Objective To examine the association between habitual sleep duration and obesity among Chinese adults.Methods The association of sleep duration and obesity was investigated among 7,094 community-dwelling Chinese adults. Sleep duration was self-reported. In this study, obesity was defined as follows: body mass index (BMI) ≥ 28 kg/m2, waist circumference (WC) ≥ 85 cm in men and ≥ 80 cm in women, and percent body fat (% BF) ≥ 25 in men and ≥ 35 in women. Logistic and quantile regressions were employed to examine relationships of interest.Results Overall, 6.42% of the participants reported short sleep durations (<6 h/d) while 14.71% reported long (≥ 9 h/d) sleep durations. Long sleepers (≥ 9 h/d) represented a greater frequency of women with obesity [odds ratio (OR): 1.30; 95% confidence interval (CI), 1.02-1.67] and high body fat (1.43, 1.04-1.96) than those who slept 7-8 h/d. An association between long sleep times and higher BMI estimations was found across the 10th-75th percentile of the BMI distribution. Among men, long sleepers (≥ 9 h/d) presented lower risks of developing abdominal obesity compared with individuals who slept 7-8 h/d (OR=0.79, 95% CI: 0.44-0.99).Conclusion Our study suggests that long sleep durations are associated with general obesity in Chinese women but reduced waist circumferences in men. Confirmatory studies are needed to determine the heterogeneous association of sleep time and obesity by gender.
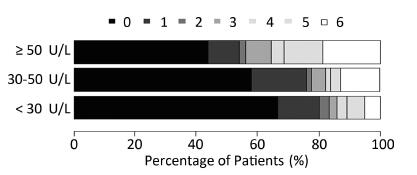
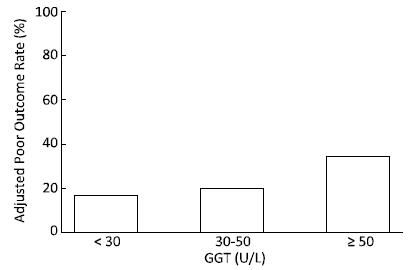
Objective We aim to explore the potential association between serum gamma-glutamyl transferase levels and functional outcome after aneurysmal subarachnoid hemorrhage in a Chinese population.Methods A total of 386 aneurysmal subarachnoid hemorrhage patients were included in the study from September 2007 to February 2015. Baseline serum gamma-glutamyl transferase levels and 6-month follow-up functional outcomes were determined. A poor outcome was defined as a modified ranking scale score of ≥ 3. The multivariable logistic model was used to analyze the relationship between serum gamma-glutamyl transferase and clinical outcomes after aneurysmal subarachnoid hemorrhage.Results The adjusted poor outcome rates of patients with gamma-glutamyl transferase levels of <30 U/L, 30-50 U/L and ≥ 50 U/L were 16.7%, 19.6%, and 34.4%, respectively (P<0.01). The age-sex and multivariable adjusted odds ratios (95% confidence intervals) of poor prognosis comparing the top group (≥ 50 U/L) with the lowest group (<30 U/L) were 5.76 (2.74-12.13), 6.64 (2.05-21.52), and 6.36 (1.92-21.02). A significant linear trend existed between gamma-glutamyl transferase level and aneurysmal subarachnoid hemorrhage prognosis. This association was also observed among nondrinkers.Conclusion Patients with higher gamma-glutamyl transferase levels were more likely to have a poor prognosis. Serum gamma-glutamyl transferase can be considered to be an independent predictor of functional outcomes after aneurysmal subarachnoid hemorrhage.
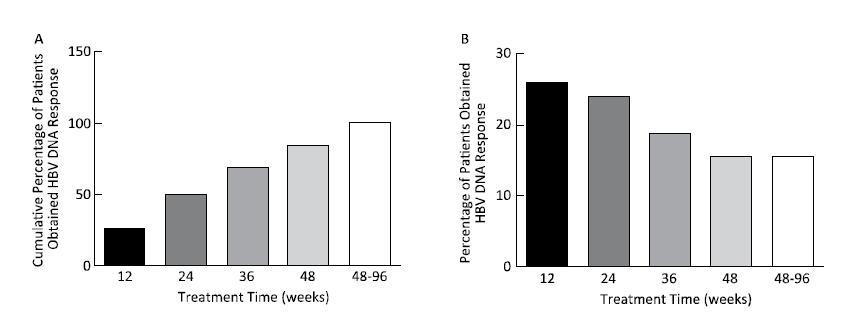
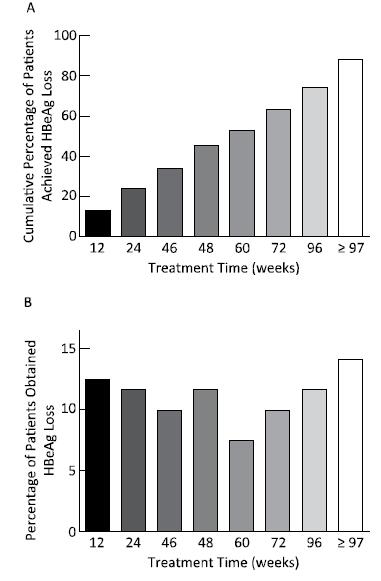
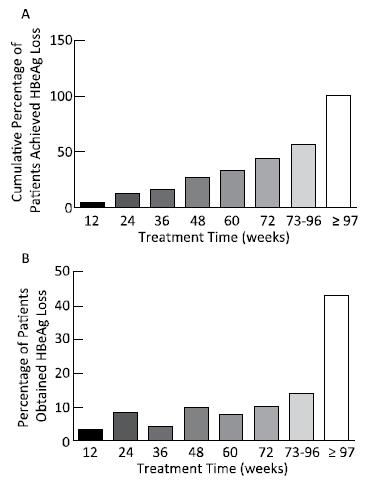
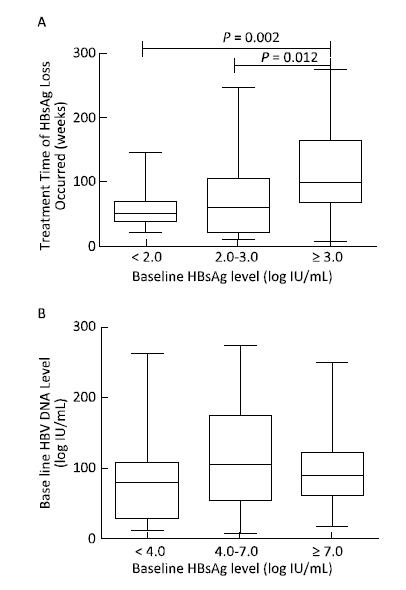
Objective To explore the predictive value of baseline HBsAg level and early response for HBsAg loss in patients with HBeAg-positive chronic hepatitis B during pegylated interferon alpha-2a treatment.Methods A total of 121 patients with HBeAg-positive chronic hepatitis B who achieved HBsAg loss were enrolled; all patients were treated with PEG-IFNα-2a 180 μg/week. Serum HBV DNA and serological indicators (HBsAg, anti-HBs, HBeAg, and anti-HBe) were determined before and every 3 months during treatment.Results The median treatment time for HBsAg loss was 84 weeks (7-273 weeks), and 74.38% (90 cases) of the patients needed extended treatment (> 48 weeks). The correlation between baseline HBsAg levels and the treatment time of HBsAg loss was significant (B=14.465, t=2.342, P=0.021). Baseline HBsAg levels together with the decline range of HBsAg at 24 weeks significantly correlated with the treatment time of HBsAg loss (B=29.862, t=4.890, P=0.000 and B=27.993, t=27.993, P=0.005).Conclusion Baseline HBsAg levels and extended therapy are critical steps toward HBsAg loss. Baseline HBsAg levels together with early response determined the treatment time of HBsAg loss in patients with HBeAg-positive chronic hepatitis B during pegylated interferon alpha-2a treatment.
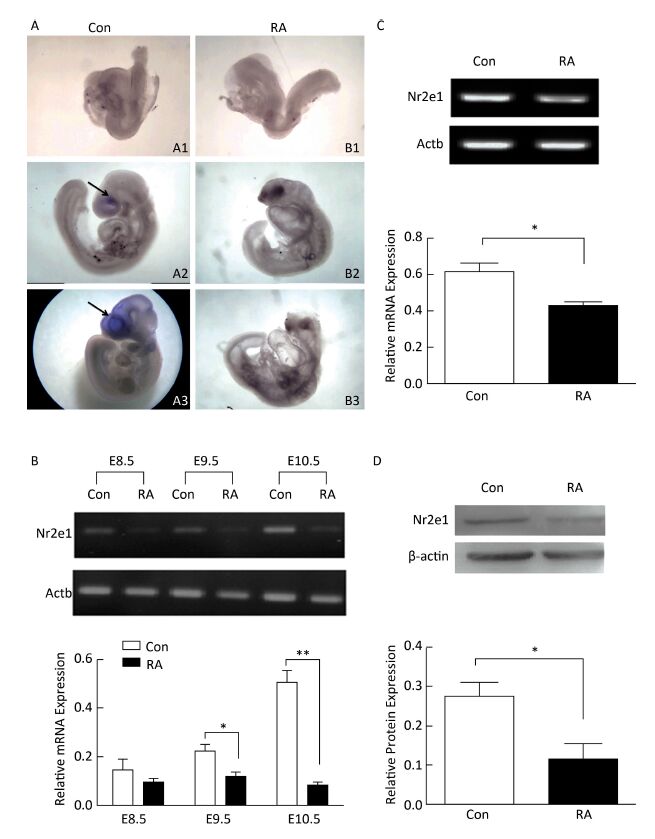
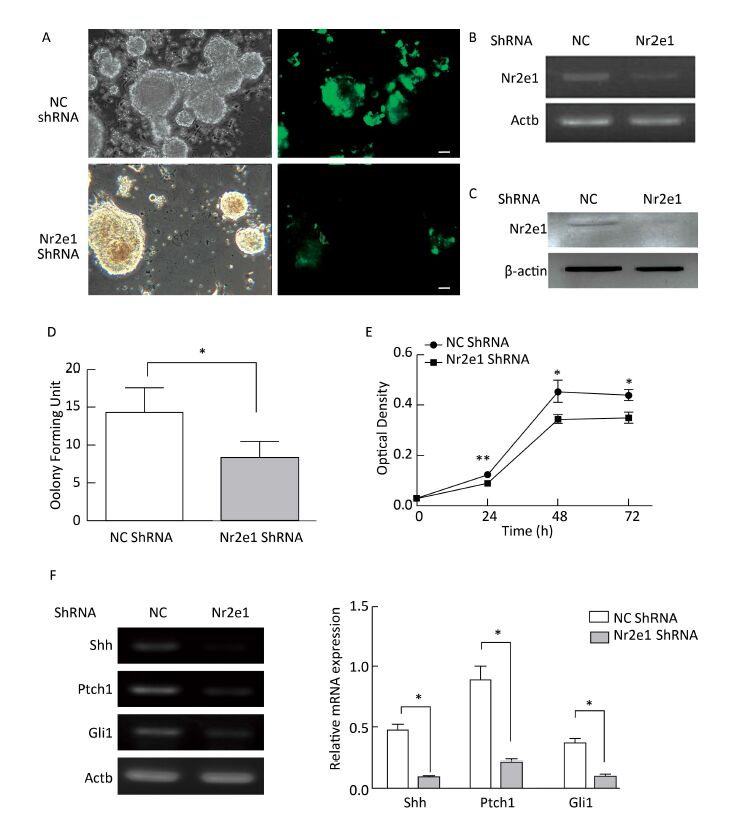
Objective This study aimed to investigate the expression pattern and function of Nuclear receptor subfamily 2 group E member 1 (Nr2e1) in retinoic acid (RA)-induced brain abnormality.Methods The mouse model of brain abnormality was established by administering 28 mg/kg RA, and neural stem cells (NSCs) were isolated from the mouse embryo and cultured in vitro. Nr2e1 expression was detected by whole mount in situ hybridization, RT-PCR, and Western blotting. Nr2e1 function was determined by transducing Nr2e1 shRNA into NSCs, and the effect on the sonic hedgehog (Shh) signaling pathway was assessed in the cells. In addition, the regulation of Nr2e1 expression by RA was also determined in vitro.Results Nr2e1 expression was significantly downregulated in the brain and NSCs of RA-treated mouse embryos, and knockdown of Nr2e1 affected the proliferation of NSCs in vitro. In addition, a similar expression pattern of Nr2e1 and RA receptor (RAR) α was observed after treatment of NSCs with different concentrations of RA.Conclusion Our study demonstrated that Nr2e1 could be regulated by RA, which would aid a better understanding of the mechanism underlying RA-induced brain abnormality.
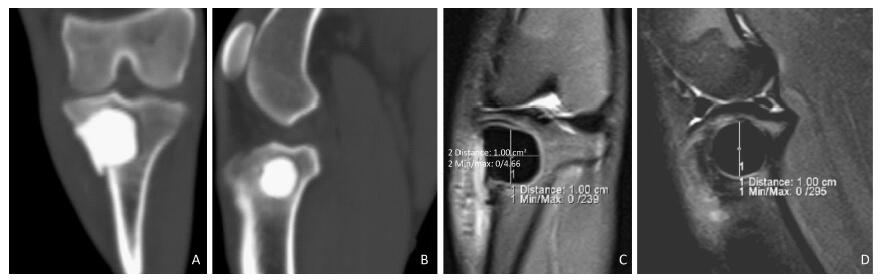
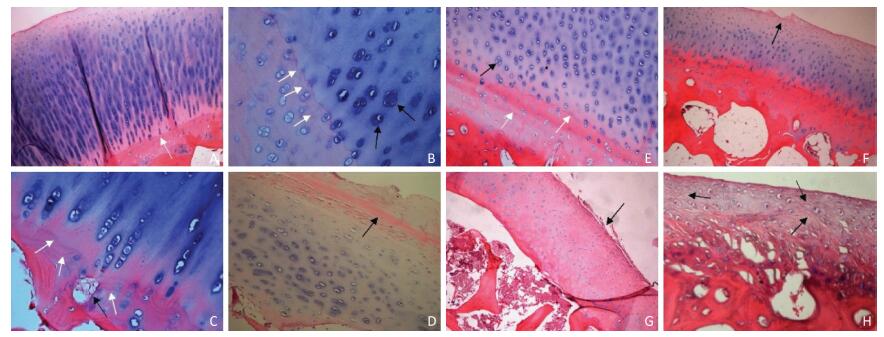
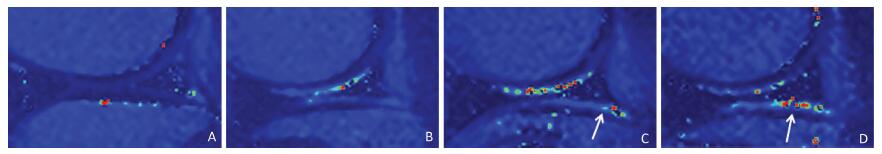
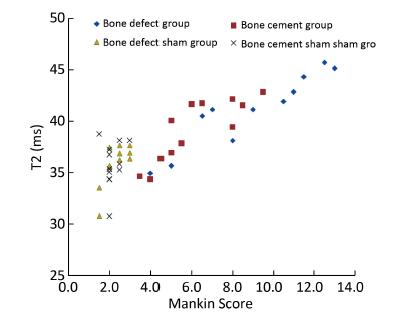
Objective Using MR T2-mapping and histopathologic score for articular cartilage to evaluate the effect of structural changes in subchondral bone on articular cartilage.Methods Twenty-four male Beagle dogs were randomly divided into a subchondral bone defect group (n=12) and a bone cement group (n=12). Models of subchondral bone defectin the medial tibial plateau and subchondral bone filled with bone cement were constructed. In all dogs, the left knee joint was used as the experimental sideand the right knee as the sham side. The T2 value for articular cartilage at the medial tibial plateau was measured at postoperative weeks 4, 8, 16, and 24. The articular cartilage specimens were stained with hematoxylin and eosin, and evaluated using the Mankin score.Results There was a statistically significant difference (P<0.05) in Mankin score between the bone defect group and the cement group at postoperative weeks 16 and 24. There was a statistically significant difference in the T2 values between the bone defect group and its sham group (P<0.05) from week 8, and between the cement group and its sham group (P<0.05) from week 16. There was significant difference in T2 values between the two experimental groups at postoperative week 24 (P<0.01). The T2 value for articular cartilage was positively correlated with the Mankin score (ρ=0.758, P<0.01).Conclusion Structural changes in subchondral bone can lead to degeneration of the adjacent articular cartilage. Defects in subchondral bone cause more severe degeneration of cartilage than subchondral bone filled with cement. The T2 value for articular cartilage increases with the extent of degeneration. MR T2-mapping images and the T2 value for articular cartilage can indicate earlycartilage degeneration.
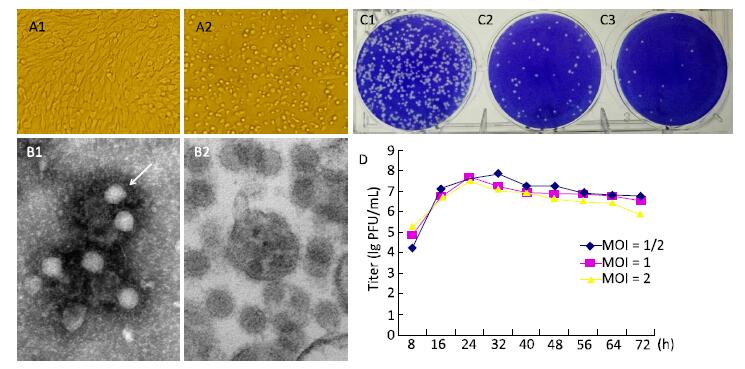
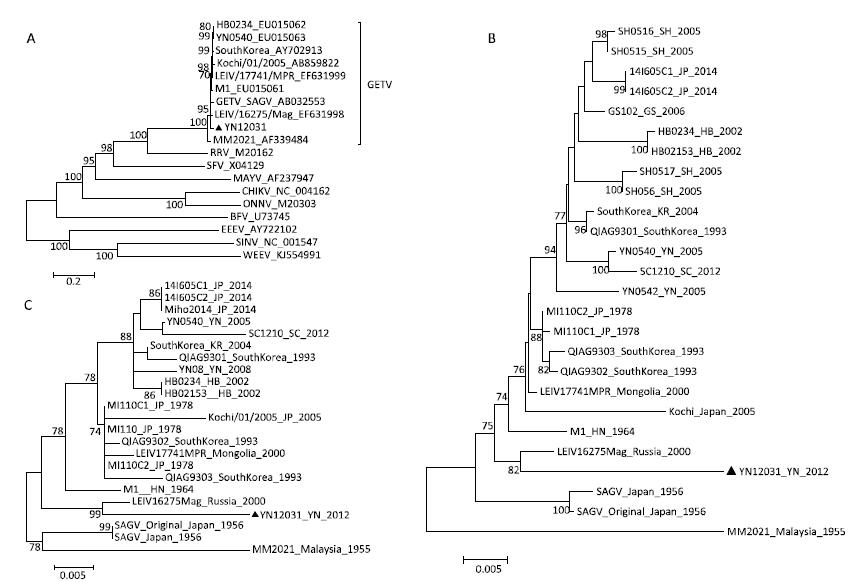
In this study, we isolated a virus strain (YN12031) from specimens of Armigeres subalbatus collected in the China-Laos border. BHK-21 cells infected with YN12031 exhibited an evident cytopathic effect (CPE) 32 h post-infection. The virus particles were spherical, 70 nm in diameter, and enveloped; they also featured surface fibers. Molecular genetic analysis revealed that YN12031 was closely related to alpha viruses such as Chikungunya virus and Sindbis virus, and located in the same clade as MM2021, the prototype of Getahvirus (GETV) isolated in Malaysia in 1955. Phylogenetic analysis of the E2 and capsid genes further revealed that YN12031 was located in the same clade as the Russian isolate LEIV/16275/Mag. Analysis of the homology of nucleotides and amino acids in the coding area and E2 gene demonstrated that the YN12031 isolated from the China-Laos border (tropical region) was related closest to the LEIV/16275/Mag isolate obtained in Russia (North frigid zone area) among other isolates studied. These results suggest that GETV can adapt to different geographical environments to propagate and evolve. Thus, strengthening the detection and monitoring of GETV and its related diseases is very crucial.




























 Quick Links
Quick Links