0.42 1.00 M16 236.69 234.14 4.94 0.94 0.97 M17 158.75 161.50 1.61 0.59 1.00 M18 90.01 65.09 1.84 0.59 1.00 Mean 144.95 138.25 2.21 0.61 0.99 Ticks Free-living ticks T01 231.30 188.00 1.93 0.64 0.99 T02 197.59 189.14 3.91 0.87 0.99 T03 250.63 240.40 0.81 0.14 0.99 T04 174.00 172.18 0.82 0.16 0.99 T05 111.58 117.27 0.78 0.16 1.00 T06 157.93 137.91 0.45 0.08 0.99 T07 164.58 175.83 1.42 0.28 0.99 Mean 183.94 174.39 1.45 0.33 0.99 Ticks parasitizing on animals T08 68.12 70.11 2.13 0.65 0.99 T09 112.26 122.20 0.41 0.08 0.99 T10 157.00 102.00 0.11 0.02 0.99 T11 116.51 136.50 2.13 0.52 0.98 T12 104.85 128.00 2.35 0.61 0.99 T13 244.13 210.19 0.52 0.08 0.98 T14 72.66 57.00 0.81 0.20 0.99 T15 54.02 54.25 1.33 0.48 0.99 Mean 116.20 110.03 1.22 0.33 0.99 Table 1. The main alpha-diversity indices of microbial community structure in all samples
-
According to the size of the individual arthropods, a certain number of arthropods were divided into a sample pool. The individuals in each sample pool were collected from the same site. Approximately 50–300 biting midges were grouped into one sample, while 7–20 ticks were sorted into one sample. Finally, we had 18 midge samples and 15 tick samples.
Each sample was rinsed with 70% ethanol for 5 minutes, then rinsed with sterile water, and repeated the above steps twice[21]. Before DNA extraction, two blank control groups without samples were set up for midge and tick samples, respectively. A total of 33 arthropod mixed samples and 4 blank control groups were individually homogenized in 1X phosphate buffer solution (PBS) with a pH of 7.4 using the TissueLyser II system (Qiagen, Germany) and were centrifuged at 13,000 × g for 10 min. The total DNA was extracted according to the manufacturer’s instructions, using the DNeasy Blood and Tissue Kit (Qiagen). The DNA concentration of each sample was estimated by a NanoDrop 2000 (Thermo, USA). Amplification of full-length 16S rRNA gene (V1-V9 region) was conducted using universal primers 27F (50-AGAGTTTGATCCTGGCTCAG-30)/1492R (50-GNTACCTTGTTACGACTT-30) with 16 nt symmetric (reverse complement) barcodes tagged at the 5’ end, as described in previous research[22]. We obtained PCR products of the 16S rRNA gene by repeatedly amplification (25 µL/tube, amplify 8 tubes for each sample pool) rather than increasing the number of cycles to reduce error and bias. Libraries of PCR products were generated, followed by sequencing on the PacBio Sequel platform at Tianjin Biochip Corporation, China. Finally, the blank control groups did not pass the step for “sequencing library preparation”, because the concentration of the PCR amplicon of the negative controls did not get the required input.
-
Filtering and quality control of the raw sequences was conducted with previous pipeline[22,23], i.e., all the full-length 16S rRNA gene sequences were clustered into operational taxonomic units (OTUs) at a 98.7% identity threshold[24], and all the OPUs were determined using the Arb tool by the visual inspection of the final phylogenetic trees. The only difference was that the reference database used was LTP132 (the latest version at the time of this study). OPU was defined as the smallest monophyletic branch of the phylogenetic tree, consisting of one or multiple OTU sequences and adjacent reference sequences[25,26]. An OPU was considered to be a member of the same species with more than 98.7% similarity to the sequences of nearly type strains. For OPUs that represented an independent lineage within a genus, amplicons were considered to be unclassified new species within the genus. When unique lineages were classified into other known genera, sequences were clustered into uncultured lineages of known families, orders, or classes.
-
The alpha diversity of each sample was calculated using the Vegan package in R software (version 4.1.0). PAST v4.09 software[27] was used to plot rarefaction curves based on OPU abundances. The beta diversity was performed to identify the differences in the composition of the arthropod communities. Permutation multivariate analysis of variance (R: vegan: Adonis)[28] used both Jaccard and Bray‒Curtis distance matrices to determine the reliability of differential analysis. The DESeq2 package[29] was used to differentially analyze the OPU composition in the arthropod species.
-
To further verify the distribution of high-abundance bacteria in the samples, genus-specific PCR was performed to screen for Pantoea spp. based on the atpD gene[30] in midge samples and Coxiella spp. based on the groEL gene[31] in tick samples. The sequences of all primers in this study and their expected amplification fragment sizes are shown in Supplementary Table S2. All PCR mixtures were composed of 1 µL DNA template and 19 µL reaction mix containing 10 µL of 2 × Es Taq MasterMix (CoWin Biotech), 7 µL deionized distilled water and 1 µL of each primer. PCR amplifications were executed on a Labcycler (SensoQuest, Germany) by using the following program settings: one cycle of 94 °C (5 min) for initial denaturation, 30 cycles of 94 °C (30 s) for denaturation, 52 °C (30 s) for annealing temperature, and 72 °C (2 min) for extension, and a final extension period (72 °C, 10 min) for Pantoea-specific amplification. Alternatively, we conducted a nested PCR assay to screen for Coxiella infection in tick samples, whereby 1 µL of the 1st PCR product was used as a DNA template for the second reaction. Both reaction conditions followed as previously described, except for adjustments in annealing temperatures to 56 °C according to the primer Tm value. Each PCR-based assay included a DNA-free negative control. PCR products were electrophoresed in 1.5% agarose gels stained with GoldenView and visualized under a UV transilluminator. The positive amplicons were sent to Ruiboxingke Biotechnology (Beijing, China) for sequencing. The results were then compared with other sequences available in the GenBank nucleotide sequence database.
-
Representative sequences of designated OPUs were aligned with almost full-length 16S rRNA gene sequences of related species using ClustalW, and maximum-likelihood phylogenetic trees were constructed with MEGA X[32]. Bootstrap analysis was conducted with 1000 replicates to evaluate the reliability of branches.
-
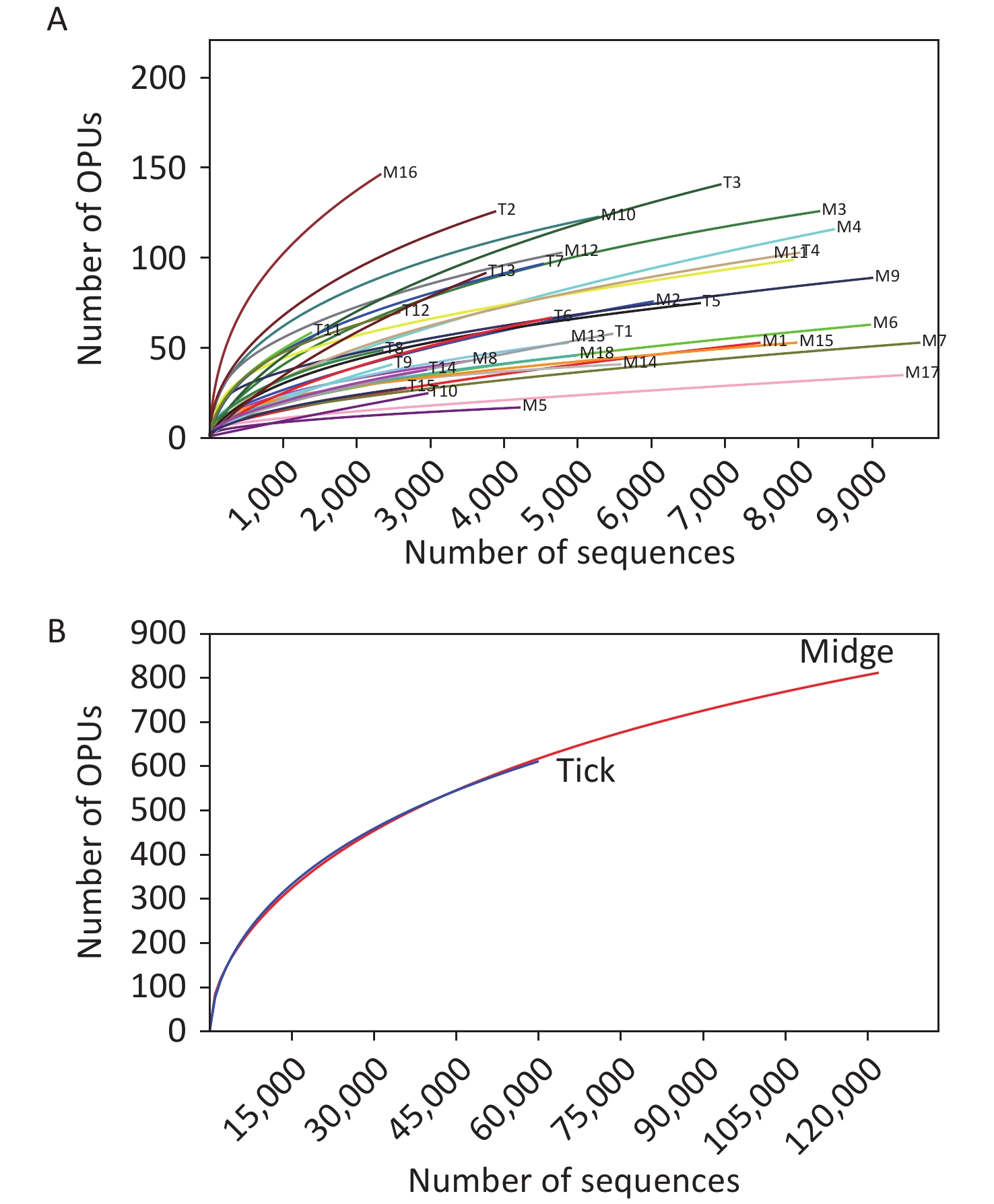
We sampled arthropod specimens from areas around Poyang Lake, namely, Pengze and De’an counties in Jiangxi Province. Midges and ticks were collected from environment and three animal sheds, namely, Qunlu Practice Base, Peach Blossom Garden, and Huangtong Animal Husbandry (Supplementary Table S1). In total, the PacBio Sequel platform rendered 237,292 raw 16S rRNA gene sequence reads from 33 samples including 3,885 individuals. After filtering out the low-quality reads, we obtained 119,165 and 61,578 valid reads for midge and tick samples, with an average of 6,620.28 ± 2,170 reads and 4,105.2 ± 1,872.99 reads per sample, respectively. The sequences were on average 1,476 ± 5.97 base pairs (bp) in length. High-quality reads were clustered into 61,258 unique OTUs at 98.7% identity, and their representative sequences were annotated into 1,039 OPUs, including 662 in midges, 618 in ticks, and 241 shared OPUs (Supplementary Table S3, available in www.besjournal.com). Rarefaction curve analysis showed high coverage but incomplete saturation (Supplementary Figure S1, available in www.besjournal.com).
-
Through the calculation of diversity indices (Table 1), we discovered that the ACE (Abundance-based Coverage Estimator) index and Chao1 index were lowest in ticks parasitizing on animals, and significantly higher in free-living ticks than in all the other groups. On average, the Shannon index and Simpson index of midges were higher than those of ticks. Additionally, the Good's coverage of all samples was greater than 97%, indicating that the sequencing depth was sufficient to uncover the biodiversity of the sequencing samples.
-
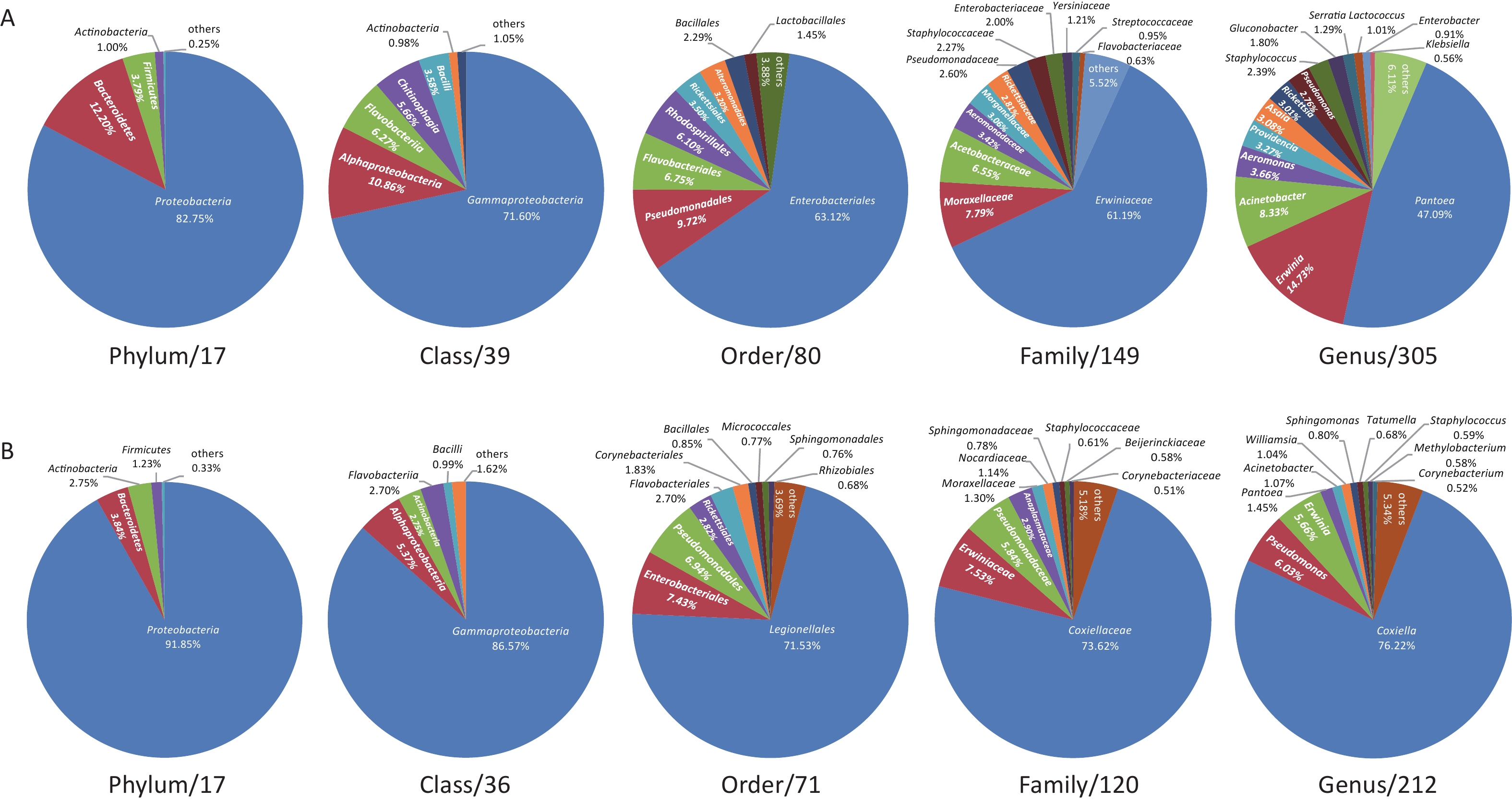
The midge microbiota was affiliated with 17 phyla, 39 classes, 80 orders, 149 families, and 305 genera, while the tick microbiota was affiliated with 17 phyla, 36 classes, 71 orders, 120 families, and 212 genera in total (Figure 1). Only four of the 17 phyla were detected in all midge samples, namely, Proteobacteria, Bacteroidetes, Firmicutes, and Actinobacteria. Correspondingly, Gammaproteobacteria, Alphaproteobacteria, Bacilli, and Actinobacteria were shared among midge samples at the class level. The common bacterial taxonomic units also included Enterobacteriales and Erwiniaceae, whereas no common genus was detected. Their genera with high abundances were Pantoea (47.09%), Erwinia (14.73%), and Acinetobacter (8.33%) (Figure 1A). Likewise, three phyla were detected in all the tick samples, namely, Proteobacteria, Actinobacteria, and Firmicutes. Three of the 36 classes were shared in ticks, including, Gammaproteobacteria, Actinobacteria, and Alphaproteobacteria. Different from midges, we did not recognize a common order, family, or even genus in ticks. Their genera with high abundance were Coxiella (76.22%), Pseudomonas (6.03%), and Erwinia (5.66%) (Figure 1B).
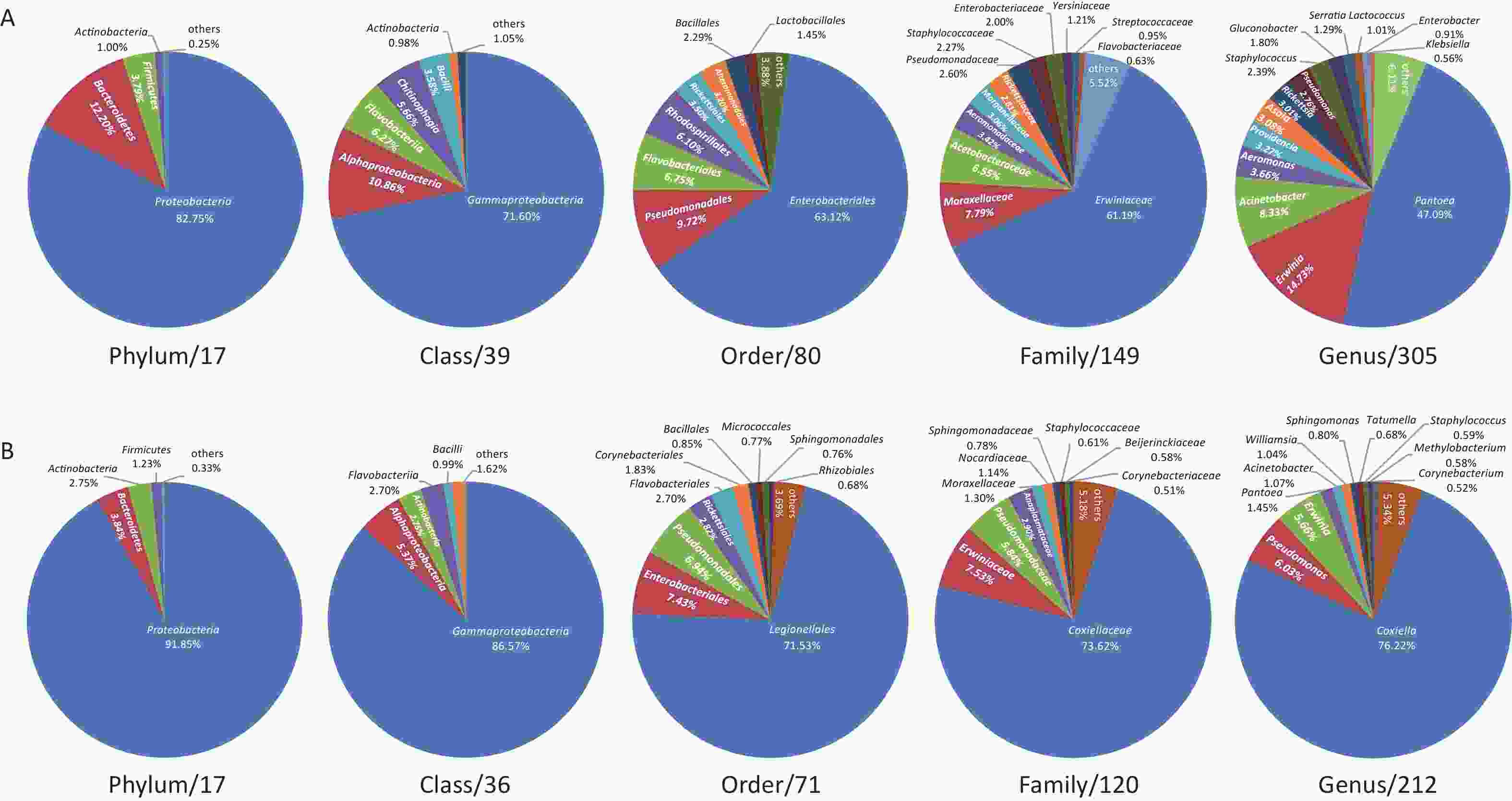
Figure 1. Microbiota profile at Phylum, Class, Order, Family, and Genus categories of (A) midges and (B) ticks. (A) Phylum: of 17 phyla classified, only 4 with > 0.5% total reads were displayed. Class: of 39 classes classified, only 6 with > 0.5% total reads were displayed. Order: of 80 orders classified, only 8 with > 0.5% total reads were displayed. Family: of 149 families classified, only 12 with > 0.5% total reads were displayed. Genus: of 305 genera classified, only 14 with > 0.5% total reads displayed. (B) Phylum: of 17 phyla classified, only 4 with > 0.5% total reads were displayed. Class: of 36 classes classified, only 5 with > 0.5% total reads were displayed. Order: of 71 orders classified, only 10 with > 0.5% total reads were displayed. Family: of 120 families classified, only 10 with > 0.5% total reads were displayed. Genus: of 212 genera classified, only 11 with > 0.5% total reads displayed.
-
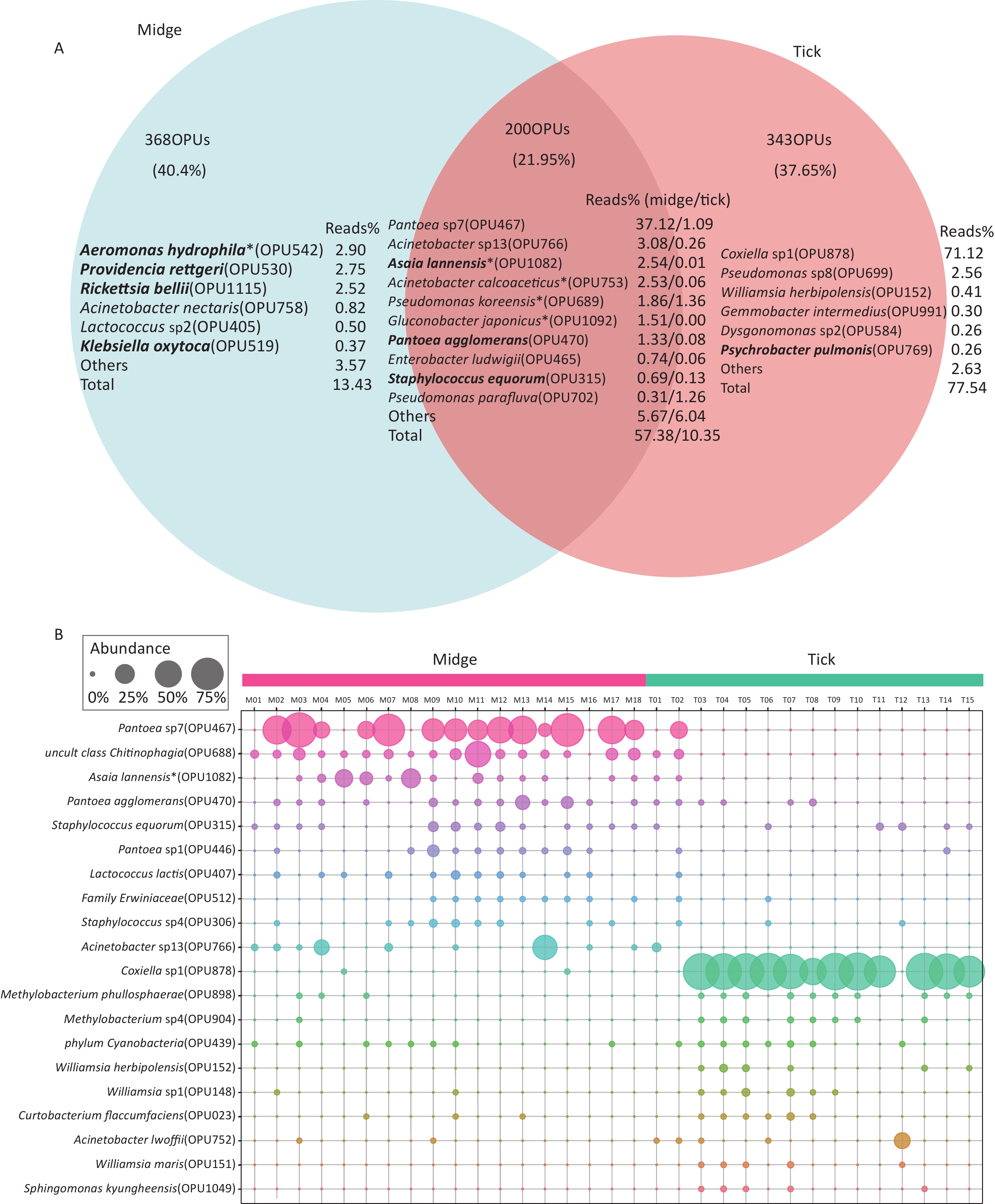
Of the 662 OPUs detected in midges, 195 (29.5%) were classified as known species (> 98.7% identity), which represented 25.37% of the total reads (Supplementary Table S4, available in www.besjournal.com). Unexpectedly, 66 of these known species were potential pathogenic bacteria, accounting for 19.22% of the total reads. The potential pathogens contained Asaia lannensis (OPU1082, 2.54% of the total reads) and Rickettsia bellii (OPU1115, 2.52%) (Supplementary Table S4). A total of 373 (56.34%) OPUs were identified as potential new species, comprising 45.43% of the total reads (Supplementary Table S5, available in www.besjournal.com). The vast majority of potential new species were of low abundance, accounting for less than 1% of total reads. The exception was Pantoea sp7 (OPU467), which comprised imbalanced 37.12% of the total reads.
By contrast, of the 618 OPUs detected in ticks, 217 (35.11%) and 326 (52.75%) OPUs were assigned as known species and potential new species, which gathered 8.83% and 79.05% of the total reads, respectively (Supplementary Tables S6, S7, available in www.besjournal.com). There were 50 potential pathogens in the 217 bacterial species, occupying 2.16% of the total reads. All the potential pathogens detected in tick samples were low in abundance, and the pathogens with relatively high abundances included Acinetobacter lwoffii (OPU752, 0.65%) and Staphylococcus sciuri (OPU313, 0.33%) (Supplementary Table S6).
In addition, there were 368 (40.4%) and 343 (37.65%) species unique to midges and ticks, respectively, while 200 (21.95%) species were shared (Figure 2A). The species with relatively high abundances unique to midges were Aeromonas hydrophila (OPU542, 2.9%), Providencia rettgeri (OPU530, 2.75%) and Rickettsia bellii (OPU1115, 2.52%), all of which are pathogens, while ticks was Coxiella sp1 (OPU878, 71.12%). The proportion of shared reads was imbalanced between midges and ticks; reads of shared species accounted for 57.38% of the total reads in midges but accounted for only 10.35% in ticks.
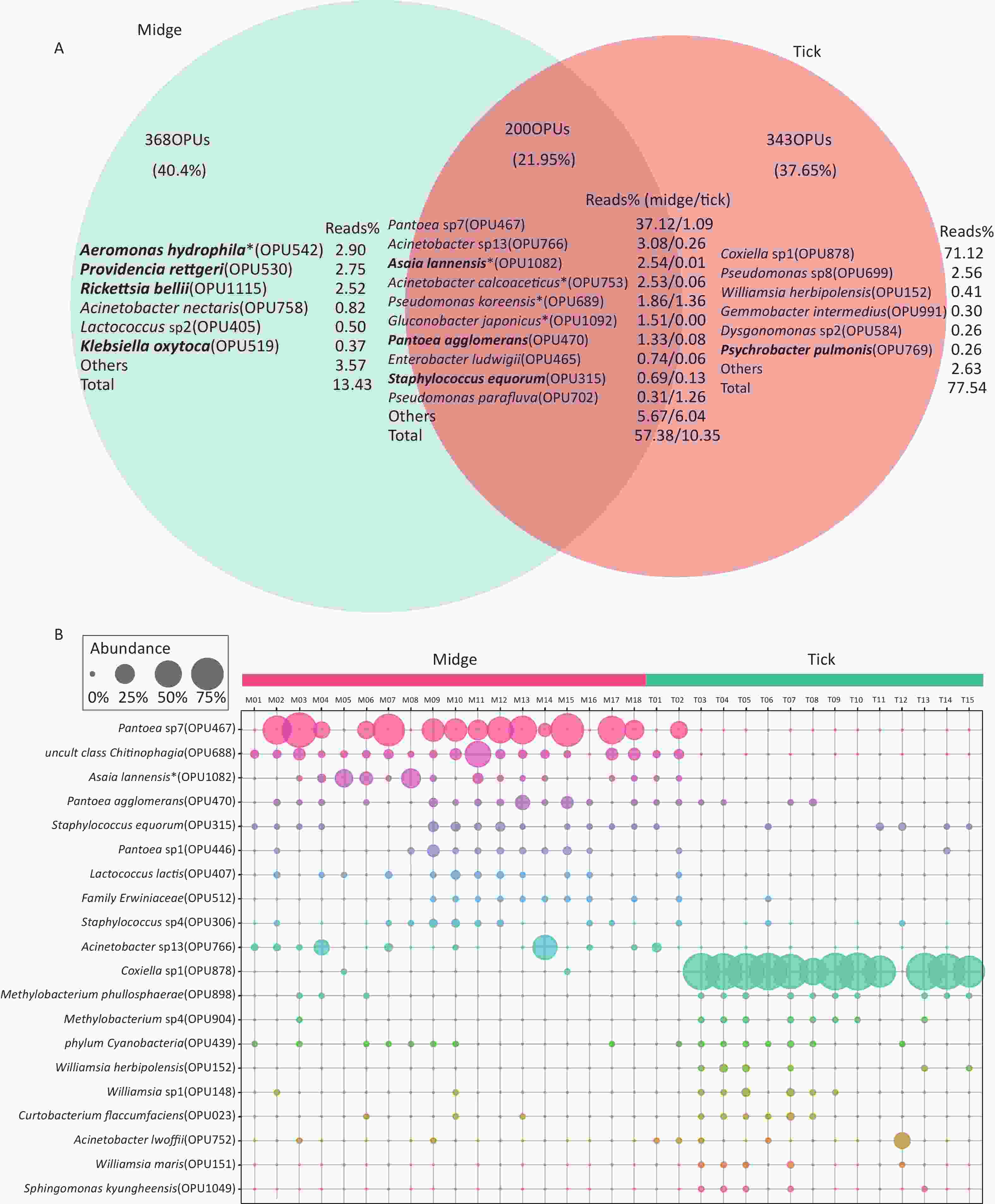
Figure 2. Abundance of shared and differential OPUs in midges and ticks. (A) Venn diagram of OPUs corresponding to shared and unique species between midges and ticks, and the percentage of reads in the top abundance species. Potentially pathogens are indicated in bold. (B) Representation of the top 10 differentially enriched OPUs in each sample type. Each color represents an OPU, and the abundance of a total of 20 OPUs was expressed as a percentage. The abundance is shown as percentages of reads in each sample. Pantoea sp7 (OPU467) and Coxiella sp1 (OPU878) are the most differentially abundant bacteria for midges and ticks, respectively. OPU, operational phylogenetic unit.
-
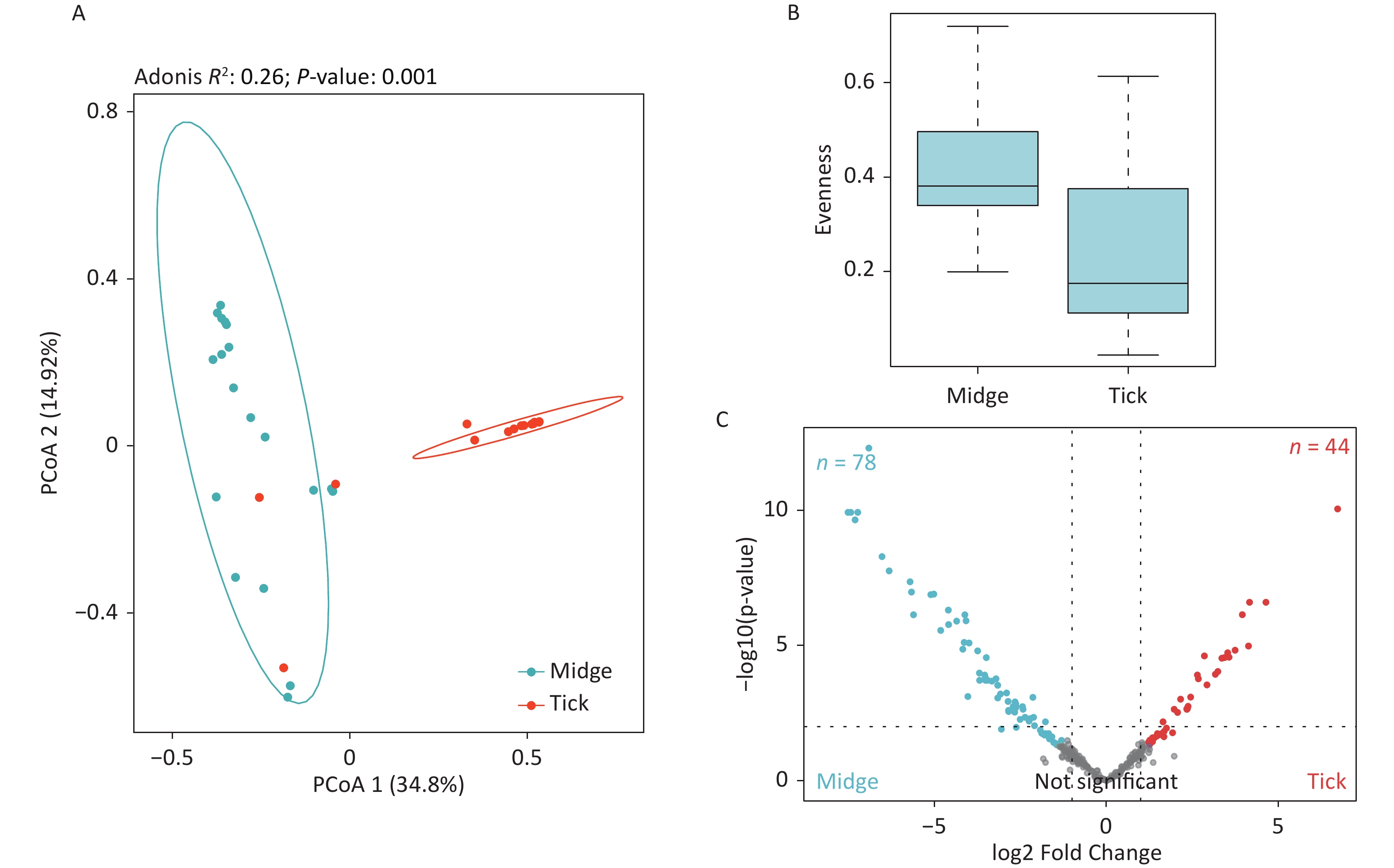
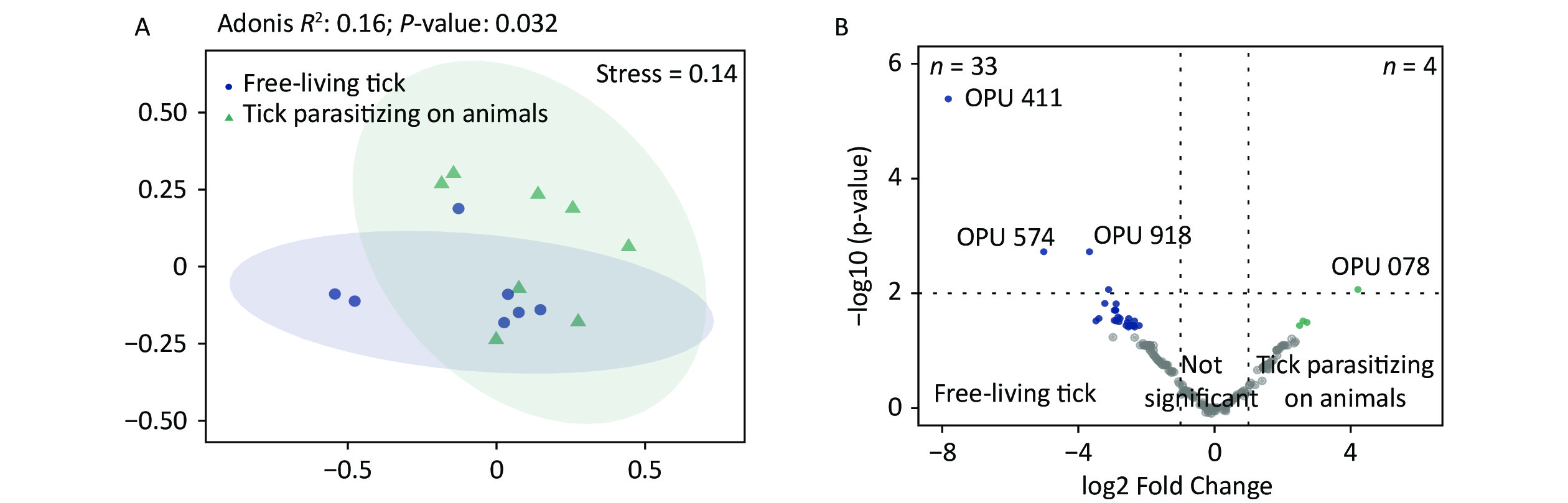
The PCoA revealed significant differences in species-level bacterial communities between the midge and tick groups (Supplementary Figure S2A, available in www.besjournal.com). Intriguingly, a significant decrease in sample evenness can be observed, with less even OPUs distribution in ticks (Mdn = 0.18, IQR = 0.09–0.39) than in midges (Mdn = 0.38, IQR = 0.33–0.5) (Supplementary Figure S2B). In terms of the free-living ticks and ticks parasitizing on animals, there were differences in the bacterial component of microbiota (Adonis, R2 = 0.16, P = 0.032) (Supplementary Figure S3A, available in www.besjournal.com).
To recognize differentially enriched OPUs for both vector species, we compared the composition of OPUs using DESeq2. We identified 78 OPUs in midges and 44 OPUs in ticks that were differentially abundant at a threshold of α < 0.05 (Supplementary Figure S2C). The abundance of differentially enriched OPU between the two vectors varied significantly. It was clearly observed that Pantoea sp7 (OPU467) and Coxiella sp1 (OPU878) were the most differentially enriched OPUs for midges and ticks, respectively, as well as the most abundant taxa in their bacterial community (Figure 2B). When identifying differentially expressed OPUs among ticks, there were 33 OPUs in free-living ticks and 4 OPUs in ticks parasitizing on animals that were differentially abundant at a threshold of α < 0.05 (Supplementary Figure S3B). Erwinia (OPU411), Flavobacteriales (OPU574), and Methylobacterium goesingense (OPU918) were the three most differentially enriched OPUs for free-living ticks, while in ticks parasitizing on animals it was Glutamicibacter nicotianae (OPU078).
-
There were 5.56% (1/18, sample M10) of midge samples positive for Pantoea spp., and the amplified sequences exhibited 99.32% identity to complete genome sequences from Pantoea agglomerans strain ASB05 (accession number CP046722). A nested PCR approach was used to detect Coxiella infection in ticks. Unexpectedly, all 15 tick samples were positive for Coxiella spp.. Specific amplification of DNA sequences from sample T15 exhibited 99.46% identity to a partial DNA sequence from Coxiella sp. isolate TON_L9_39 chaperone protein encoding gene (groEL) (accession number MZ327926) and sequences of other tick samples exhibited approximately 98% identity to complete genome sequences from the Coxiella-like endosymbiont strain 580 (accession number CP084737).
-
OPU467 with 44,214 reads clustered as a separate branch within the genus Pantoea, probably representing a novel bacterial species distinct from known Pantoea species (Figure 3A). The representative sequences of OPU467 showed < 97% 16S rRNA gene sequence identity to Pantoea hericii (KU189725) and the potential pathogen Pantoea stewartii (Z96080).

Figure 3. Phylogeny of OPU467 assigned as Pantoea sp7 and OPU878 assigned as Coxiella sp1. The numerals (values > 50% are noted) indicate the percentage of bootstrap samplings as derived from 1,000 replications. Potential pathogens are indicated with asterisks. (A) Maximum-likelihood tree rooted with E. coli ATCC 11775T based on the representative 16S rRNA gene sequences of OPU467 defined as Pantoea sp7, other strains of species in the genus Pantoea, two type strains of Tatumella and seven type strains of Erwinia. The tree shows the taxonomic positions of OPU467 in the genus Pantoea. The sequence of E. coli ATCC 11775T serves as an outgroup. Bar, 0.010 substitutions per nucleotide position. (B) Maximum-likelihood tree rooted with Thioalkalivibrio denitrificans ALJDT and Rickettsia rickettsia based on the representative 16S rRNA gene sequences of OPU878 defined as the genus Coxiella sp1, and other strains of species in the genus Coxiella. The tree shows the taxonomic positions of OPU878 in the genus Coxiella. The sequences of Thioalkalivibrio denitrificans ALJDT and Rickettsia rickettsia serve as an outgroup. Potential pathogens are indicated with asterisks. Bar, 0.020 substitutions per nucleotide position.
OPU878 with 43,792 reads affiliated as an independent clade within the Coxiella genus (Figure 3B), closely related to Coxiella in Haemaphysalis hystricis S002T and Coxiella endosymbiont of Haemaphysalis hystricis HHTK1T, with identities of 96.6% ± 1.2% and 96.7% ± 2%, respectively. Therefore, OPU878 might represent a novel species in genus Coxiella. In addition, it showed a maximum identity of 95.6% to the human pathogen Coxiella burnetiid (HM208383).
-
In this study, we investigated the composition and pathogen distribution in microbial communities between biting midges and ticks collected from areas around Poyang Lake. We discovered that biting midges and ticks carry large numbers of known and potentially novel bacteria. Moreover, potentially pathogenic OPUs (mainly belonging to Asaia lannensis, Pantoea agglomerans, Staphylococcus equorum and Enterobacter ludwigii) are found in both vector arthropods, suggesting that these vector arthropods may increase the risk of various human and animal diseases when transmitting pathogens.
Previous studies of bacterial community composition in arthropods mainly used short-read amplicon sequencing based on partial 16S rRNA genes (for example, V3-V4), and were limited to genus-level taxonomical profiles. Here, we applied a metataxonomics approach with full-length 16S rRNA gene amplicons to elucidate the species-level structure and variation in the microbial communities between midges and ticks in the same area. Unlike most community studies based on OTU methods, this study used the comparison of OPU composition, which can yield more diverse data with high confidence, especially for low-abundance microbiota[23].
Similar to previous studies[16,33,34], the most dominant taxa found in biting midges and ticks are Proteobacteria, which are likely to be the prevalent bacterial members in multiple vector arthropods. Interestingly, the microbes of both arthropods are dominated by a few highly abundant bacteria. For instance, OPU in the genus Coxiella is the most abundant in ticks, and this result is generally concordant with those reported by Machado et al., and Guizzo et al.[35,36]. Coxiella, present within multiple tick species, is a vertically transmitted tick endosymbiont that maintains a steady commensal pattern with ticks and has successfully diversified within the communities[35,37]. These endosymbiotic bacteria in ticks have lost a mass of genes critical for a free-living lifestyle while reserving genes encoding major vitamin and cofactor biosynthetic pathways[38,39]. Ticks are obligate blood feeders and require endosymbionts for the contribution of cofactors and B vitamins that are scarce in blood to overcome the deficiencies dependent of using vertebrate blood as a primary nutrient source[36,40]. Therefore, Coxiella is essential for the survival and even overall fitness of some tick species. In our study, Coxiella was widely distributed among ticks and detected in all tick samples, further supporting this view. In the lone star tick Amblyomma americanum, Coxiella spp. which are widespread in the body tissues, was thought to be relevant to larval hatching success and viability[41]. Nevertheless, the underlying mechanism remains unclear. A better understanding of the patterns in which these Coxiella-like symbionts employ to colonize and survive on ticks will advance our comprehension of tick-microbiome interactions.
In the case of midges, Pantoea was the most common bacterial member, representing a large proportion of their abundances. However, we found no relevant reports of the dominance of Pantoea among species associated with midges. As reported elsewhere, Pseudomonas and Cardinium were some of the core bacterial communities in midges[42], distinct from our findings. But in wild mosquito Aedes albopictus, in the same order as midges, Pantoea was likewise found to be the most prevalent genus. A study has found that Pantoea enhances insect viability benefiting from the breakdown of toxic substances[43]. Many strains of Pantoea spp. show striking competition and fitness in diverse environments, making them particularly valuable for both biocontrol and bioremediation. In our study, according to phylogenetic analysis, the representative sequences of OPU467 affiliated with Pantoea were close to the potential pathogen Pantoea stewartii, implying a possible association with pathogenicity. However, much uncertainty remains regarding the host association and pathogenicity of individual strains, which deserves further investigation.
Considering that both midges and ticks were collected from the same habitat (area) and host animal source, we compared the composition of OPUs at the species level and observed tremendous divergence in the community composition of microbes for midges and ticks. This could be explained by their different diet structures. In comparison to ticks, midges are subject to a wider dietary spectrum, feeding on the blood of mammals and birds, or survive on plant sap and nectar[44]. In contrast, ticks have a single food source, bloodmeal from animal hosts alone, and therefore are subject to a narrower dietary diversity. Ticks spend their entire life stages dependent on their hosts[45], and it is expected that the bulk of tick microbiota are derived from these hosts, while certain symbiotic bacteria are genetically inherited maternally[5]. This would explain the lower community diversity in ticks, which may also be related to the dominance of their endosymbionts, possibly designed for the exclusion of potentially competing bacteria[46]. Further research on the association of arthropods and their feeding preferences would require a broader sampling of different arthropods. In addition, we also found the bacterial structure between free-living and ticks parasitizing on animals differed. Compared to ticks parasitizing on animals, there were higher ACE index and chao1 index in free-living ticks, indicating greater bacterial diversity. Ticks acquire microbes primarily from hosts and habitats, and these microbes are highly variable among ticks[47]. Given that these ticks parasitizing on animals were in engorged stages, we speculated that blood sucking serves to reset the microbiota. Another hypothesis is that the bacteria they obtain by feeding on host blood are influenced by their habitat environment and own metabolism, allowing some recolonization of the bacteria. Such heterogeneity could also result from random variation among arthropod individuals, along with distance, quantitative and ecological characteristics of diverse habitats, or methodological approaches regarding experimental processing and data analysis. It is worth mentioning that the bacterial component of the vector microbiota in our results had no significant associations with environmental and host characteristics (Supplementary Figure S4, available in www.besjournal.com), although midges and ticks were gathered over a range of locations and environmental conditions. Our samples were mostly collected from hosts, and animal hosts may provide a buffer effect to stabilize against potential environmental alterations.
Congruently, potentially pathogenic bacterial species were detected in both arthropods. Pantoea agglomerans, for instance, is not an obligate infectious agent in humans, but it could be a cause of opportunistic human infections, mainly related to penetrating trauma by vegetative material and hospital-acquired infection[48]. Next, Asaia lannensis is highly resistant to most antibiotics and can cause bacteremia[49]. Some species of the genus Acinetobacter are important pathogens of nosocomial infections; for instance, Acinetobacter lwoffii is emerging as the cause of neonatal sepsis[50]. These potential pathogens associated with epidemiological functions may lead to an increased risk of infectious diseases and thus deserve in-depth analysis to prevent and control vector-borne diseases.
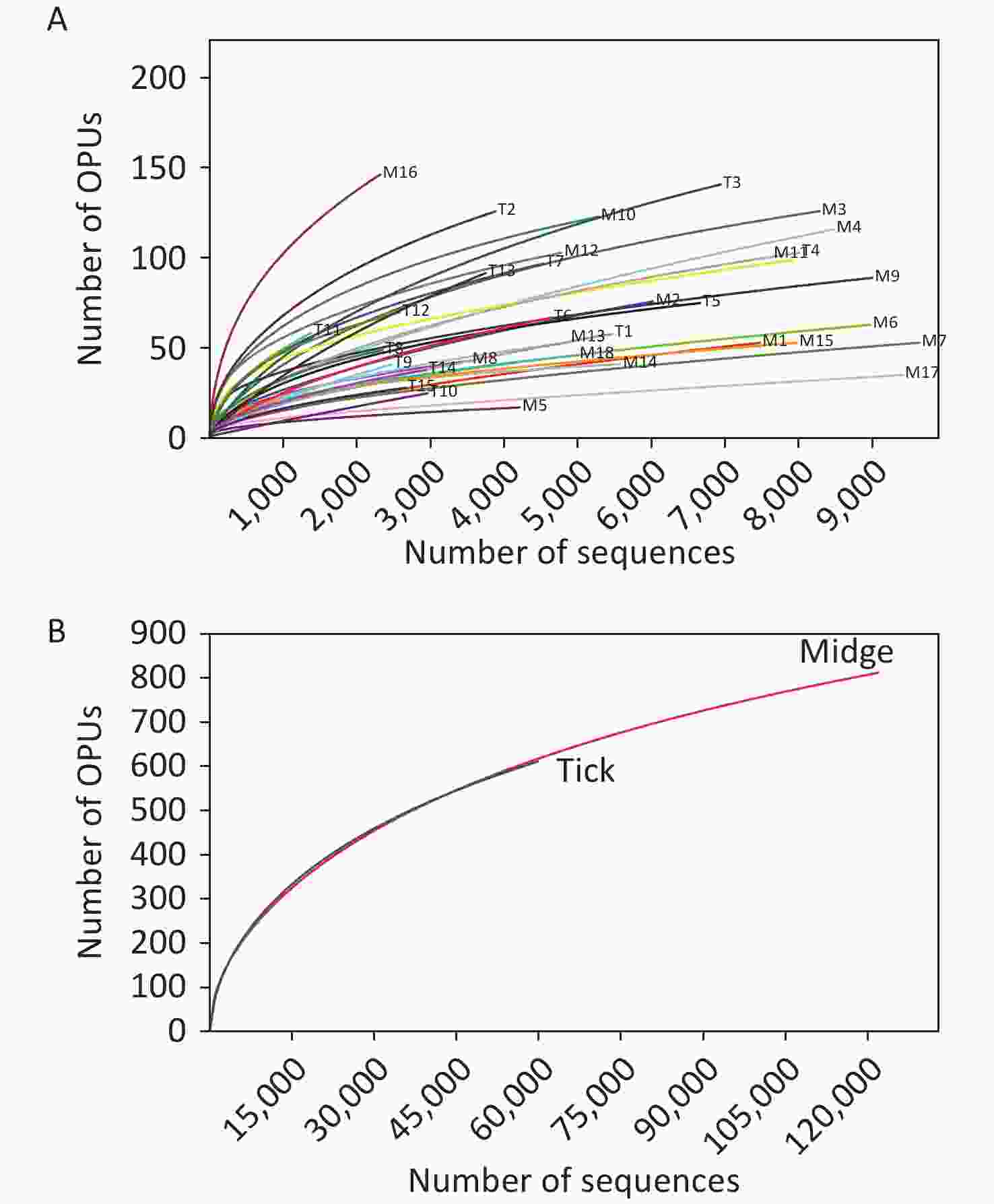
Figure S1. Rarefaction curves for 16S rRNA gene sequences of (A) all samples and (B) both vectors. Each line represents an independent sample.
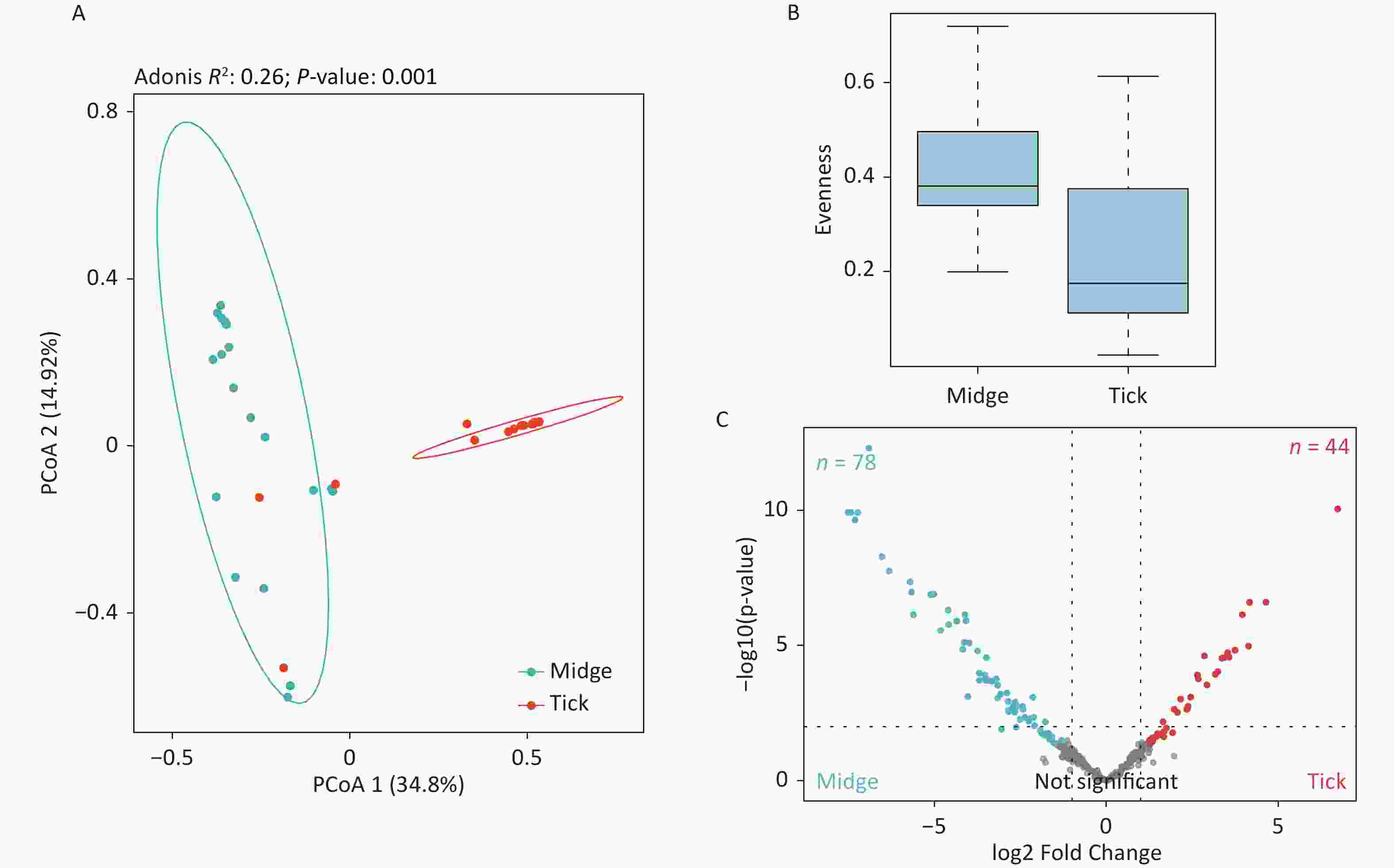
Figure S2. Assessment of microbial diversity in midge and tick groups. (A) Principal coordinate analysis (PCoA) of midge and tick bacterial communities. Each dot represents a collective sample. All samples found inside the circle constitute biological replicates at a confidence level of 99.95%. The samples presented two distinct clusters, as indicated by Adonis test (R2 = 0.24, P = 0.001). (B) Boxplots representing the OPU (Operational phylogenetic Unit) evenness by vector type. (C) Volcano plot for identifying differentially expressed OPUs using a negative binomial generalized linear model. Midges and ticks were differentially enriched for 78 and 44 OPUs, respectively, at a threshold of α < 0.05.
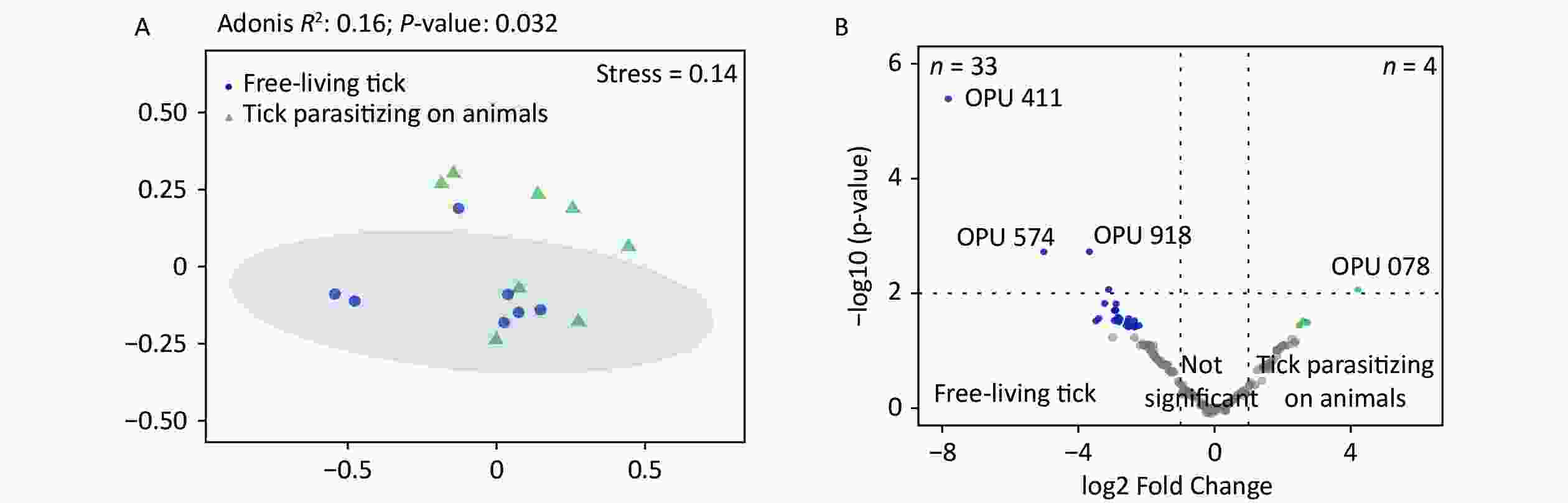
Figure S3. Comparisons of bacterial microbiota among ticks. (A) Non-metric multidimensional scaling ordination—NMDS—of the bacterial communities of free-living ticks and ticks parasitizing on animals. The samples presented two distinct clusters, as indicated by Adonis test (R2 = 0.16, P = 0.032). (B) Volcano plot for identifying differentially expressed OPUs between free-living ticks and ticks parasitizing on animals using a negative binomial generalized linear model. Free-living ticks and ticks parasitizing on animals were differentially enriched for 33 and 4 OPUs, respectively, at a threshold of α < 0.05.
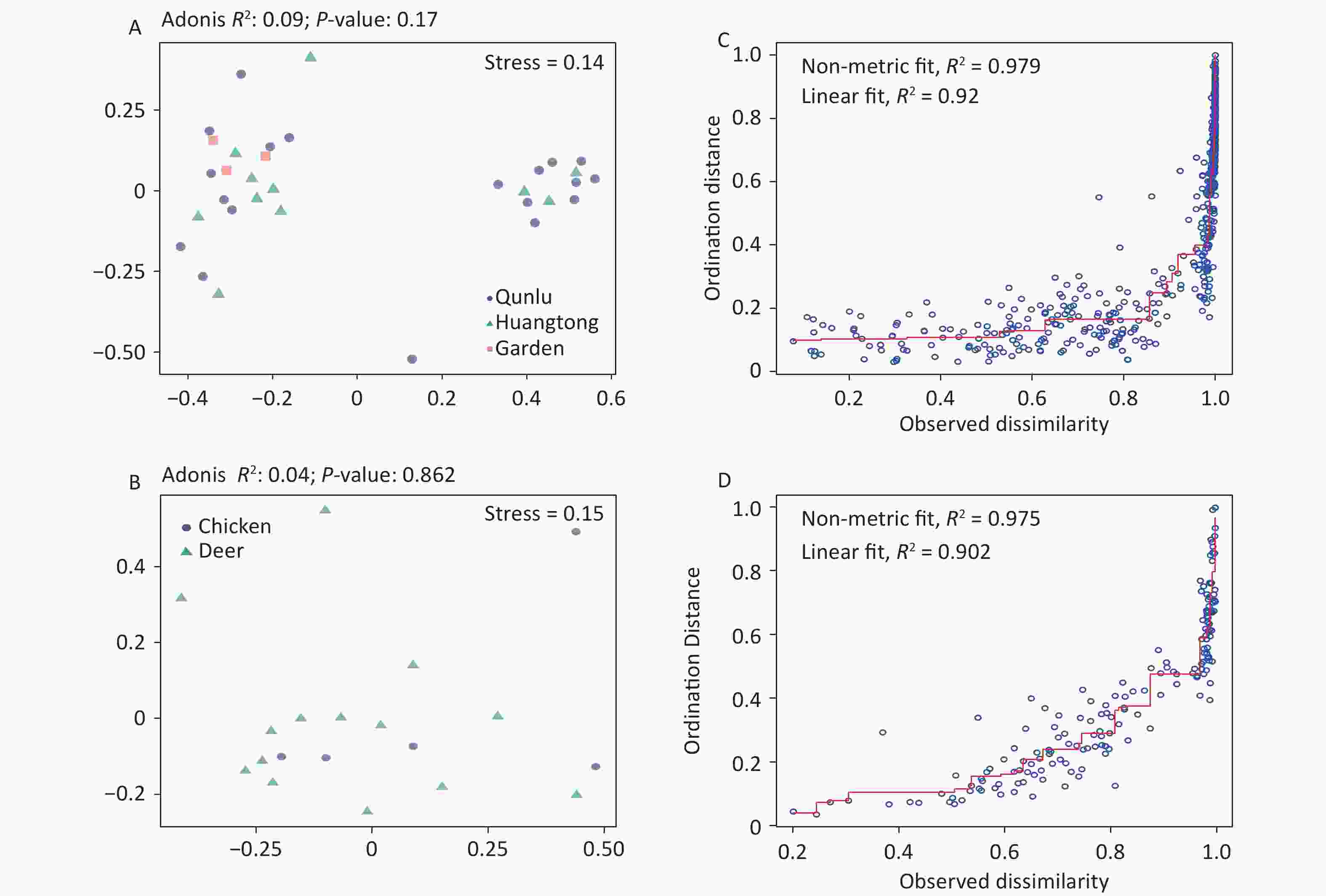
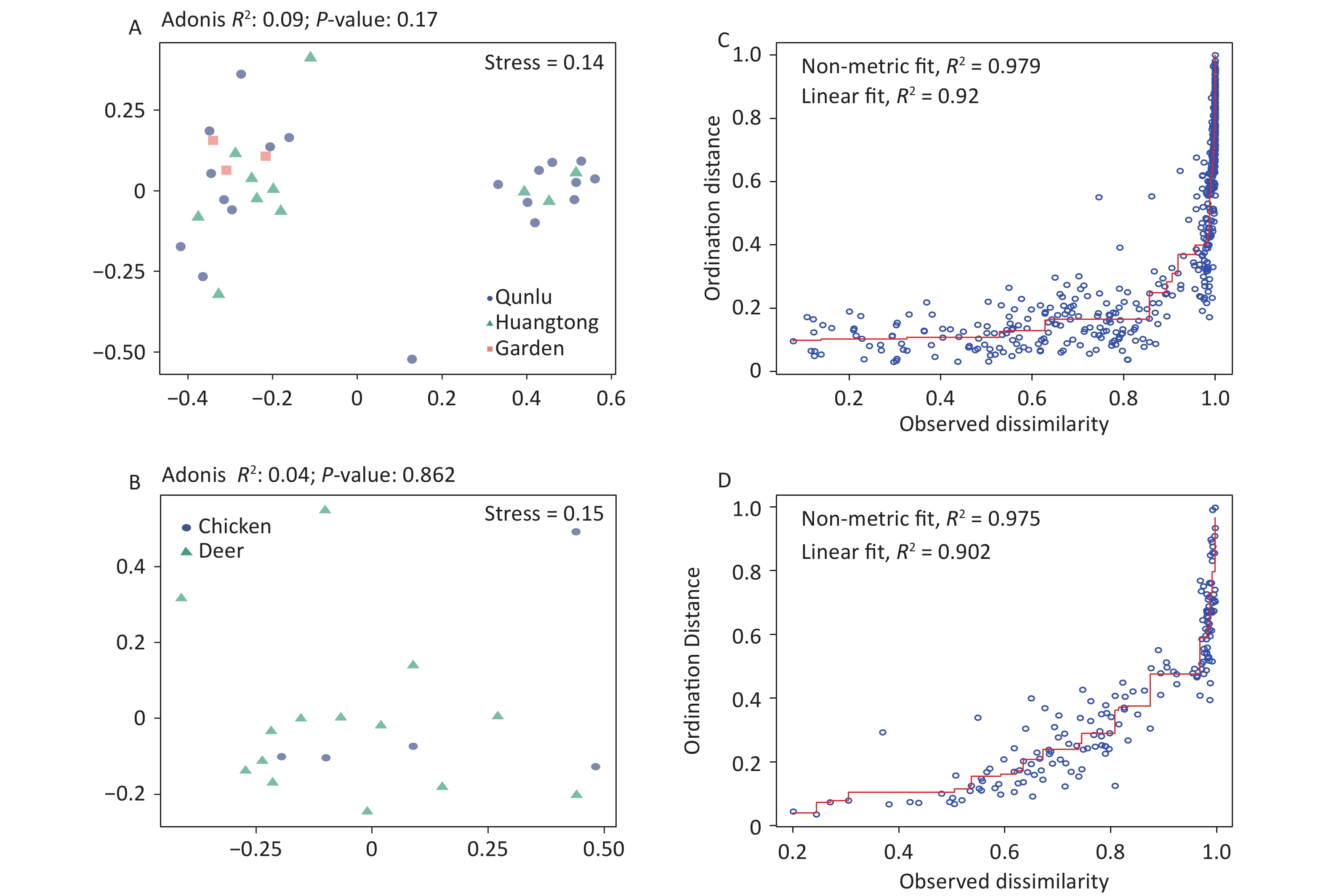
Figure S4. Non-metric multidimensional scaling ordination—NMDS—of the bacterial communities of all OPUs with respect to (A) geography and (B) hosts of midges. There was no significant difference in the bacterial community structure between geography (Adonis, R2 = 0.09, P = 0.17) and hosts of midges (Adonis, R2 = 0.04, P = 0.86). Shepard stress diagrams for all OPUs with respect to (C) geography and (D) hosts of midges.
-
By using an almost full-length of 16S rRNA genes amplicon sequencing approach, we determined the species-level bacterial community for biting midges and ticks. The results revealed that Proteobacteria was the most dominant group in all samples. We observed the presence of potentially pathogenic OPUs in both vector arthropods. Furthermore, the bacterial components of the vector types (biting midge vs. tick) tend to be governed by a few highly abundant bacteria, where Pantoea sp7 was enriched in biting midges, while Coxiella sp1 was more abundant in ticks and Coxiella spp. were detected in all tick samples. We also found that Coxiella may be essential for the survival of Haemaphysalis longicornis. Overall, the identification of dominant species in midges and ticks in this study is relevant in advancing our comprehension as to the microbes of arthropod vectors. However, we propose to capture a larger and more diverse sample of individuals in extensive sampling across multiple sites and to finely characterize the key structural and functional features of vectorial taxa in future studies. Then, the evolutionary processes and determinants of regional patterns driving vectorial biodiversity will be more precisely unraveled. Our research highlights the necessity for knowledge of the basic structure and evolution of vector-borne microbial communities, as they are potentially associated with the infection and transmission of pathogenic and infectious diseases in humans.
-
The Ethical practice was approved by Ethical Committee of National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention, China (No. ICDC-2019012).
HTML
Sample Collection
Total DNA Extraction and Full-Length 16S rDNA Amplification and Sequencing
Species-level Taxonomy by Operational Phylogenetic Unit (OPU) Analyses
Statistical Analysis
Screening for Pantoea and Coxiella
16S rRNA Gene Based Phylogenetic Analysis
Data Collection
Bacterial Diversity and Richness in Arthropods
Microbiota Profile at High Taxon Levels
Species-level Taxonomic Annotation
Bacterial Communities Vary in Arthropods
Distribution of Pantoea in Midges and Coxiella in Ticks
Identification of Novel Species in the Genera Pantoea and Coxiella
AVAILABILITY of DATA and MATERIALS All data generated or analyzed during this study are included in this article and its supplementary information files. Sequence files and metadata for all samples used in this study have been deposited under project PRJNA898083 in NCBI. The GenBank accession numbers for sequencing data of genus-specific amplified sequences are OR060692-OR060698.
DISCLOSURE STATEMENT The authors declare that they have no competing interests.
 23158+Supplementary Materials.pdf
23158+Supplementary Materials.pdf
|

|

 Quick Links
Quick Links
 DownLoad:
DownLoad: