-
Coxsackievirus (CV) B belongs to the species Enterovirus B, genus Enterovirus of the family Picornaviridae. Enterovirus B (EV-B) includes 63 serotypes: CVB1–6; CVA9; echoviruses E1–7, 9, 11–21, 24–27, and 29–33; EV-B69, EV-B 73–75, EV-B 77–88, EV-B 93, EV-B 97–101, EV-B 106–107, and EV-B 110–114 (www.picornaviridae.com/ensavirinae/enterovirus/ev-b/ev-b.htm). Like other EV-Bs, CVBs are single-stranded, small non-enveloped and positive-sense RNA viruses. The CVB genome is ≥ 7,400 nucleotides long and includes a large open reading frame (ORF), encoding a single pre-polyprotein that is further processed into P1, P2, and P3. P1 is cleaved to VP4, VP2, VP3, and VP1 structural proteins and P2, and P3 is cleaved into seven nonstructural proteins (2A, 2B, 2C, 3A, 3B, 3C, and 3D)[1]. Mature structural proteins can be used for viral assembly, while nonstructural proteins can be used for replication, induction of apoptosis, suppression of innate immunity, and shutting down host cell translation[2].
CVB1 is commonly associated with mild infections [fever, irritation, and hand, foot and moth disease (HFMD)]; however, some patients may develop serious illness, such as aseptic meningitis, myocarditis, acute flaccid myelitis, and necrotizing hepatitis. CVB1 is also associated with type 1 diabetes[3]. Since 2008, CVB1 has frequently been detected in HFMD cases and associated with severe disease[4]. In 2019, CVB1 was one of the most frequent causative pathogens of HFMD in Yunnan, China[5]. Eleven complete genomes of CVB1 are currently accessible in GenBank, and three strains, CVB1/XM0108 (MG780414), MSH/KM9/2009 (JN596588), and CVB1SD2011CHN (JX976769), were isolated in China between 2009 and 2011. Hence, in the study, the six full-length genome sequences of CVB1 strains were determined and compared with those of other EV-Bs.
During the HFMD surveillance in 2019, 242 stool samples were collected from children with HFMD under 5 years old in Yunnan, Southwest China. The green monkey kidney (Vero), the human rhabdomyosarcoma (RD) and human embryonic lung diploid fibroblast (KMB17) cell lines were used to isolate the samples, respectively. Viral RNA was extracted from positive culture supernatants that produced typical EV-like cytopathic effect (CPE) using the QIAamp Viral RNA Mini Kit (Qiagen, CA, USA). Reverse transcription polymerase chain reaction (RT-PCR) was performed using the PrimeScript™ One Step RT-PCR Kit Ver.2 (TaKaRa, Dalian, China). The primers BOAS and BOS were used, and the positive RT-PCR products were sequenced on an ABI 3130 Genetic Analyzer (Applied Biosystems, USA) at Kunming Qingke Biotechnology Co. (Yunnan, China). Six CVB1 strains (7V3/YN/CHN/2019, 46V3/YN/CHN/2019, 53V3/YN/CHN/2019, 55V3/YN/CHN/2019, 62V3/YN/CHN/2019, and 64V3/YN/CHN/2019) were identified as CVB1 by enterovirus typing online software (https://www.rivm.nl/mpf/typingtool/enterovirus/). However, the CVB1 isolates were only isolated from Vero cells, whereas none were from RD and KMB17 cells. This indicated CVB1 is sensitive to Vero cells. Then, two long-fragment PCR amplifications of viral RNA were performed using PrimeScriptTM One Step RT-PCR Kit Ver.2 (TaKaRa) and the following primers: E201F (5’ TTAAAACAGCCTGTGGGGTTG 3’) and B12R (5’ TTAGTGGGCCGTGAACTAAT 3’) and B13F (5’ AACCCAAGCACGTGAAGGCG 3’) and EV8R (5’ CACCGAATGCGGAGAATTTA 3’). The sequencing primers for the complete genome were designed according to the primer walking strategy[6]. All primers used in this study are shown in Supplementary Table S1 (available in www.besjournal.com). Nucleotide and amino acid sequence comparisons were carried out using Geneious Basic 5.4.1 Beta software. Phylogenetic analyses were performed using MEGA 7.037. Recombination Detection Program (RDP) 4.101 software in default mode was used to determine recombinant sequences using seven algorithms (RDP, GENECONV, BootScan, Maxchi, Chimaera, SiScan, and 3Seq). The complete genome sequences of the six CVB1 strains isolated in this study were submitted to GenBank under accession numbers OQ259520–OQ259525.
Primer Sequence (5ʼ→3ʼ) Nucleotide position Orientation BOAS#* GGTGCTCACTAGGAGGTCYCT RTTRTARTCYTCCCA GNAYRWACAT 3,505–3,484 Reverse BOS#* GGYTAYATNCANTGYTGGTAYCARAC NGARAYNGG 2,324–2,329 Forward E201F#* TTAAAACAGCCTGTGGGTTG 1–20 Forward B12R TTAGTGGGCCGTGAACTAAT 2,519–2,500 Reverse B11r CCTGAGCTCCCATTTCG 751–735 Reverse B11f CAGTGACAAACAGAGCTA 636–653 Forward B12f CCAGGTAAGTTCACTGAA 886–903 Forward B13F AACCCAAGCACGTGAAGGCG 3,157–3,176 Forward EV8R#* CACCGAATGCGGAGAATTTA 7,444–7,426 Reverse B14f GGCTGGAGGACGATGCAAT 3,707–3,726 Forward B14f1 CTGTTCTCCAATGTGCAGTAC 4,255–4,275 Reverse B14f2 TCACTCAAGGCATTAGTG 3,453–3,471 Forward B15f AGATGTTYAGAGARTACAAC 4,955–4,974 Forward B15f2 AGGCTAAGGTTCAAGGGC 5,332–5,349 Forward B16f CGCATGTTGATGTACAAC 5,749–5,766 Forward B17f* AAGGGGTATGGTCTGAT ACCTTCCTTAAAAGATACTTTA 6939–6955 Forward Table S1. Primers of amplification and sequencing for the complete genomes
The whole VP1 sequences (834 nucleotides) of the six isolates showed the highest identity (97.7%–97.8%) with CVB1 strain CV-B1/SWG69/SD/CHN/2018 (MN541037), isolated from sewage in Shandong, China, 2018. The six isolates shared 79.3%–80.2% nucleotide identity and 93.9%–94.2% amino acid identity with the whole VP1 sequence of the CVB1 prototype strain Conn-5 (M16560) and 80.1%–97.7% nucleotide and 93.9%–100% amino acid identity with other CVB1 strains. The complete VP1 nucleotide and amino acid identities among the six isolates were 94.4%–100% and 98.2%–100%, respectively.
The 131 whole VP1 sequences available in GenBank were contained in this study (Supplementary Figure S1, available in www.besjournal.com). The VP1 sequence divergences between EV genotypes was 15% at the nucleotide level[7]. All CVB1 strains were divided into genotypes A–E. D and E genotypes were further divided into D1 and D2, and E1 and E2 subgenotypes, respectively (Supplementary Table S2, available in www.besjournal.com). The Chinese CVB1 strains belonged to D1 (2003–2018), E1 (1989–1994), and E2 (1999–2019) subgenotypes. Along with most Chinese strains, the six isolates in the study belonged to E2 subgenotype. In addition, the average differences between the VP1 nucleotide and amino acid sequences of these six isolates and all full VP1 gene sequences of other Chinese strains in GenBank were 10.65% (2.20%–19.10%) and 3.25% (0.4%–6.1%). Compared with the three Chinese CVB1 strains in GenBank, CVB1/XM0108 (MG780414), MSH/KM9/2009 (JN596588), and CVB1SD2011CHN (JX976769), the differences between their whole genome nucleotide and amino acid sequences were 8.90% (4.70%–13.10%) and 6.15% (1.50%–10.80%), respectively. This indicates that CVB1 strains have evolved in China.
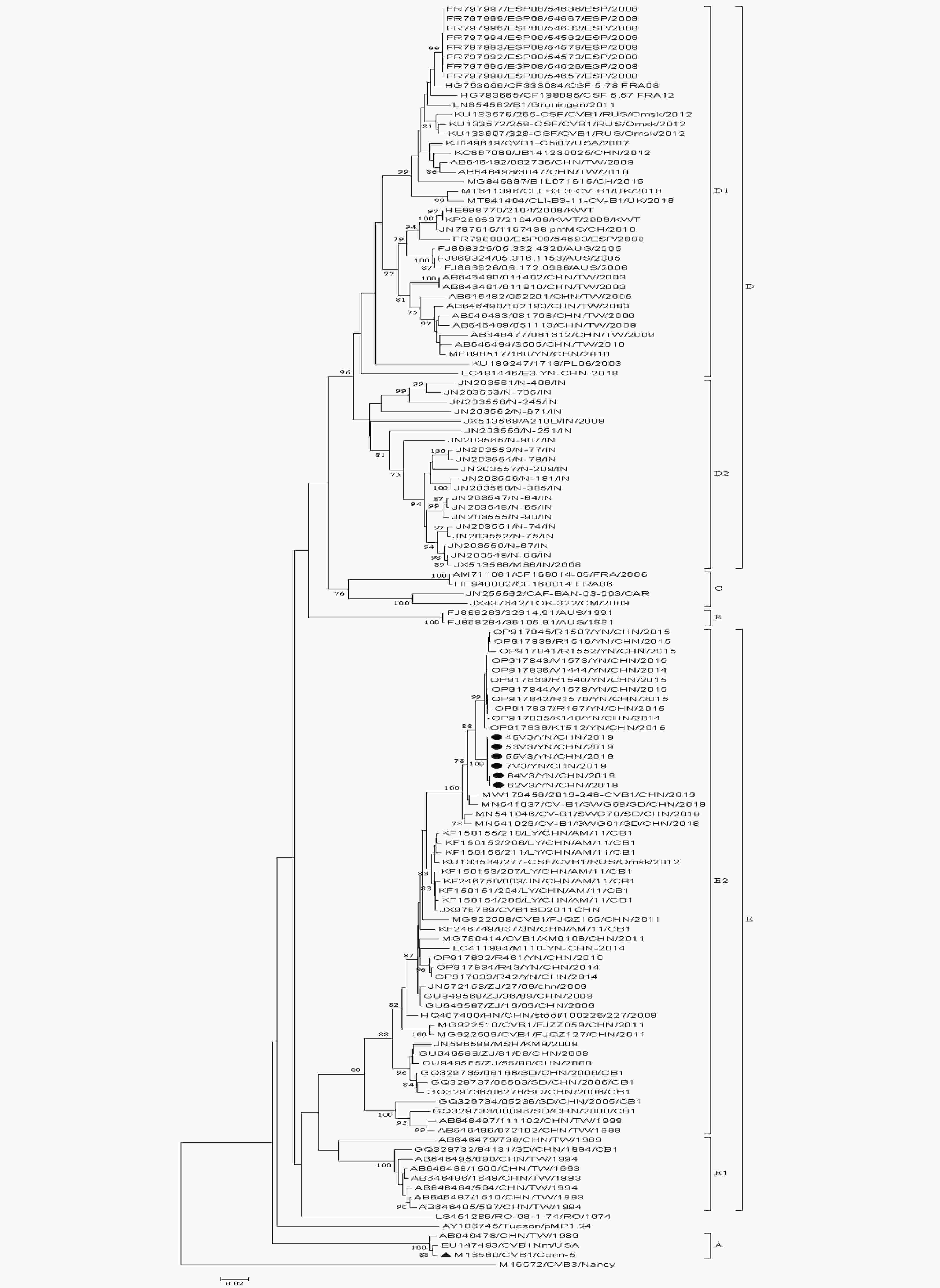
Figure S1. Phylogenetic tree based on complete VP1 sequences (834 bp) of all CVB1 isolates available in GenBank. λStrains isolated in this study. ▲Prototype strain.
Genotype A B C D1 D2 E1 E2 A 80.5–80.7 80.7–81.7 78.4–79.0 78.1–79.0 81.3–80.3 79.0–80.3 B 96.0–96.4 82.1–83.7 83.3–83.9 83.8–84.1 82.5–82.9 81.3–83.1 C 95.3–96.4 96.0–97.8 84.3–85.0 83.2–86.6 83.3–84.1 80.7–82.9 D1 92.8–95.7 97.5–98.2 96.4–97.5 88.4–90.0 83.7–85.6 81.1–83.0 D2 96.0–97.1 97.5–98.2 97.1–98.2 97.8–98.1 83.7–83.9 80.0–84.1 E1 95.3–95.7 95.7–96.0 96.0–98.6 96.8–97.1 96.8–97.1 85.4–87.1 E2 92.8–95.0 93.2–96.0 93.9–96.0 95.0–97.1 94.2–96.4 96.0–98.6 Note. the data in the upper right corner was for nucleotide homology analysis and the data in the lower left corner was for amino acid homology analysis. Table S2. The VP1 nucleotide and amino acid homology comparison between CVB1 genotypes or subgenotypes
The whole genomes of the six CVB1 strains (7V3/YN/CHN/2019, 46V3/YN/CHN/2019, 53V3/YN/CHN/2019, 55V3/YN/CHN/2019, 62V3/YN/CHN/2019 and 64V3/YN/CHN/2019) isolated in Yunnan, Southwest China, 2019 were obtained. The length of their genomes was 7,402–7,412 nucleotides, containing a single ORF of 6,549 nucleotides, and encoding a polyprotein of 2,182 amino acids. The ORF was flanked by a 5′-untranslated region (UTR) of 750–757 nucleotides and 3′-UTR of 105–113 nucleotides. The whole genome nucleotide and amino acid identities of the six strains were 99.6%–99.8% and 99.1%–99.8%, respectively. The overall base compositions were 28.1%–28.2% A, 22.9%–23.0% G, 22.6%–24.7% C, and 24.2%–24.3% U. The whole genome nucleotide sequences and the deduced amino acid sequences of the six strains had > 99.6% mutual identity; therefore, the 46V3/YN/CHN/2019 strain was selected as a representative strain for further analysis. The 46V3/YN/CHN/20193 strain showed 79.0% and 79.0%–95.2% whole genome nucleotide identities and 95.0% and 95.0%–98.8% amino acid sequence identities with the prototype strain Conn-5 and other CVB1 strains, respectively (Table 1).
Genomic
regionPrototype strain Conn-5, % Other CVB1 strains, % Nucleotide identity Amino acid identity Nucleotide identity Amino acid identity 5′-UTR 84.0 84.0–96.8 VP4 82.1 94.2 79.7–94.7 92.8–98.6 VP2 80.0 96.2 80.0–94.6 96.2–99.6 VP3 77.7 95.1 77.7–95.5 93.1–98.7 VP1 79.8 94.2 79.8–95.3 94.6–99.6 2A 74.4 89.3 74.0–94.2 88.7–94.0 2B 75.1 93.9 74.7–96.6 93.9–99.0 2C 80.5 98.9 79.6–94.8 97.6–99.1 3A 77.9 95.5 77.5–94.8 94.4–97.9 3B 71.2 86.4 71.2–95.5 81.8–90.9 3C 77.0 94.5 77.0–96.5 94.5–98.9 3D 78.3 95.7 77.7–95.2 91.6–99.1 3′-UTR 76.6 72.7–93.5 Genome 79.0 95.0 79.0–95.2 95.0–98.8 Note. UTR, non translation area. Table 1. Nucleotide and amino acid sequence identity between six isolates and the prototype strain and other CVB1 strains in all sequenced genomic regions
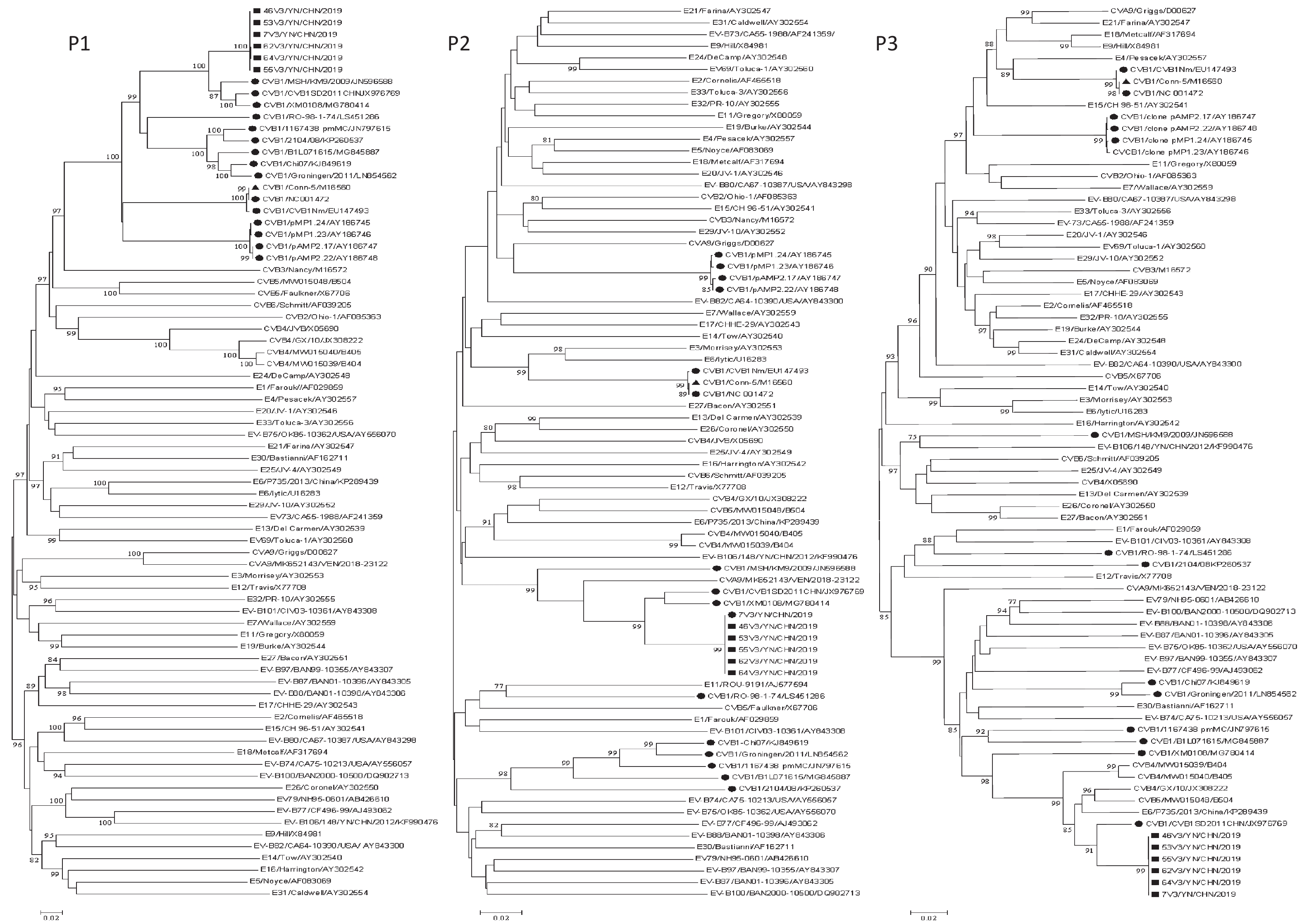
Phylogenetic analyses were performed on the P1, P2, and P3 regions of all available CVB1 strains and EV-B prototype strains in GenBank, as well as the six Yunnan CVB1 isolates and six EV-B epidemic strains (CVA9/VEN/2018-23122/MK652143, E6/P735/2013/China/KP289439, CVB4/B405/MW015040, CVB4/GX/10/JX308222, CVB4/B404/MW015039, and CVB5/B504/MW015048) (Figure 1). In the P1 coding region, the six isolates clustered with all CVB1 strains including the CVB1 prototype Conn-5 strain, which confirmed the initial VP1 typing result. The CVB1 strains clustered with different EV-B strains to form different clusters in the P2 and P3 coding regions (Figure 1). In the P2 coding region, the six isolates clustered with three CVB1 isolates (CVB1SD2011CHN/JX976769, XM0108/MG780414, and MSH/KM9/2009/JN596588) and CVA9/MK652143/VEN/2018-23122 strain. CVB1 strain RO-98-1-74/LS451286 clustered with E11 strain ROU-9191/AJ577594. CVB1 Nm/EU147493, Conn-5/M16560, and CVB1 strain NC 001472 clustered with E6 prototype lytic/U16283. E3 prototype Morrisey/AY302553 clustered with CVA9 prototype strains Griggs/D00627, pMP1.24/AY186745, pMP1.23/AY186746, pAMP2.17/AY186747, and pAMP2.22/AY186748. The five CVB1 strains, CVB1-Chi07/KJ849619, Groningen/2011/LN854562, 1167438 pmMC/JN797615, B1L071615/MG845887, and 2104/08/KP260537, exhibited clustering. In the P3 region, the six isolates only clustered with one CVB1 strain, namely CVB1 SD2011CHN/JX976769, along with five EV-B strains (E6/P735/2013/China/KP289439, CVB5 /B504/MW015048, CVB4/GX/10/JX308222, CVB4 /B404 /MW015039, and CVB4//B405 /MW015040). The prototype Conn-5 strain formed a single lineage with the E4 prototype strain Pesacek/AY302557. The findings suggest that multiple instances of recombination have taken place among the CVB1 strains (including this study) and other EV-B strains in the nonstructural coding regions. Furthermore, it is plausible that these CVB1 isolates represent distinct recombinant strains. The online BLAST tool was utilized to screen for sequences that exhibited the top three identities with strain 46V3/YN/CHN/2019 (Supplementary Table S3, available in www.besjournal.com). The 3A to 3D regions of strain 46V3/YN/CHN/2019 shared the highest identity with that of CVB1SD2011CHN, and followed with other EVBs (CVB5 strain B504 and E6 strain E6/E6SD11CHN). In the 3′-UTR, strain 46V3/YN/CHN/2019 shared the greatest identity (93.58%) with E6 strain E6/P735/2013/China.
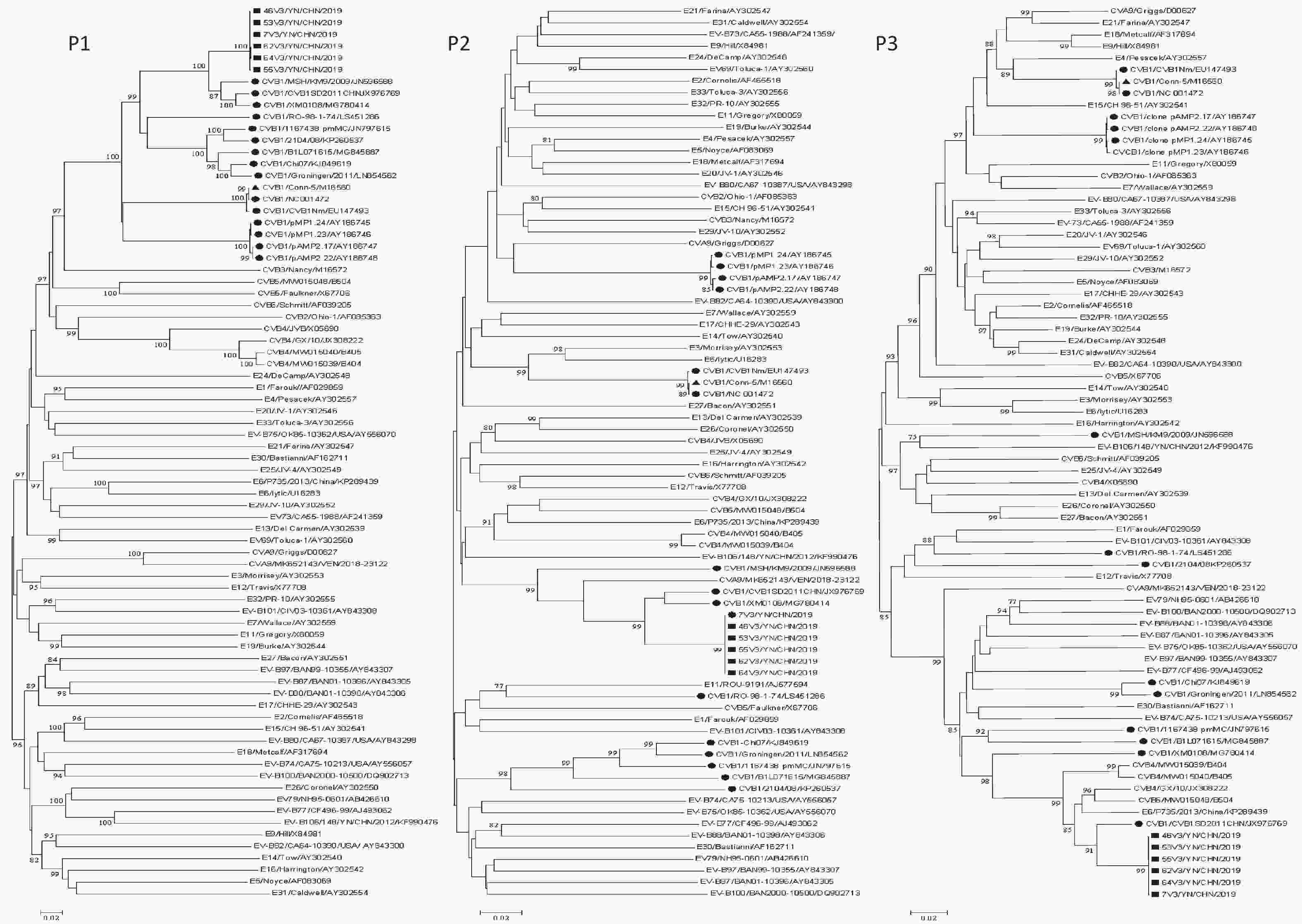
Figure 1. Phylogenetic trees based on the P1, P2, and P3 regions of all CVB1 strains and EV-B prototype strains available in GenBank, the six Yunnan isolates, and five EV-B strains: one CVB5 strain B504, three CVB4 strains (GX/10, B405, and B404), and one E6 strain E6/P735/2013/China). Nucleotide sequences were analyzed using the neighbor-joining algorithm (1,000 bootstrap repeats) in MEGA 7.0.26. The scale bars indicate the genetic distance. Numbers at the nodes indicate bootstrap support. Only high boot values (> 75%) are displayed. νStrains isolated in this study. λOther CVB1 strains. ▲CVB1 prototype strain.
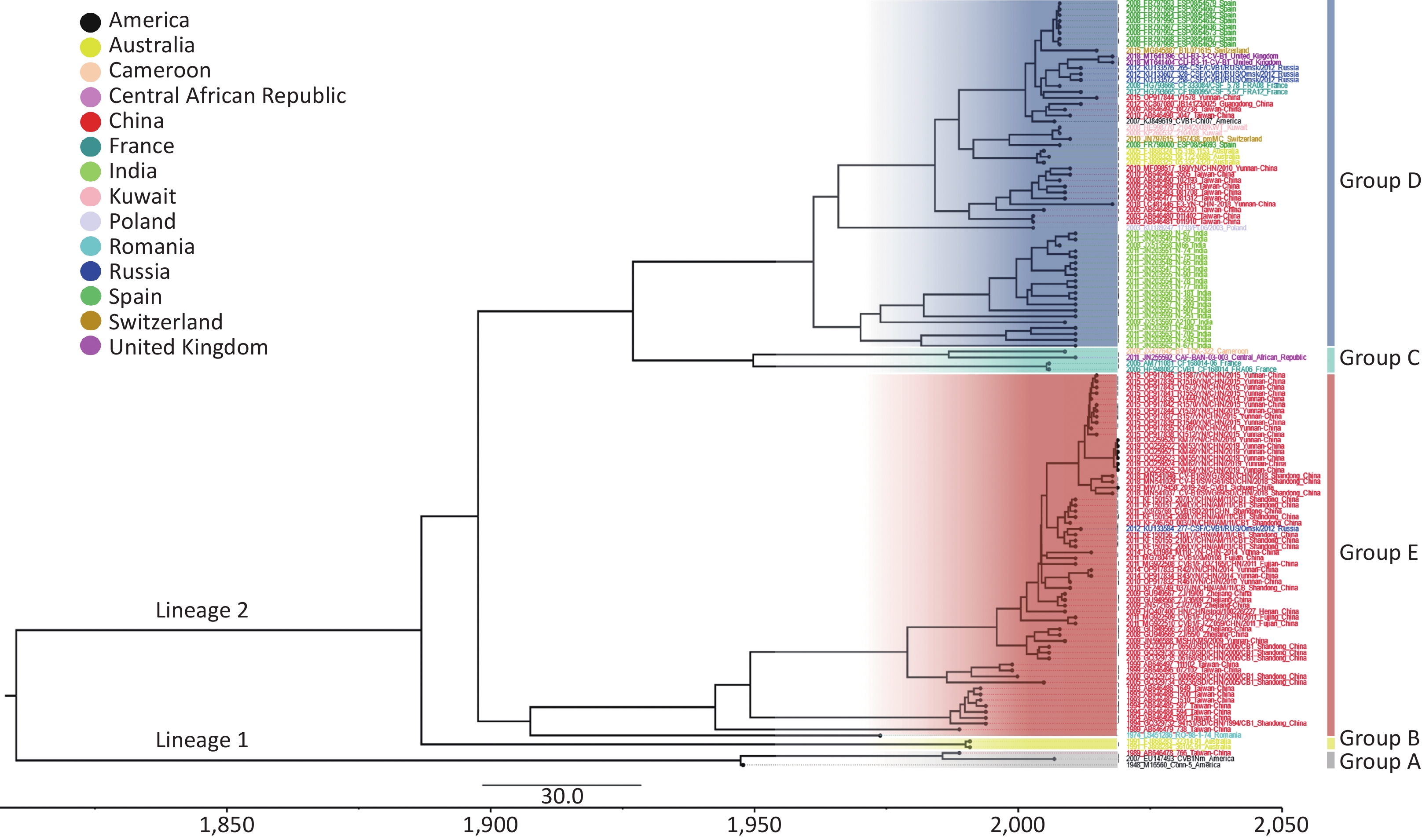
Bayesian Markov Chain Monte Carlo (MCMC) analysis of the worldwide CVB1 VP1 sequences was carried out with the combination of the strict clock model, GTR + Gamma nucleotide substitution model and a Bayesian skyline tree prior. The MCMC chain length was set to 200 million states, and sample was taken every 20000 states. After produced through TreeAnnotator v1.10.4, the Maximum Clade Credibility (MCC) tree was visualized by FigTree v1.4.2 (Figure 2). The clustering structure of the MCC tree was highly similar to that of the phylogenetic analysis (Supplementary Figure S1). The mean base substitution rate of CVB1 VP1 was estimated to be 2.0 × 10–3 substitutions/site/year, with the 95% highest posterior density (HPD) was 1.5 × 10–3–2.5 × 10–3. The most recent common ancestor (tMRCA) of CVB1 worldwide could date back to 1810.1 (95% HPD: 1755.2–1852.9).
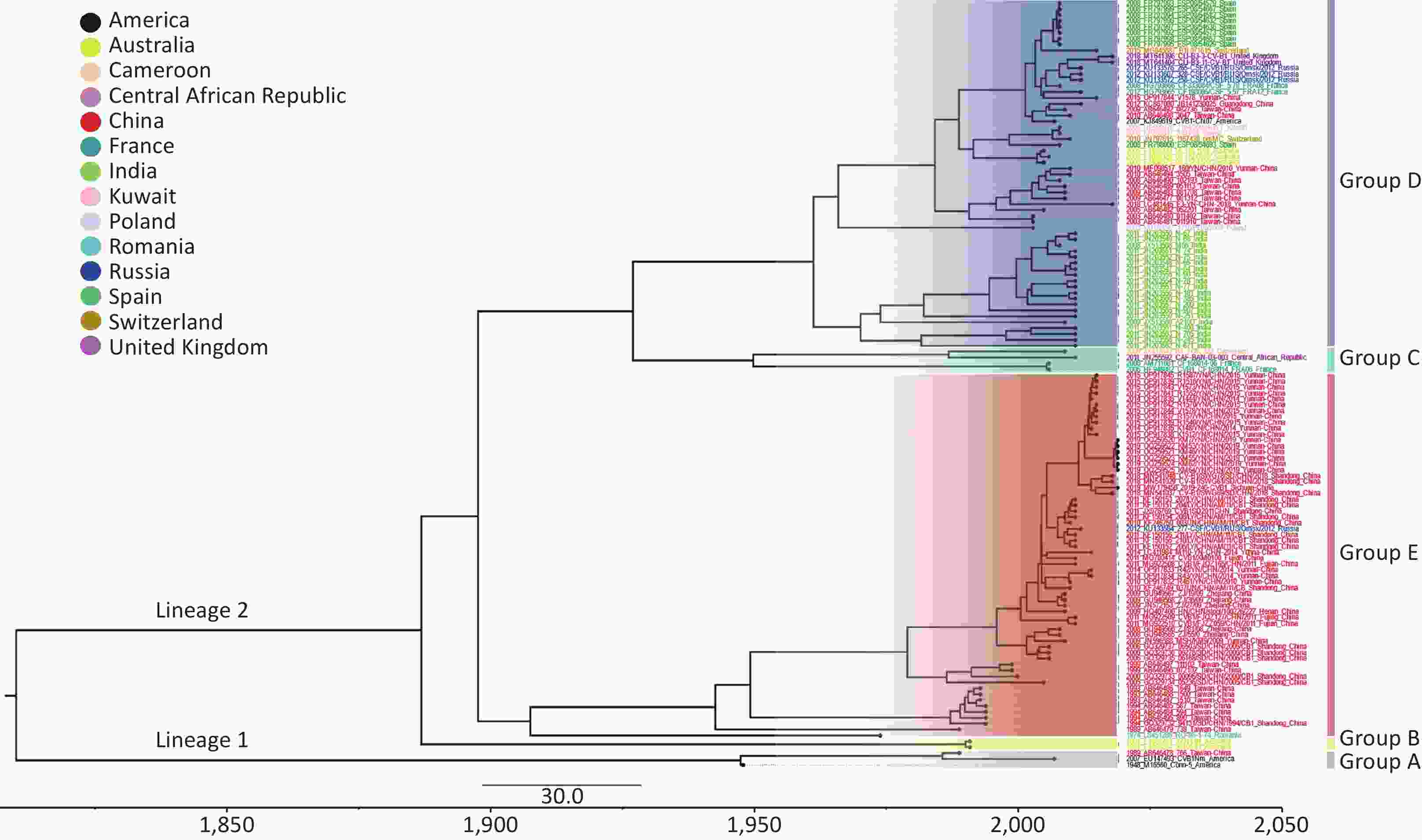
Figure 2. Maximum clade credibility (MCC) tree based on the complete VP1 sequences of global CVB1 strains available in GenBank. The different colours of the tip labels corresponded to the countries where the strains were collected, and the colour panel is placed at the top left part. The time scale is given at the bottom of Figure 2.
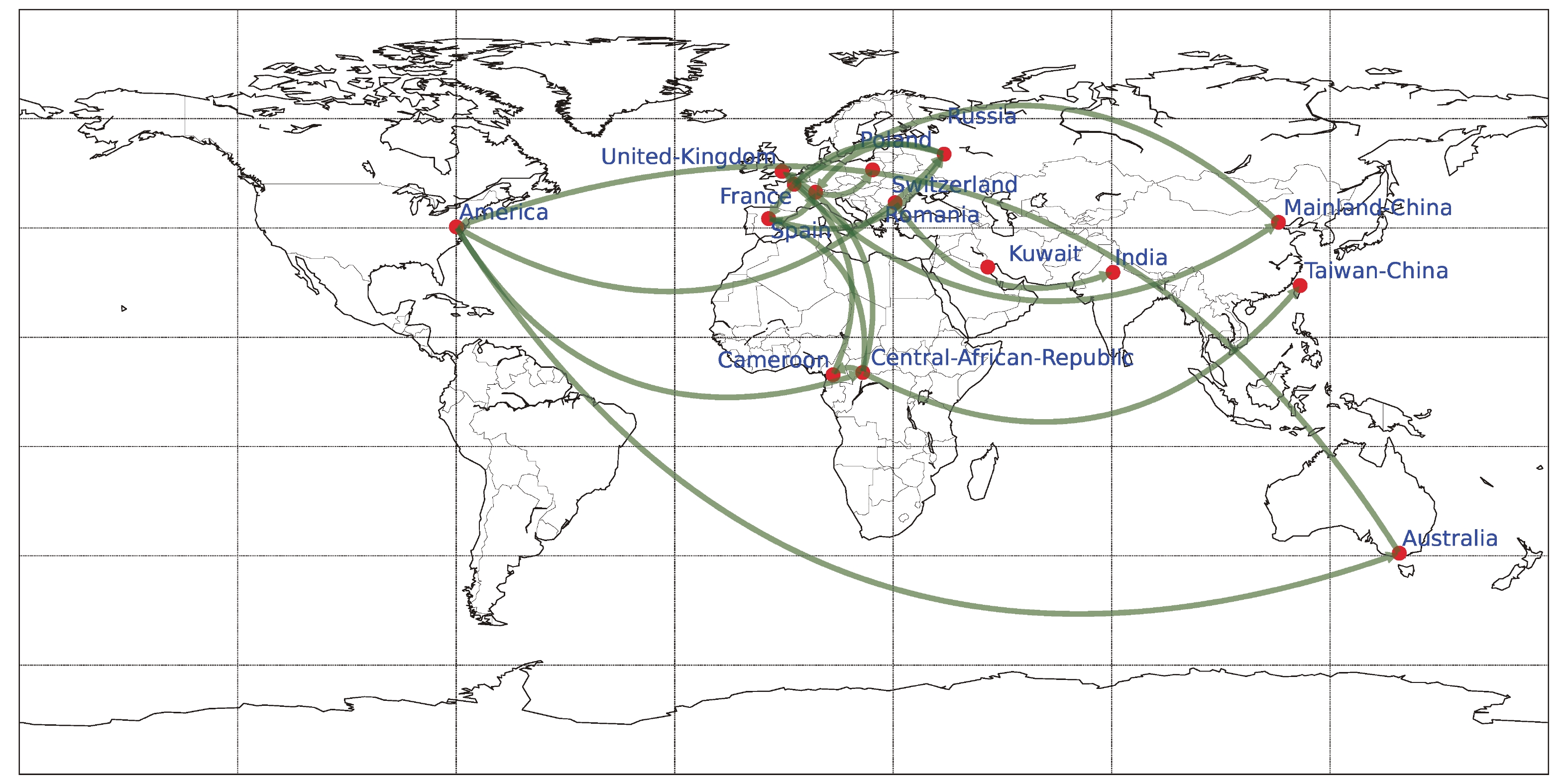
Phylogeographic analysis was carried out using BEAST v1.10.4, employing Bayesian stochastic search variable selection (BSSVS) and the Asymmetric Substitution Model. The migration pathways and their Bayes factors (BF) were assessed utilizing SpreaD3 v0.9.6, and subsequently visualized on a map. (Supplementary Figure S2, available inwww.besjournal.com). The results showed that there were 17 major CVB1 migration routes (BF > 5) worldwide from 1948 to 2019. Notably, transmission originating from the United States, either directly or via Africa, to Europe, as well as inter-country transmission within Europe, predominates. Additionally, bidirectional transmission of CVB1 was observed between the United States and Australia, as well as between Mainland China and Europe.
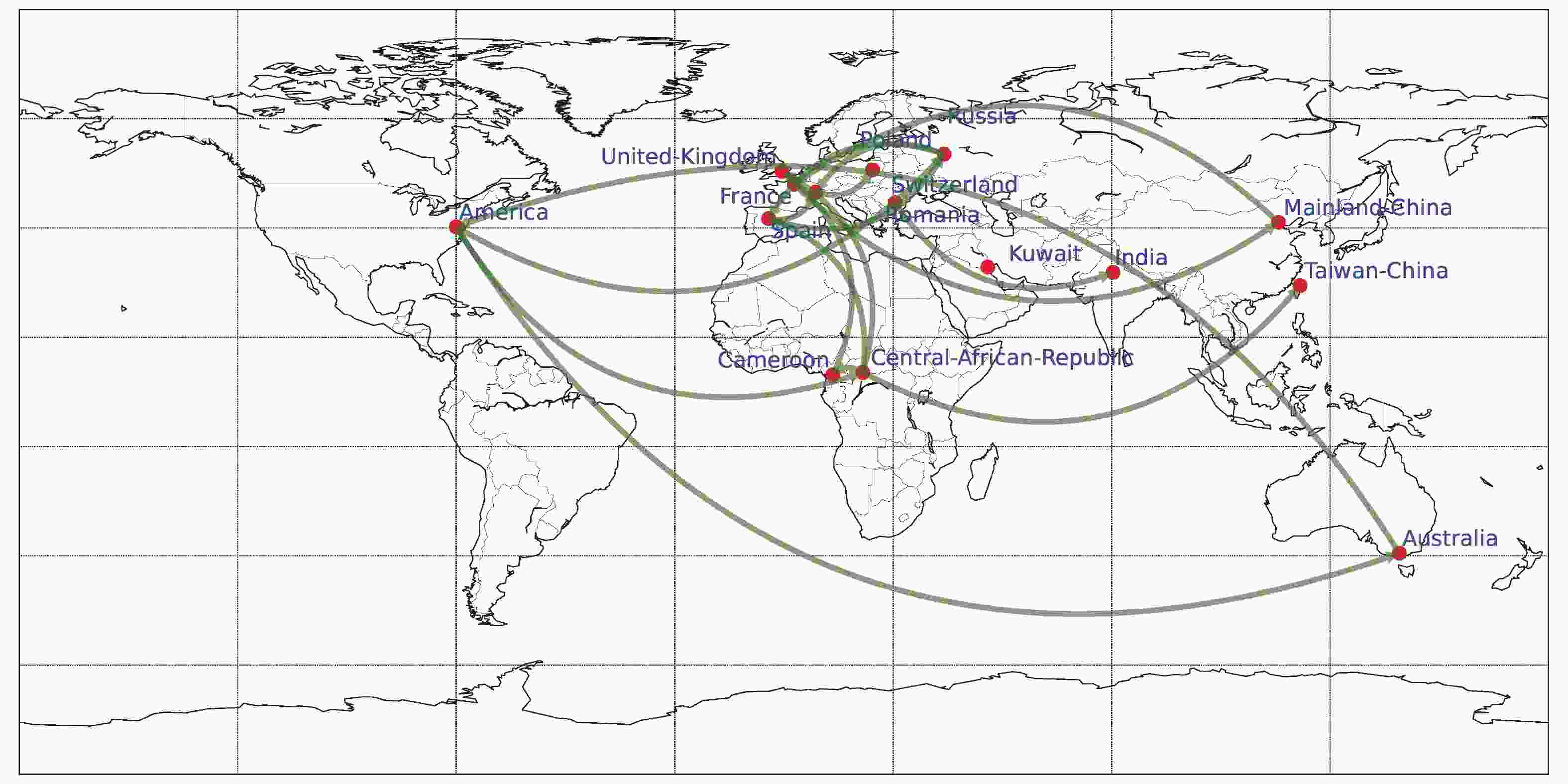
Figure S2. The spatial transmission of CVB1 worldwide. The green arrows indicate transmission paths with BF > 5.
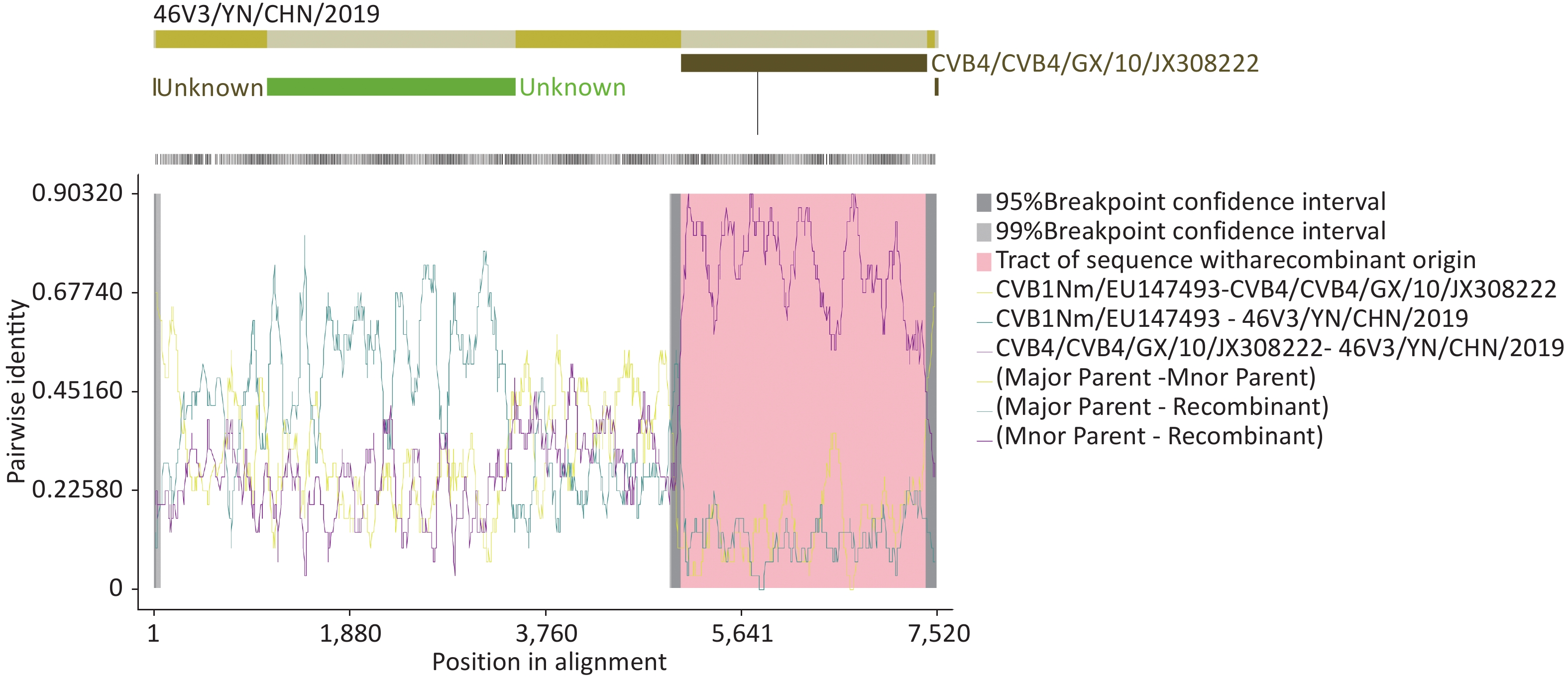
RDP4 analysis revealed that this isolate was involved in a recombination event with CVB4/GX/10/JX308222. The recombination event occurred between 5,000 and 7,500 nucleotides (Supplementary Figure S3, available in www.besjournal.com). Recombination is one of the major mechanisms of EV-B evolution[8]. Sequence analysis revealed that the P2 and P3 regions of the 46V3/YN/CHN/2019 strain had the highest homology with the CVB1SD2011CHN strain, which was isolated from a patient with HFMD in Shandong, China in 2010. The CVB1SD11 CHN shares sequences with E6SD11 CHN, CVA9-Alberta-2010, COXB5/Henan/2010, CVB4/GX/10, and E1-HN serotypes in the P2 and P3 regions, which indicates that multiple recombination events have occurred in the nonstructural region of CVB1SD2011CHN. In this study, we found several putative recombination events in nonstructural regions between strain CVB1 46V3/YN/CHN/2019 and CVB5 B504/MW015048, CVB4 strain (GX/10, B405, and B404) and EV-B E6/P735/2013/China. Based on the substantial sequence identity (≥ 90%), it can be inferred that these viruses serve as potential recombination parents. Among them, E6/P735/2013/China and CVB4 strain GX/10 were isolated from patients with HFMD in 2013 and 2010 in China, respectively, CVB4 strains B405 and B404 were isolated from raw sewage in Japan in 2010. CVB infections are often associated with HFMD[4]. The numbers of HFMD cases infected with CVB4 and CVB5 are increasing in China. These viruses also often co-circulate to cause epidemics or outbreaks of HFMD in China. Yunnan, in southwestern China, is a major tourist province, with hundreds of people from all over the world (including other provinces of China) visiting every year. These characteristics may provide ample opportunities for recombination of these viruses. However, the recombination can make new variants with altered pathogenicity, and new variants have serotype-specific structural protein sequences but share nonstructural protein sequences from different sources (different viruses or identical viruses from different sources)[9]. In addition, frequent recombination events have been found between CVB4 and EV-B multiple serotypes (CVB5, E9, CVA9, and E20) in the 3Dpol region. This recombination resulted in differences in the 3Dpol, which affected the fidelity of replication and ultimately the virus's environmental adaptability and survival[10]. Thus, the new variant might cause outbreaks and epidemics. Attention should be paid to cases caused by CVB1 and its derivatives.
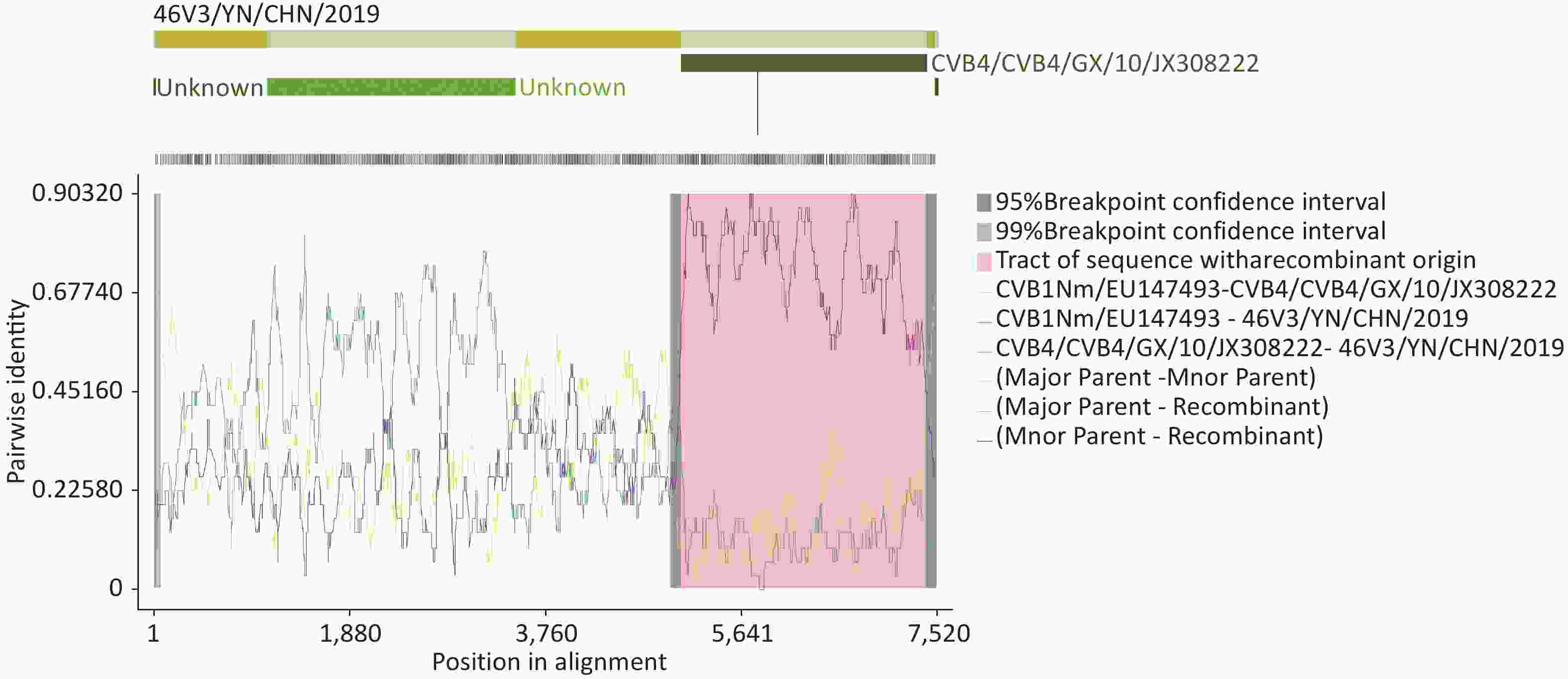
Figure S3. RDP4 analysis of strain 46V3/YN/CHN/2019 with closely related strains. Recombination analysis was performed by seven algorithms: RDP, GENECONV, BootScan, Maxchi, Chimaera, SiScan, and 3Seq.
In conclusion, although the six CVB1 isolates in this study were not isolated from samples collected from throat swabs, blood, and other lesions of HFMD patients, the current prevalence of CVB1 suggests that CVB1 is closely related to HFMD. Recombination and mutation have driven the evolution of CVB1. The six CVB1 strains in this study were isolated only from patients with HFMD who did not present with aseptic meningitis, myocarditis, and acute flaccid myelitis, but systematic epidemiological surveillance is needed to evaluate the association between CVB1 and these diseases. The findings contribute to the expansion of the complete genome of CVB1 in the GenBank database. Nevertheless, it would be beneficial to elaborate on the potential implications of these findings for the understanding of CVB1 epidemiology and the development of preventive measures.
-
Genomic KM46 region strain %Nucleotide identity Accession number 5′UTR CVB1SD2011CHN 96.84 JX976769 CVB1/XM0108 95.60 MG780414 CVB1/MSH/KM9/2009 95.06 JN596588 VP4 CVB1/SWG78/SD/CHN/2018 95.65 KU574629 E30/XM 95.17 MF177222 CVB1/XM0108 95.17 MG780414 VP2 CVB1/SWG69/SD/CHN/2018 98.10 MN541037 CVB1/SWG61/SD/CHN/2018 97.97 MN541029 CVB1/SWG78/SD/CHN/2018 97.72 MN541046 VP3 CVB1/SWG61/SD/CHN/2018 98.18 MN541029 CVB1/SWG78/SD/CHN/2018 97.90 MN541046 CVB1/SWG69/SD/CHN/2018 97.76 MN541037 VP1 CVB1/SWG69/SD/CHN/2018 97.84 MN541037 CVB1/2019–246–CVB1 97.72 MW179458 CVB1/SWG78/SD/CHN/2018 97.48 MN541046 2A CVB1/SWG61/SD/CHN/2018 96.89 MN541029 CVB1/SWG78/SD/CHN/2018 96.67 MN541046 CVB1SD2011CHN 94.22 JX976769 2B CVB1/SWG78/SD/CHN/2018 98.65 KU574629 CVB1/SWG61/SD/CHN/2018 98.32 MN541029 CVB1SD2011CHN 96.96 JX976769 2C CVB1SD2011CHN 94.83 JX976769 CVB1/XM0108 91.39 MG780414 CVA9 /VEN/2018–23122 90.98 MK652143 3A CVB1SD2011CHN 94.76 JX976769 CVB5/B504 94.35 MW015048 CVA9 /VEN/2018–23122 94.01 MK652143 3B CVB1SD2011CHN 95.45 JX976769 CVB4/B405 95.45 MW015040 CVB4/B404 95.45 MW015039 3C CVB1SD2011CHN 96.54 JX976769 CVB4/GX/10 94.35 JX308222 E1/precuror 94.17 JQ979292 3D CVB1SD2011CHN 95.17 JX976769 CVB4/B404 92.29 MW015039 E6/E6SD11CHN 92.21 JX976771 3′’UTR E6/P735/2013/China 93.98 KP289439 CVB5/Chzj–1 93.90 KF311743 CVA9/CV–A9/P56/2013 92.93 KP289434 P1 CVB1/SWG61/SD/CHN/2018 97.56 MN541029 CVB1/SWG78/SD/CHN/2018 97.52 MN541046 CVB1/SWG69/SD/CHN/2018 97.21 MN541037 P2 CVB1SD2011CHN 94.98 JX976769 CVB1/XM0108 91.98 MG780414 A9 /VEN/2018–23122 87.68 MK652143 P3 CVB1SD2011CHN 95.46 JX976769 CVB5/B504 92.60 MW015048 E6/E6SD11CHN 92.46 JX976771 genome CVB1SD2011CHN 95.29 JX976769 CVB1/XM0108 90.63 MG780414 CVB1/MSH/KM9/2009 86.96 JN596588 Table S3. Top three sequences with the highest nucleotide sequence similarities between all sequenced individual genomic regions of strain 46V3/YN/CHN/2019 and enteroviruses in the GenBank in the BLAST online tool
HTML
Competing Interests The authors declare no competing financial interests.
&These authors contributed equally to this work.
Reference
 23422-S.pdf
23422-S.pdf
|

|

 Quick Links
Quick Links
 DownLoad:
DownLoad: