
-
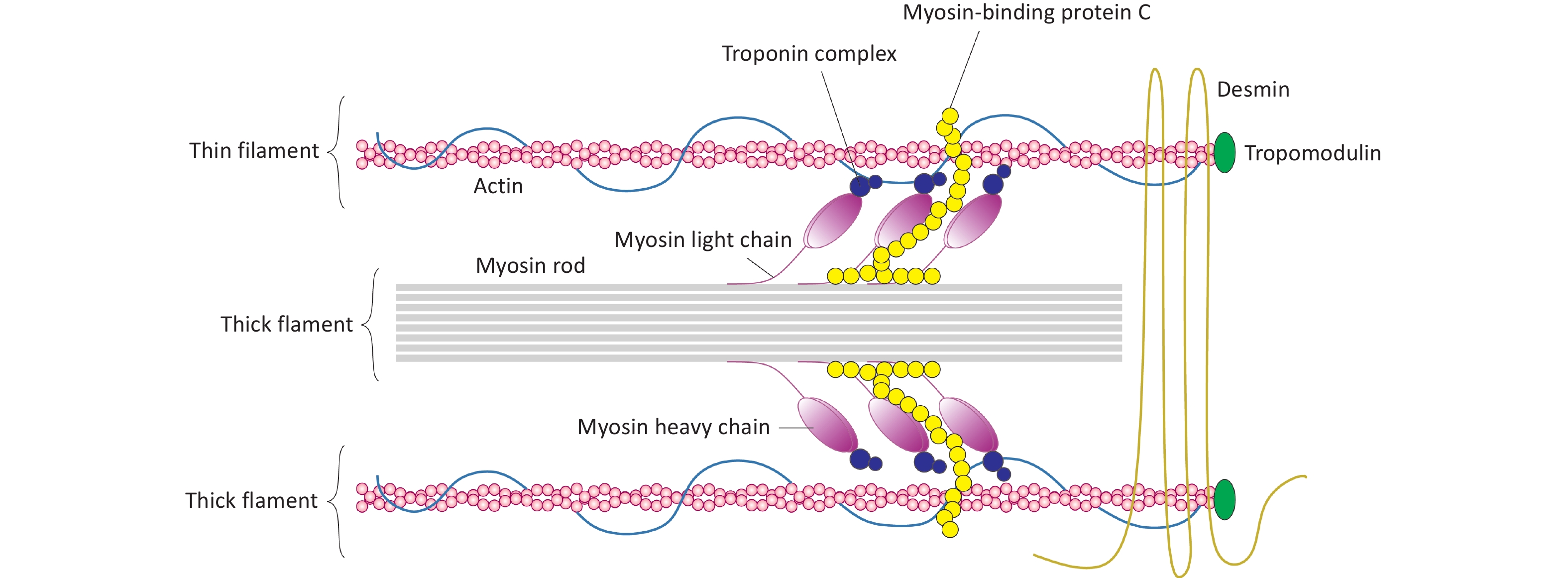
In mammalian hearts, the MYH7 (myosin heavy chain 7) gene encodes the β-myosin heavy chain (β-MyHC), and MYH6 (myosin heavy chain 6) encodes the α-isoform (α-MyHC). These myosin isoforms are the primary contractile proteins within the thick filaments of the sarcomere, a unit responsible for muscle contraction (Figure 1)[1-4]]. Specifically, in cardiac muscles, MYH7 is predominantly expressed in the ventricles, while MYH6 is mainly expressed in the atria[5-8]. Uniquely in cardiac muscle, the MYBPC3 (myosin-binding protein C3) gene encodes the cardiac myosin-binding protein C (cMyBP-C)[9,10]. This protein plays a crucial role in maintaining sarcomere structure and regulating contraction and relaxation in cardiac myocytes[9].
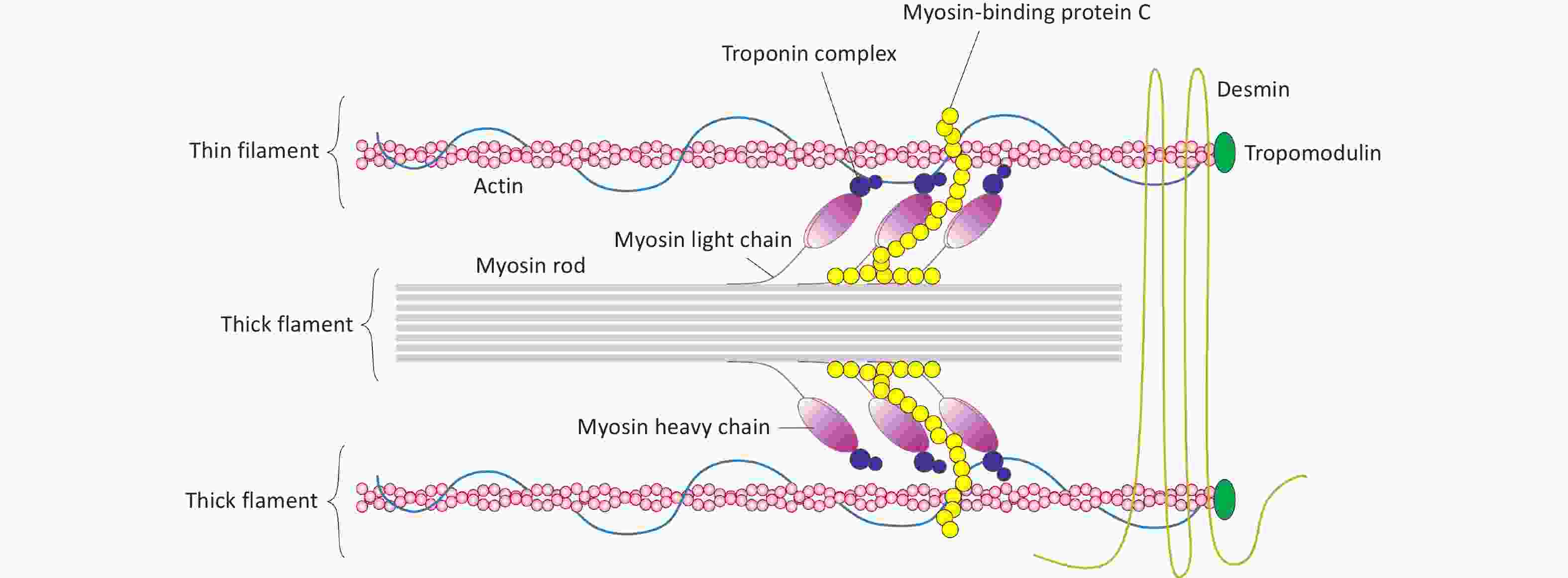
Figure 1. Myosin and actin in thick and thin filaments of sarcomere in cardiomyocytes.
It is well established that mutations in several sarcomere-associated genes, as listed in Table 1, are strongly linked to HCM[11,12]. Among these, mutations in MYH7 and MYBPC3 account for approximately 50% of HCM cases[13,14]. Notably, p.Arg403Glu was the first identified mutation in the MYH7 gene, while p.Arg502Trp and p.Val762Asp are among the most prevalent MYBPC3 mutations in patients with HCM (Figure 2)[15-17]. Mutations in the MYH7 gene primarily result in defects in the hinge region or globular head of β-MyHC, although certain mutations may also affect the rod domain[18-21]. Mutations in other sarcomere-associated genes such as TNNT2 (troponin T2, cardiac type), TNNI3 (troponin I3, cardiac type), and TPM1 (tropomyosin 1) are relatively uncommon, accounting for less than 10% of HCM cases[22-25]. Although missense mutations constitute the majority of HCM-associated mutations, MYBPC3 mutations are predominantly frameshift mutations, including insertions, deletions, and premature truncations[22,23,26]. Importantly, not all loss-of-function (LoF) or missense mutations in these genes are pathogenic, indicating the presence of additional modifiers that influence the development of HCM[27].
Table 1. Most common mutations involved in HCM
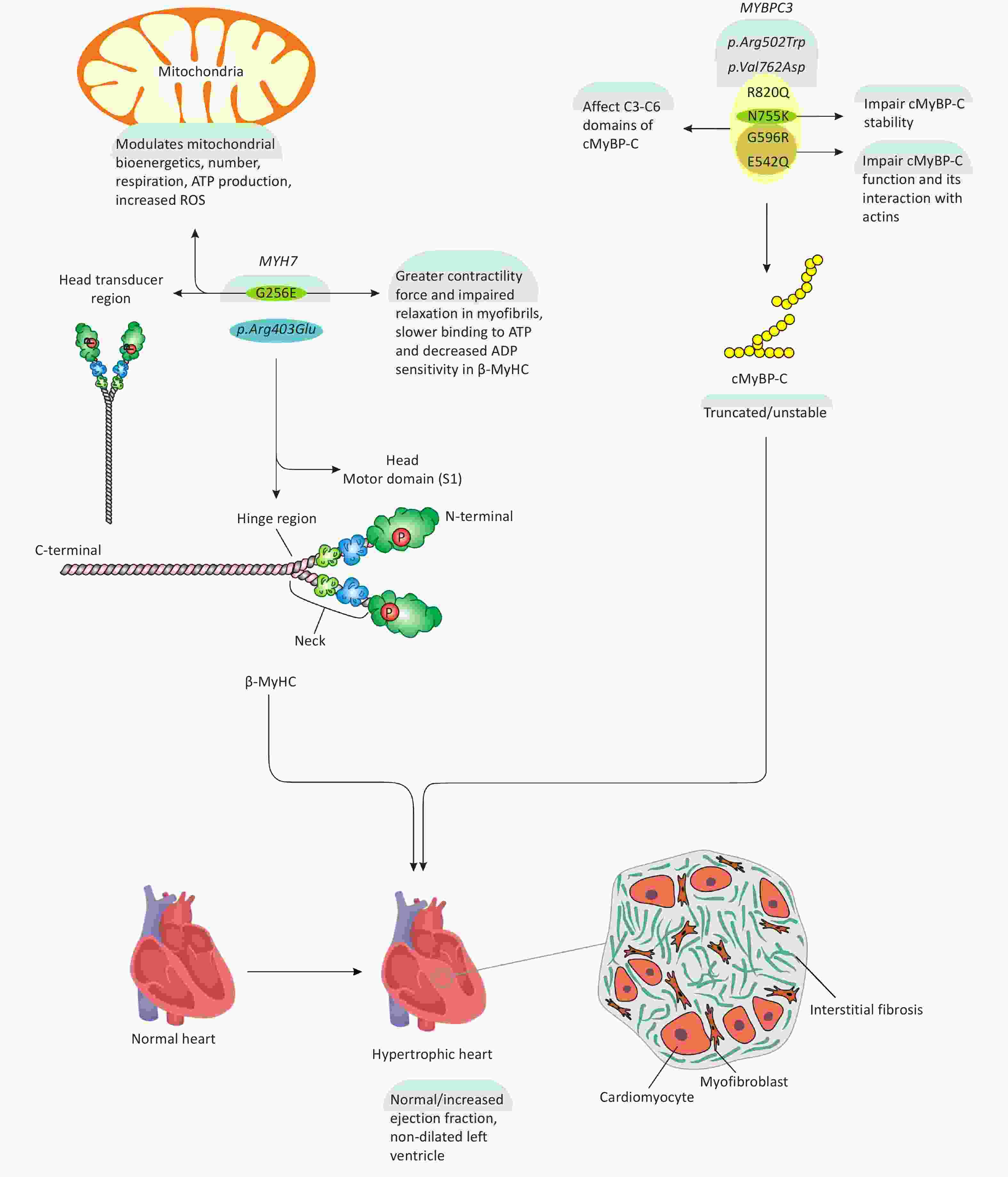
Figure 2. MYH7 and MYBPC3 mutations, their impacts on myofibrils, and HCM development. As shown in the figure, G256E and p.Arg403Glu mutations in MYH7 gene mainly affect the head and hinge region of β-MyHC, thereby influencing myofibril contractility and mitochondrial homeostasis associated with HCM development. 6 predominant mutations in MYBPC3 gene including p.Arg502Trp, p.Val762Asp, R820Q, N755K, G596R, E542Q, somehow modulate cMyBP-C function and stability, ultimately, leading to HCM.
HCM is a heterogeneous genetic disorder, predominantly inherited in an autosomal dominant pattern within cardiomyocytes[13,28-32]. However, 5-10% of HCM cases, particularly in children, are phenocopies of storage diseases—a group of disorders caused by lysosomal dysfunction, leading to excessive accumulation of substances within various cellular organelles[30,33,34]. In such cases, mutations responsible for storage diseases can result in HCM-mimetic phenotypes, which may include X-linked genetic disorders such as Fabry disease[35-38]. Pathologically, HCM is characterized by cardiac hypertrophy, which occurs independently of cardiac loading conditions, and typically presents with a normal or increased ejection fraction and a non-dilated left ventricle (LV). At the microscopic level, cardiomyocytes appear hypertrophied and disorganized, often interspersed with interstitial fibrosis (Figure 2)[13,28-30,39-41].
Above, we outlined the fundamental pathological concepts of HCM. In the subsequent sections, we will closely examine some of the most recent discoveries related to HCM pathogenesis at both the molecular and cellular levels. These analyses may provide insights into novel molecular targets and pave the way for new research directions in the field of HCM.
-
A missense mutation is a point mutation that alters a single nucleotide within a DNA codon, leading to the incorporation of a different amino acid into the resulting protein[42]. While most mutations in the MYBPC3 gene result in a truncated and unstable version of cMyBP-C, missense mutations specifically affect the central domains[43]. Structurally, cMyBP-C comprises 11 domains, labeled C0 to C10, starting from the N-terminal[10,44,45]. Functionally, cMyBP-C regulates cardiomyocyte contractility by restraining myosin activity/mobility and governing myosin-actin interactions in the sarcomere[46]. Consequently, impaired cMyBP-C function due to MYBPC3 mutations disrupts its interaction with binding partners, compromising cardiomyocyte contractility and triggering hypertrophic responses that lead to the development of HCM[46]. A recent study evaluated the functional impact of four missense mutations—R820Q, N755K, G596R, and E542Q—affecting the C3 to C6 domains of cMyBP-C (Figure 2)[43]. These domains are crucial as they interact with the myosin head domain [myosin subfragment 1 (S1)][47]. The study found that the N755K and R820Q mutations in cMyBP-C did not significantly affect ATPase activity or interaction with myosin S1, exhibiting behavior similar to wild-type cMyBP-C[43]. However, the G596R and E542Q mutations increased interaction with myosin S1 and elevated its ATPase activity by 30% and 50%, respectively[43]. These mutations impair cMyBP-C function and its ability to regulate myosin activity, thereby constraining cardiomyocyte contractility[43]. Additionally, the G596R and E542Q mutations reduced cMyBP-C interaction with actin in thin filaments of the sarcomere[43]. Moreover, the N755K mutation was found to modulate myofibril structure, altering cMyBP-C stability and its localization within the sarcomere (Figure 2)[43].
Collectively, these findings indicate that a set of missense mutations in the MYBPC3 gene either impair cMyBP-C function or alter its localization within the sarcomere, potentially leading to aberrant function. Based on this, we hypothesize that such mutations modulate myosin activity in a way that enhances cardiomyocyte contractility, thereby triggering intracellular signaling pathways that may ultimately contribute to HCM pathogenesis or manifest phenotypically as HCM. Despite these important findings, this study and the existing literature do not sufficiently explore the underlying signaling pathways and associated molecular mechanisms. As a result, our current understanding is largely confined to identifying mutations and their functional impacts on sarcomere proteins, without deeper mechanistic insights. Therefore, future studies should aim to bridge these critical gaps by employing innovative basic research approaches, which may not only deepen our understanding of HCM pathogenesis but also identify novel molecular targets with potential therapeutic and translational value.
-
In a unique study, human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) were gene-edited using CRISPR-Cas9 technology to develop an in vitro model of HCM. The study found that the MYH7 G256E mutation induced a greater contractile force in hiPSC-CMs compared to control myofibrils[48]. Additionally, during the myofibril relaxation phase, the initial kinetics declined and the cross-bridge detachment rate was significantly slower[48]. Furthermore, myofibrils carrying the MYH7 G256E mutation exhibited decreased ADP sensitivity, which was associated with impaired relaxation[48]. Moreover, a stopped-flow kinetic assay revealed that MYH7 G256E-mutated myofibrils exhibited slower ATP binding, further impacting their functional dynamics[48]. To gain deeper mechanistic insights, the study employed molecular dynamics simulations to compare the structural behavior of wild-type and mutated β-MyHC. The results demonstrated that the MYH7 G256E mutation destabilized the β-MyHC transducer region by reducing hydrogen bonding between adjacent β-sheets, ultimately impairing its structural integrity[48]. Collectively, these in silico findings suggest that the MYH7 G256E mutation modulates the transducer region of β-MyHC, thereby affecting nucleotide binding and actin-myosin interactions, leading to excessive hypercontractility and impaired relaxation in hiPSC-CM myofibrils (Figure 2). This study provides in-depth biochemical and structural analyses of β-MyHC function following the MYH7 G256E mutation, elucidating its impact on myofibril contractility and HCM pathogenesis. However, despite these valuable insights, the study did not investigate whether the MYH7 G256E mutation triggers alterations in cellular signaling pathways or transcriptional regulation that could contribute to HCM development. Thus, while this study significantly enhances our understanding of biophysical and biochemical changes resulting from myosin mutations, further research is warranted to elucidate the underlying molecular mechanisms driving increased hypercontractility. Such investigations could reveal novel therapeutic targets with potential clinical and translational relevance.
-
Incomplete-penetrant gene variants are mutated variants that do not consistently develop the expected disease or exhibit phenotypic disease traits[49]. The MYH7 G256E variant exemplifies this, as its role in HCM pathogenesis remains elusive due to its variable penetrance and clinical severity[50]. A recent study demonstrated that the MYH7 G256E mutation induces hypercontractility and alters mitochondrial bioenergetics in cardiomyocytes. To ascertain the functional and biomechanical effects of the MYH7 G256E variant, comprehensive analyses were conducted at the cellular, tissue, myofibril, and protein levels[50]. The findings indicated that the MYH7 G256E mutation impairs the myosin S1 transducer domain, enhancing hypercontractility[50]. Consequently, in MYH7 G256E mutated hiPSC-CMs and engineered cardiac tissues, myofibrils developed stronger, faster, and more sustained contractile tensions[50]. Additionally, metabolic profiling and single-cell transcriptomic analyses identified upregulated mitochondrial genes linked to increased respiration, suggesting bioenergetic alterations due to the MYH7 G256E variant[50]. Despite these significant findings regarding the altered biomechanical properties of myosin S1 due to the MYH7 G256E mutation, the study did not delve into the underlying molecular mechanisms of this process. For instance, the specific intracellular mechanisms or signaling pathways involved in MYH7 G256E-induced myosin modulation and subsequent HCM development remain unidentified. Therefore, further in vivo and in vitro studies are essential to bridge this critical gap. In-depth investigations focusing on molecular mechanisms could not only deepen our understanding of myofibril hypercontractility but also pave the way for practical therapeutic interventions aimed at modulating this pathological process.
On the other hand, the aforementioned findings support the general involvement of mitochondrial dysfunction, particularly altered mitochondrial energetics, in HCM pathogenesis[51]. In a multiomics study using myocardial samples from HCM patients and healthy controls, it was shown that dysregulation of fatty acid metabolism in the mitochondria, mitochondrial accumulation of free fatty acids, and reduced acylcarnitines (precursor metabolites for β-oxidation of fatty acids in the mitochondria) were closely linked with HCM development[52]. Other metabolic dysregulations included the downregulation of mitochondrial genes related to the ATP synthesis complex and creatine kinase[52]. Moreover, electron microscopic findings revealed an increase in damaged mitochondria accompanied by decreased cristae density and suppressed oxidative phosphorylation in the respiratory chain[52]. These events also led to an increase in reactive oxygen species (ROS), further disturbing cellular homeostasis and cardiomyocyte health, thus significantly contributing to HCM development[52]. Nonetheless, this study lacks a thorough elucidation of the molecular mechanisms linking disturbed mitochondrial homeostasis and energy metabolism with HCM pathogenesis, thereby necessitating further research in this area. In an independent key study, septal myectomy tissue samples from HCM patients were used to conduct respirometry analyses of mitochondrial oxidative phosphorylation and the β-oxidation process of fatty acids[53]. It was shown that NADH (nicotinamide adenine dinucleotide)-linked functions of the respiratory chain were impaired (likely involving complex I)[53]. However, the mitochondria were neither damaged nor fragmented, as transmission electron microscopic data suggested[53]. Additionally, mitochondria were disorganized in relation to myofibrils, which may be considered a form of mitochondrial dysfunction[53]. Interestingly, pharmacological increase of mitochondrial NAD+ levels (using elamipretide) significantly improved mitochondrial respiratory function, suggesting the potential of mitochondria-targeting therapies in managing HCM[53]. Notably, these findings were obtained after excluding the influence of common genetic mutations observed in HCM patients.
Lastly, this study briefly examined the role of mitochondria in HCM pathogenesis concerning the MYH7 G256E mutation[50]. Though not fully delineated by this study[50], it appears that mitochondria warrant further investigation to elucidate their crucial involvement in HCM development under conditions of myosin mutations[52,54-56]. In a related study, mitochondrial dysfunction was implicated in HCM development and associated complications[57]. This research used hiPSC-CMs in vitro, where it was found that the number of mitochondria was significantly reduced, alongside impaired respiration and decreased ATP production. It was hypothesized that mitochondrial depolarization, increased ROS (reactive oxygen species) production, autophagy dysfunction, and the downregulation of respiratory genes (Figure 2) (encoding mitochondrial complexes) all contribute to mitochondrial dysfunction in hiPSC-CMs[57]. However, this study did not define the underlying molecular mechanisms of mitochondrial dysfunction in HCM and overlooked the potential impact of HCM-associated mutations on mitochondrial dysfunction. These limitations, together with growing evidence from recent studies[58-61], highlight the need for further investigation into the role of mitochondria in cardiac pathophysiology and the progression of HCM.
On the other hand, mitochondria are increasingly recognized as a promising target for drug development, with significant efforts historically dedicated to their therapeutic modulation across various diseases, including cardiovascular disease (CVD)[62-66]. For instance, the supplementation of hiPSC-CMs with acid alpha-glucosidase (a human recombinant enzyme) has been shown to restore mitochondrial function, thereby alleviating HCM pathology[57]. However, despite these advancements, the identification of reliable molecular targets and therapeutic agents that effectively modulate mitochondrial involvement in HCM pathogenesis remains a challenge.
-
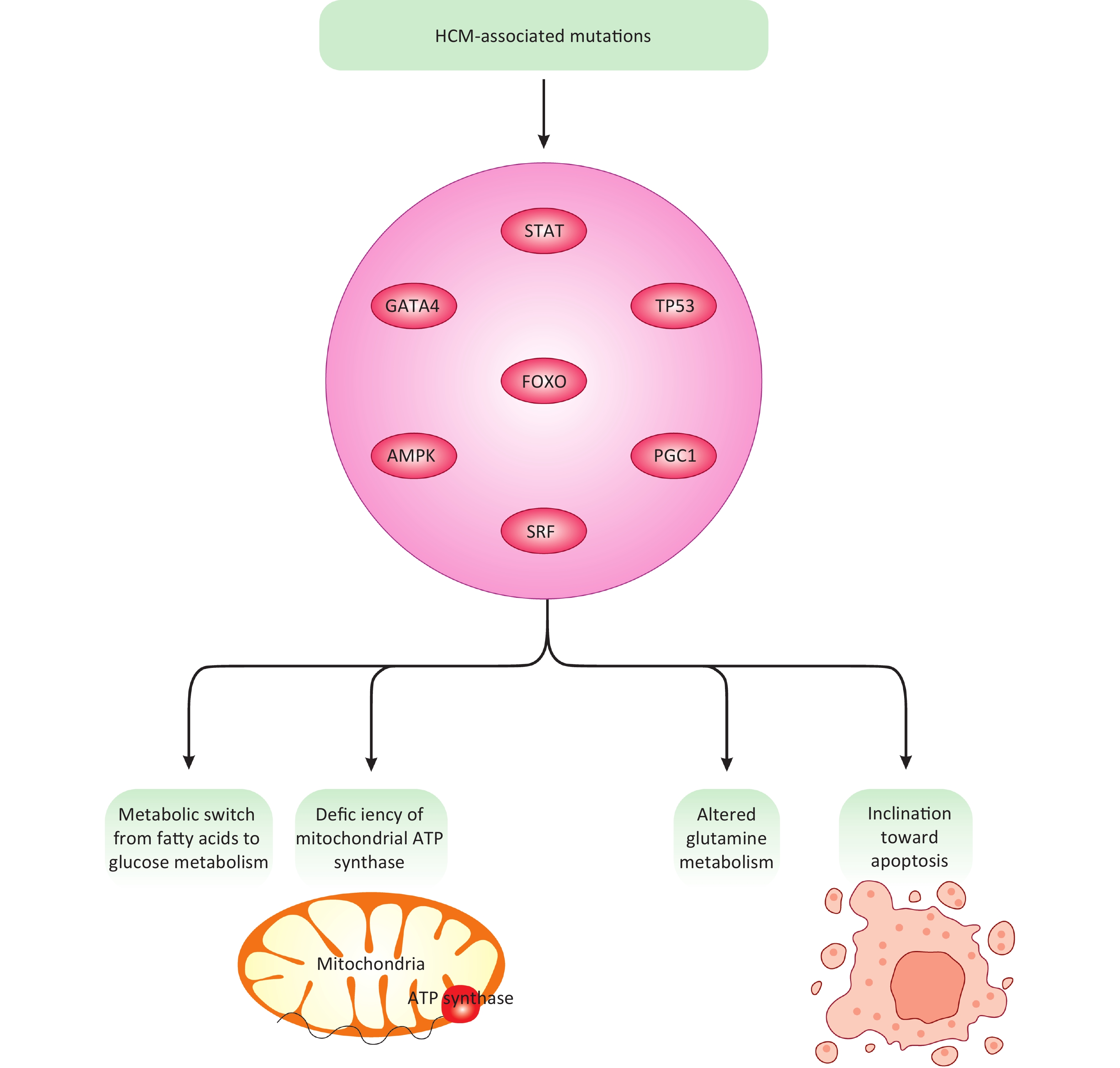
In a recent computational study, an HCM model was developed to mimic the signaling pathways associated with HCM and subsequent alterations in cardiomyocyte metabolism[67]. This computational model utilized a mathematical equation, specifically a stochastic logic-based ODE approach, to closely illustrate the interconnection between cardiomyocyte signaling pathways, metabolic network, and alterations[67]. HCM mutations were shown to induce metabolic shifts such as ATP synthase deficiency and a switch from lipid/fatty acid metabolism to carbohydrates[67]. Based on these results, HCM-associated mutations were interpreted to manipulate cellular metabolism, triggering a metabolic switch from fatty acids to glucose/carbohydrates and mitochondrial ATP synthase deficiency[67]. Additionally, altered glutamine metabolism and an inclination toward apoptosis[68] due to ATP synthase deficiency were also documented (Figure 3)[67]. These events are largely associated with cardiomyocyte hypertrophy, primarily through transcription factors like FOXO (forkhead box protein O), STAT (signal transducer and activator of transcription)[69], TP53 (tumor protein p53), GATA4 (GATA binding protein 4), and SRF (serum response factor)[67]. Moreover, common mechanisms exist across different HCM mutations and model species, including the AMPK (adenosine monophosphate-activated protein kinase)-PGC1 (peroxisome proliferator-activated receptor gamma coactivator-1) signaling pathway and the TTN (titin)-FHL1 (four and a half LIM domains) axis[67]. Collectively, these findings shed light on HCM-associated signaling pathways and their subsequent impact on cardiomyocyte metabolism, thereby laying the groundwork for more integrative and effective therapeutic strategies. This emerging framework holds promise for the development of targeted therapies aimed at mitigating HCM pathogenesis. However, the current literature remains insufficient to fully enable such interventions, highlighting the need for further mechanistic research to bridge this gap. While this study has made appreciable efforts, it has largely neglected in-depth experimental exploration of the involved molecular mechanisms, instead relying on mathematical, statistical, and bioinformatics approaches and analyses. Although these techniques are helpful, they may not adequately replace basic experimental research. Therefore, more efforts are needed to make meaningful advances in this new research line of HCM pathogenesis and cardiomyocyte metabolism.
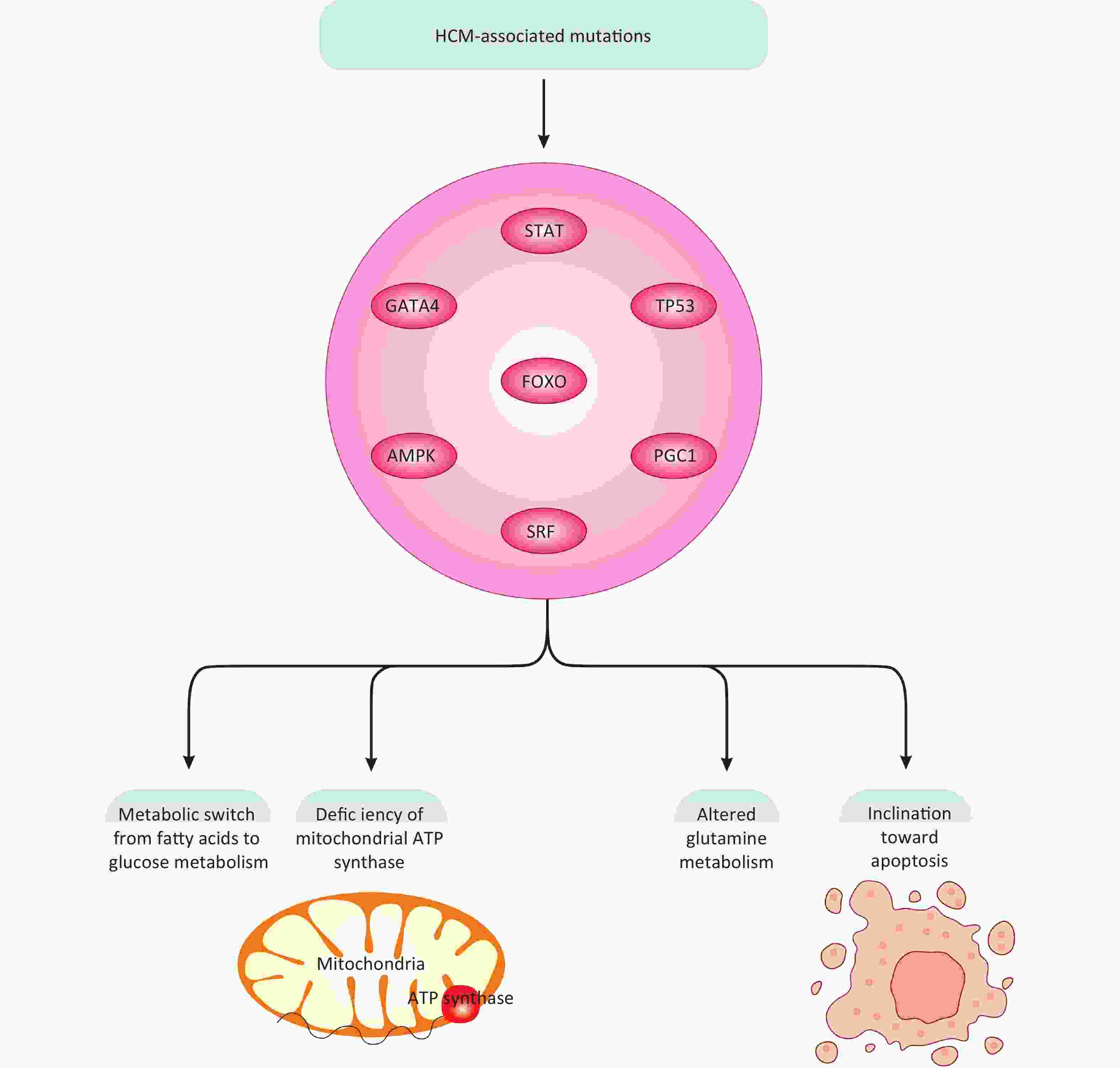
Figure 3. HCM-associated mutations, signaling molecules, and transcription factors. HCM-associated mutations through modulation of multiple transcription factors and signaling molecules can ultimately change glucose, fatty acids, and amino acid metabolism, mitochondrial ATP production, and apoptotic fate in cardiomyocytes, all linked with HCM development.
-
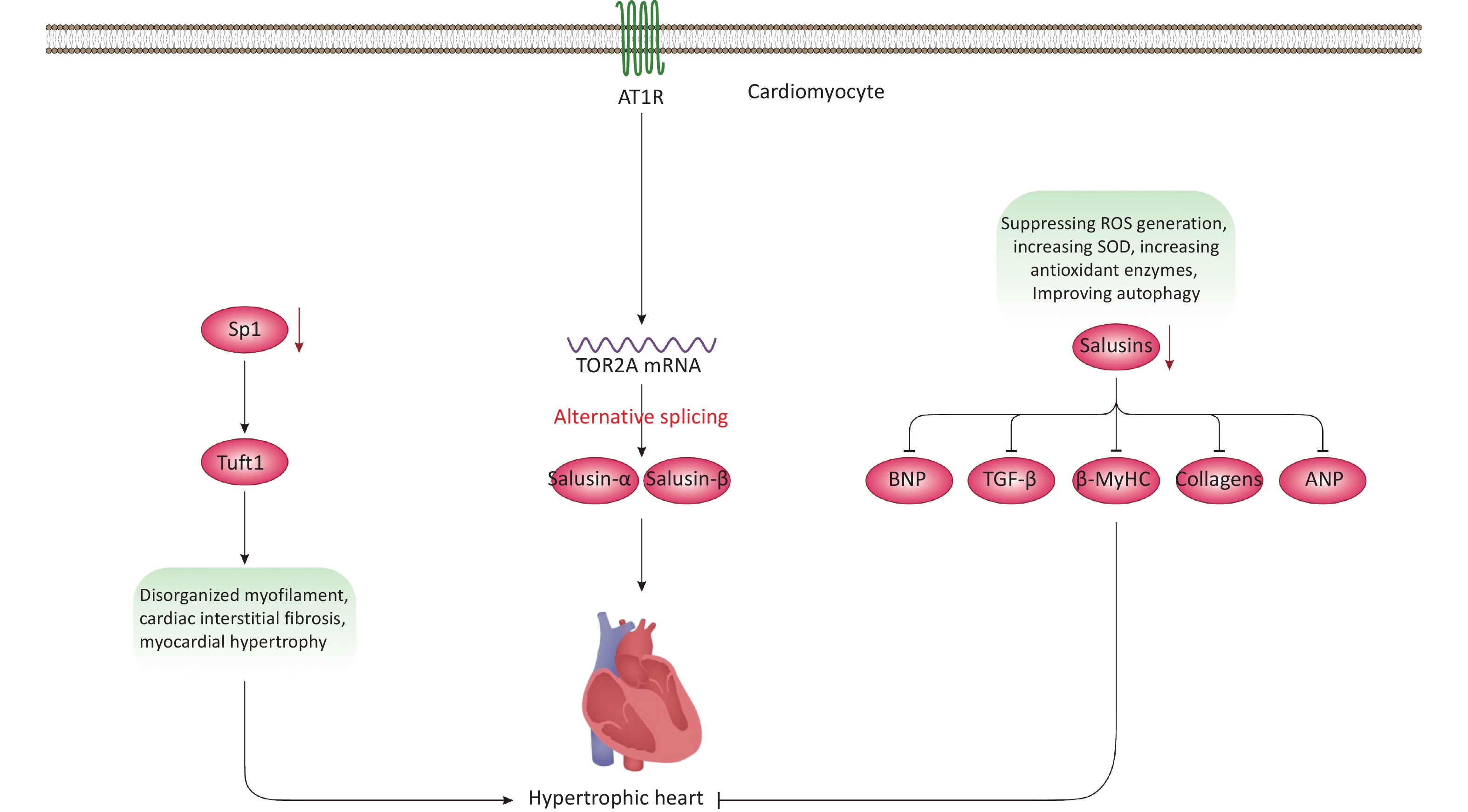
SP1 (Sp1 transcription factor) regulates the transcription of several genes involved in cellular processes such as DNA repair, homeostasis, cell growth and proliferation, apoptosis, migration, as well as invasion and metastasis[70-72]. It is hypothesized that Sp1 contributes to HCM pathogenesis due to its role in cardiomyocyte growth[72-75]. In a recent study, cardiac-specific Sp1 knockout in mice was associated with HCM development, presenting phenotypes that included disorganized myofilaments, cardiac interstitial fibrosis, and myocardial hypertrophy (Figure 4)[74]. The cardiac phenotype was analyzed using transmission electron microscopy and histochemical experiments, as well as echocardiography. Experimentally, techniques such as chromatin immunoprecipitation, RNA sequencing (cornerstones of current biomedical research), and in vivo adenoviral transfections were conducted to explore the downstream mechanisms of Sp1 and its therapeutic impact on HCM in mice[74]. Additionally, hiPSC-CMs were used for in vitro examination of Sp1’s role and therapeutic potential in HCM[74]. In hiPSC-CMs, Sp1 knockdown significantly disorganized the intracellular myofibrillar structure, resembling hypertrophic cardiomyocytes indicative of HCM phenotype[74]. From a mechanistic viewpoint, Tuft1 (tuftelin 1) was identified as a target gene of Sp1. Indeed, Tuft1 overexpression reversed the hypertrophic phenotypes induced by Sp1 ablation, both in vivo and in vitro[74]. In mice, Sp1 overexpression also prevented HCM development and alleviated hypertrophic phenotypes in hiPSC-CMs[74]. These findings suggest that Sp1 ablation in mouse hearts can lead to HCM development primarily by modulating Tuft1 expression and mediating its downregulation. We hypothesize that the Sp1-Tuft1 axis may also instigate other cellular and molecular alterations in cardiomyocytes that ultimately result in HCM development (Figure 4). However, this study did not elucidate these mechanisms at the molecular level. Therefore, additional experimental studies are essential to unravel these elusive pathways, providing deeper insights into the role of the Sp1-Tuft1 axis in HCM pathology. Such investigations could also identify potential molecular targets for therapeutic intervention and clinical translation. Overall, this promising research area is still in its early stages, necessitating further exploration to fully understand its implications in HCM.
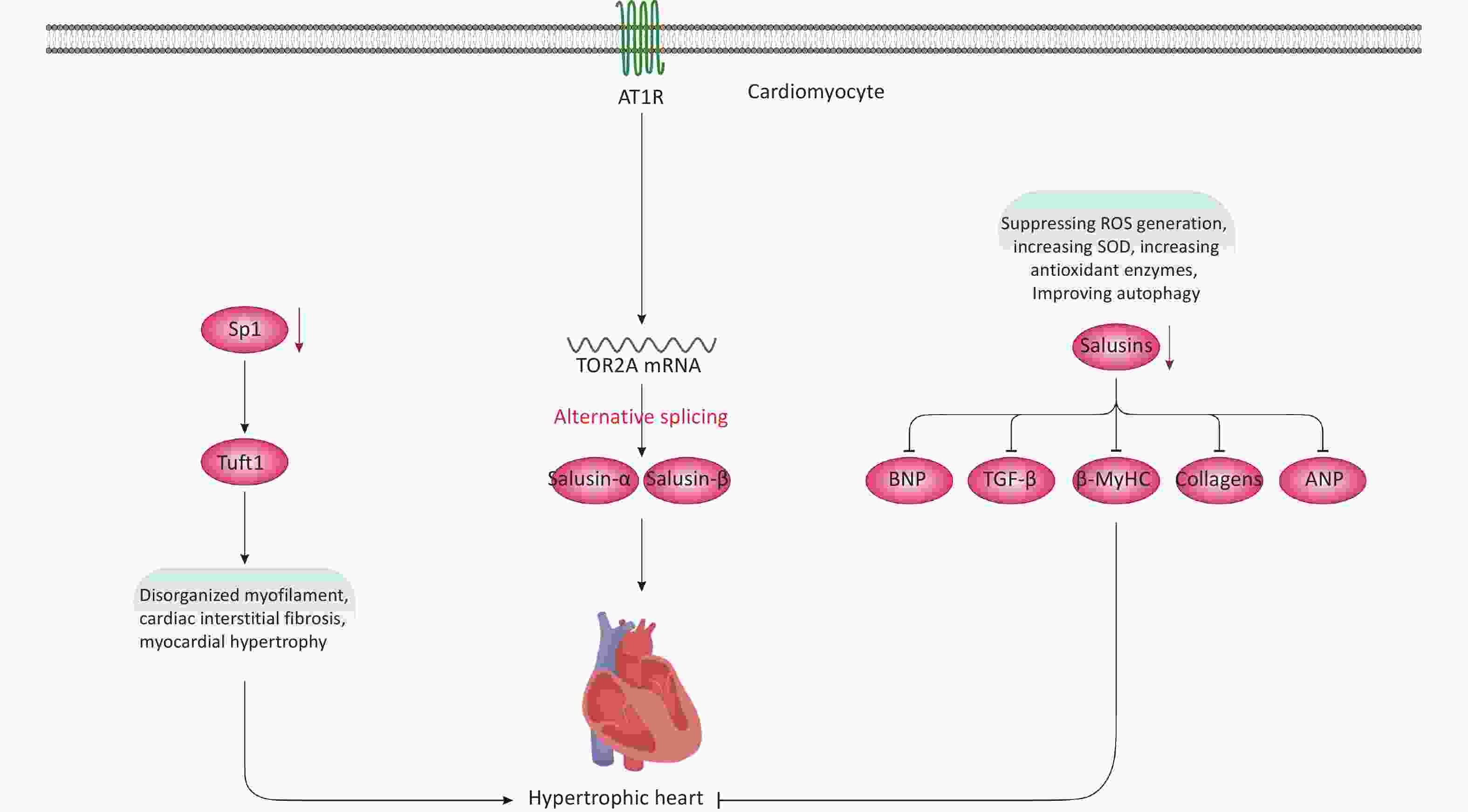
Figure 4. Non-genetic factors and molecules involved in HCM development. Modulation of the Sp1-Tuft1 axis is associated with HCM development mainly due to increased interstitial fibrosis and disorganized myofilament. Besides, Ang II-induced activation of AT1R signaling can lead to the upregulation of salusins, which is closely linked with HCM development via largely unknown mechanisms. Moreover, downregulation of salusins is associated with suppression of multiple factors, resulting in prevention/alleviation of HCM. AT1R, angiotensin II type I receptor;
-
Salusins are peptides consisting of 20-28 amino acids with mitogenic and hemodynamic activities[76]. These proteins result from the alternative splicing of the TOR2A (torsin family 2 member A) mRNA[77]. Both human and mouse tissues produce and synthesize salusins, detectable in urine or plasma[76,78-81]. Regarding CVD, multiple studies support the role of salusins in atherosclerosis[82] and related cardiovascular conditions[32,83-85]. A recent study[86], infused angiotensin II (Ang II)[87] into Sprague-Dawley rats to develop an in vivo model of HCM, and subsequently employed Doppler echocardiography to assess cardiac function after HCM development[86]. The in vitro component involved treating cardiac fibroblasts and primary neonatal rat cardiomyocytes (NRCMs) with Ang II[86]. Results showed that Ang II infusion upregulated levels of salusin-α and salusin-β in plasma, rat cardiac tissues, NRCMs, and cardiac fibroblasts (Figure 4)[86]. These increases were closely associated with HCM development. Mechanistically, Ang II triggered these effects by activating certain signaling pathways and ultimately modulating gene expression[86]. However, downregulating salusins reversed these effects and HCM development by suppressing the production/protein levels of β-MyHC, BNP (brain natriuretic peptide), and ANP (atrial natriuretic peptide). Downregulation of salusins also reversed cardiac fibrosis by inhibiting or reducing TGF-β (transforming growth factor-beta) and collagen types III and I. Furthermore, reducing salusin levels neutralized ROS generation, increased superoxide dismutase (SOD) activity—an antioxidant enzyme—and moderated autophagy both in vitro and in the rat model[86]. Collectively, these findings suggest that upregulated expression and salusin protein levels are linked to HCM development via largely unknown mechanisms. The study briefly revealed some of these mechanisms, such as the salusin-β-MHC/-BNP/-ANP/-TGF-β axes. However, an in-depth exploration of the molecular mechanisms and associated signaling pathways is lacking. Nonetheless, the study suggests that salusins could be potential molecular targets for alleviating HCM pathology by suppressing oxidative stress and regulating autophagy flux. Therefore, this area requires thorough investigation before advancing to practical clinical studies and interventions.
-
Accumulating evidence underscores the involvement of immune cells in the pathophysiology of HCM[88,89]. Advances in bioinformatics tools and immune cell infiltration analyses have significantly enhanced our understanding of the complex interactions between immune cells and HCM development[90]. For example, analysis of public datasets such as GSE141910 and GSE36961, which include samples from human HCM patients and controls, has identified differentially expressed genes (DEGs) and variations in immune cell infiltration. In the HCM group, the density/score of dendritic cells (DCs), monocytes, macrophages, regulatory T cells (Tregs), and type 1 T helper (Th1) cells were notably decreased, whereas levels of platelets, fibroblasts, basophils, and CD8+ T cells were significantly elevated[91]. Further studies have pinpointed STAT3 (signal transducer and activator of transcription 3) signaling and a specific subtype of macrophages (LYVE1+ CD163+ macrophages) as crucial contributors to HCM pathogenesis. These findings, consistent with those from clinical samples, underscore the functional role of LYVE1+ CD163+ macrophages in the progression of HCM[91]. This highlights the need for more research into the role of immune cells in HCM, especially given the topic's limited coverage in recent literature. Experimental models have shown that while dendritic cells (DCs), neutrophils, and mast cells can negatively affect cardiac function, natural killer T (NKT) cells and eosinophils may offer cardioprotective effects. Thus, these cells play opposing regulatory roles in HCM pathogenesis[92]. Similarly, the role of monocytes and macrophages in HCM varies depending on the cellular subset[92]. Mechanistically, in pressure overload-induced HCM, both resident innate immune cells and cardiomyocytes are thought to release chemokines, cytokines, and immune modulators that facilitate the recruitment of additional immune cells to the affected myocardium. This process intensifies local inflammation, potentially exacerbating HCM pathophysiology[92-94]. Moreover, emerging evidence suggests that certain immune cell subsets, such as CD4⁺ T cells, may also play roles in cardiac remodeling, heart failure (HF), and ischemic HF[95-98], thereby underscoring the need for further research in this context within HCM.
-
Although its name suggests a relation to genetics, epigenetics refers to a distinct process that occurs in all eukaryotic cells, involving reversible modifications of DNA that alter gene expression without changing the underlying DNA sequence[99-101]. Emerging evidence suggests that epigenetic mechanisms play a crucial role in the pathophysiology of HCM beyond genetic factors[102,103]. For instance, DNA methylation has been strongly implicated in HCM development. In a 2014 study[104], using a mouse model of left ventricular (LV) pressure overload, researchers identified 5,346 regions of DNA that were methylated in the hypertrophic myocardium compared to controls, indicating an increase in DNA methylation during HCM progression[104]. Similarly, an independent study using a norepinephrine-induced HCM rat model found that global genomic DNA methylation levels increased over the course of the disease[105], with a positive correlation between methylation levels and norepinephrine concentration[105]. However, these studies did not explore the downstream molecular consequences of DNA methylation, leaving a significant gap in understanding its functional links to HCM development and highlighting the need for further mechanistic investigations. Additionally, research on transgenic mice has shown that upregulated expression of DNA methyltransferase 3-like (DNMT3L) and DNA methyltransferase 3A (DNMT3A)—key enzymes responsible for catalyzing DNA methylation—resulted in HCM development and mortality within 20 weeks[106]. These findings suggest that DNMT3A and DNMT3L may serve as potential therapeutic targets for treating epigenetically driven HCM, necessitating further research to validate their translational potential. Beyond DNA methylation, histone modifications have also been implicated in HCM pathogenesis[103,107,108]. Genome-wide analyses have revealed that methylation of histone H3 at lysine 4 (H3K4), lysine 27 (H3K27), and the specific tri-methylation of lysine 4 (H3K4me) modulates the transcription of genes linked to pressure overload-induced HCM[109]. The functional consequences of histone methylation are highly dependent on the specific residue being modified, as it can either activate or repress gene transcription[110]. Notably, H3K9me3 (tri-methylation of lysine 9 on histone H3) and H3K9me2 (di-methylation of lysine 9 on histone H3) have been associated with gene repression and HCM pathogenesis[111]. However, further in-depth studies are required to establish the precise functional connections between histone methylation and HCM development.
Given the vast scope of epigenetics and the specific focus of our review, readers seeking a more comprehensive discussion of HCM-related epigenetic mechanisms are encouraged to refer to other specialized reviews on this topic[102,107,112].
-
Overall, the current literature provides valuable insights into the pathogenicity and development of hypertrophic cardiomyopathy (HCM) in both animal and in vitro models. Mechanistically, HCM pathogenesis is mainly driven by two primary mechanisms: genetic mutations in sarcomere-associated genes and proteins, and non-genetic factors such as altered signaling pathways and transcriptional dysregulation in cardiomyocytes. It is important to note that the underlying molecular mechanisms of HCM pathology are highly complex, requiring more than a single review to comprehensively address all aspects. Nevertheless, this review highlights some of the most recently identified crucial findings in this field. Despite the progress made, significant gaps in experimental research remain. Many studies suffer from inadequate design and limited mechanistic exploration, particularly in elucidating the broader molecular networks implicated in HCM. For instance, while Ang II and salusins have been implicated in HCM development, current knowledge is largely limited to isolated findings on specific molecules, without a detailed characterization of their downstream signaling cascades, gene regulatory effects, or how they collectively contribute to pathological functional changes. Furthermore, the existing literature disproportionately emphasizes HCM-associated mutations and their biochemical effects on myosin and sarcomere function and structure. While these genetic contributions are critical, we hypothesize that intracellular signaling alterations and broader molecular changes within cardiomyocytes play a fundamental role in HCM pathogenesis. Unfortunately, this aspect has been substantially overlooked in recent studies, underscoring the need for expanded research efforts in this domain.
Moving forward, a more comprehensive elucidation of the implicated molecular mechanisms is crucial for advancing HCM therapy. This requires a broader scope of investigation than what the current literature has achieved thus far. Additionally, genetic and molecular findings could potentially facilitate HCM patient stratification, paving the way for personalized treatments. However, realizing this goal remains challenging due to several limitations. One major obstacle is the lack of large-scale cohort studies that systematically screen HCM-related genetic variations across diverse patient populations, with appropriate exclusion of confounding factors to establish robust genetic biobanks. Furthermore, the absence of well-structured basic experimental studies focusing on molecular mechanisms significantly hinders progress toward precision medicine in HCM.
Another important limitation is the translational gap between experimental models and clinical applications. The majority of studies included in this review rely on in vitro models and animal (murine) studies, which inherently differ from human physiology. These species-specific variations raise concerns regarding the clinical applicability of preclinical findings. Thus, direct studies on human HCM patients, or the development of in vivo human-based models, would provide more relevant insights and enhance the likelihood of translating experimental discoveries into meaningful clinical interventions.
Hypertrophic Cardiomyopathy: Mechanisms of Pathogenicity
doi: 10.3967/bes2025.096
- Received Date: 2024-12-08
- Accepted Date: 2025-03-31
-
Key words:
- HCM /
- Genetic mutations /
- Molecular mechanisms /
- Cardiomyocytes /
- Myofibrils /
- Sarcomere /
- Myosin
Abstract: Hypertrophic cardiomyopathy (HCM) is a major contributor to cardiovascular diseases (CVD), the leading cause of death globally. HCM can precipitate heart failure (HF) by causing the cardiac tissue to weaken and stretch, thereby impairing its pumping efficiency. Moreover, HCM increases the risk of atrial fibrillation, which in turn elevates the likelihood of thrombus formation and stroke. Given these significant clinical ramifications, research into the etiology and pathogenesis of HCM is intensifying at multiple levels. In this review, we discuss and synthesize the latest findings on HCM pathogenesis, drawing on key experimental studies conducted both in vitro and in vivo. We also offer our insights and perspectives on these mechanisms, while highlighting the limitations of current research. Advancing fundamental research in this area is essential for developing effective therapeutic interventions and enhancing the clinical management of HCM.
Baoxi Wang, Yueting Zhou, Yipin Zhao, Yong Cheng, Jun Ren, Guanchang Tan and Xiaohu Wang were involved in manuscript drafting and editing of the study.The authors declare no conflict of interest.
&These authors contributed equally to this work.
| Citation: | Baoxi Wang, Yueting Zhou, Yipin Zhao, Yong Cheng, Jun Ren, Guanchang Tan, Xiaohu Wang. Hypertrophic Cardiomyopathy: Mechanisms of Pathogenicity[J]. Biomedical and Environmental Sciences. doi: 10.3967/bes2025.096

|

 Quick Links
Quick Links
 DownLoad:
DownLoad: