
HTML
-
The genus enterovirus is a member of the family picornavirus, a historically ancient virus species. Enteroviruses includes polioviruses, coxsackieviruses A and B, echoviruses, and recently identified enteroviruses[1, 2]. Most enteroviruses enter the body through the respiratory or gastrointestinal tract and usually cause mild diseases; however, they can cause encephalitis, paralysis, and myocarditis[3]. Coxsackievirus B3 (CVB3) is one major pathogen of viral myocarditis, a condition that may evolve into dilated cardiomyopathy. The pathogenic mechanisms of CVB3-induced myocarditis include directly attacking the heart tissue and those involving the host immune response to CVB3 infection[4].
The endoplasmic reticulum (ER) is an essential organelle of eukaryotic cells responsible for protein folding and modification, lipid biosynthesis, and calcium homeostasis[5]. When circumstances change, such as with glucose deprivation, hypoxia, aberrant Ca2+ regulation, and pathogen invasion, the unfolded or misfolded proteins accumulate in the ER, resulting in ER stress[6]. In response to ER stress, an intracellular adaptive mechanism called the unfolded protein response (UPR) is activated. UPR recovers ER homeostasis by attenuating protein synthesis, upregulating protein folding and trafficking machinery, and activating the ER-associated degradation system. However, when ER stress is so prolonged and severe that ER function cannot be restored, UPR will removes the damaged cells by activating apoptosis[7]. ER stress and ER stress-induced UPR play important roles in the course of virus infection[8]. Many plus-strand RNA viruses, including CVB3, Japanese encephalitis virus, dengue virus, and hepatitis C virus, may modify the ER membrane, exploit the ER, or reduce the ER calcium concentration during their infection. Then viral infection leads to imbalance of ER function and, subsequently, actives ER stress[9-12].
Autophagy is a highly conserved catabolic mechanism that occurs under normal conditions to maintain cellular homeostasis by clearing unwanted cytosolic materials and liberating energy. During the process, a double-membraned structure called an autophagosome is produced that engulfs damaged organelles and long-lived proteins, then fuses with lysosome to degrade and recycle the entrapped contents[13]. Many studies have focused on the relationship between autophagy and virus infection. Autophagy is a protective cellular mechanism that defends the host cells against viral infection. Autophagy directly captures and degrades intracellular viral cargo to restrict viral infection. Furthermore, autophagy activates innate and acquired immunities by delivering viral components to endosomal pattern recognition receptors and major histocompatibilty complex (MHC)[14-16]. However, to defend the antiviral role of autophagy, many viruses have evolved various ways to escape, impair or even enhance autophagy. Many RNA viruses infection, including poliovirus, CVB3, foot-and-mouth disease virus, mouse hepatitis virus, and influenza A virus, contribute autophagy and use the membranes of autophagic vacuoles for their RNA replication[17-19].
Autophagy and ER stress are 2 independent biological processes. However, an increasing number of reports indicate that ER stress may also trigger autophagy. Autophagy alleviates ER stress by removal of the aggregated protein and the portions of the expanded ER[5, 6]. Additionally, several forms of virus-activated ER stress subsequently regulates autophagy[20-22]. Previous studies have shown that CVB3 infection activates ER stress and autophagy[9, 23], but the relationship of ER stress and autophagy is still not completely understood. In our study, we have demonstrated that CVB3 infection activated all 3 pathways of ER stress contributing to autophagy.
-
HeLa cells were purchased from the American Type Culture Collection (ATCC) and grew in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum, penicillin, and streptomycin at 37 ℃ in 5% CO2. CVB3 was obtained from ATCC and propagated in HeLa cells. The virus titer was calculated by plaque assay. Briefly, HeLa cells were plated in 6-well plates [(7-9) × 105 cells/well] and grew overnight to approximately 90% confluency, washed with PBS and overlaid with 500 µL of virus-containing samples 10-fold serially diluted in serum-free DMEM. Then the cells were incubated at 37 ℃ for 1 h and shaken gently every 15 min, washed with PBS and overlaid with 2 mL DMEM supplied with 0.7% agar and 2% FBS. After 72 h, the cells were fixed with 4% paraformaldehyde and stained with 1% crystal violet. Subsequently, the plaques were counted and the viral titer was calculated.
-
HeLa cells grown to approximately 80%-90% confluency were infected with CVB3 at an multiplicity of infection (MOI) of 10 for 1 h, then washed with PBS and cultured in DMEM supplemented with 2% FBS until they were harvested at various times. To inhibit ER stress, HeLa cells were pretreated with 1 μmol/L PRK-like ER protein kinase (PERK) inhibitor GSK2656157 (S7033, Selleck), 300 μmol/L ATF6 inhibitor AEBSF (78431, Thermo Scientific) or 5 μmol/L IRE1 inhibitor STF-083010 (ab146176, Abcam) for 2 h, then the cells were infected with CVB3 at an MOI of 10. At 1 h post-infection, HeLa cells were incubated in fresh maintenance medium in the presence of GSK2656157, AEBSF or STF-083010 until being harvested.
-
The cells were lysed in lysis buffer (P0013, Beyotime) on ice for 10 min. The cell lysates were centrifuged at 13, 000 ×g for 10 min at 4 ℃ and the supernatant was preserved. Then the protein sample was separated by SDS-PAGE and transferred to nitrocellulose membranes. The membranes were blocked in PBS containing 5% nonfat dry milk and 0.1% Tween-20 at room temperature for 1 h, and subsequently incubated overnight at 4 ℃ with one of the following primary antibodies: antibodies against GRP78 (#3177), PERK (#3192), eIF2α (#5324), p-eIF2α (#3398), IRE1 (#3294), p62 (#5114), mammalian target of rapamcin (mTOR) (#2983), and p-mTOR (#5536) were purchased from Cell Signaling Technology, antibodies against p-IRE1 (ab124945), ATF6 (ab122897), and β-actin (ab8226) were purchased from Abcam, antibody against LC3 (L8918) was purchased from Sigma, and antibody against VP1 (M7064) was purchased from Dako. After washed with PBS containing 0.1% Tween-20, the membranes were then reacted with Horseradish peroxidase (HRP)-labeled anti-rabbit IgG (ab6721, Abcam) or anti-mouse IgG (ab6728, Abcam) at room temperature for 1 h. Finally, the specific protein bands were visualized using chemiluminescence detection kit (NEL103E001EA, PerkinElmer).
-
Total RNA was extracted using Trizol (15596018, Invitrogen). First-strand complementary DNAs (cDNAs) were synthesized using PrimeScript RT Master Mix (RR036A, TaKaRa). PCR was performed using Premix Taq™ (R004A, TaKaRa) with the corresponding primer pairs: primers for XBP1 (human) are 5'-CTGGAACAGCAAGTGGTAGA-3' and 5'-CTGGATCCTTCTGGGTAGAC-3'; primers for β-actin (human) are 5'-TTAGTTGCGTTACACCCTTTCTTG-3' and 5'-TCACCTTCACCGTTCCAGTTT-3'. The condition of PCR was 30 cycles of 98 ℃ for 10 s, 55 ℃ for 30 s, and 72 ℃ for 1 min.
-
HeLa cells were seeded in 12-well plates and grown to 60%-70% confluency. Then the cells were transfected with plasmid GFP-LC3 (11060, Addgene) using X-tremeGENE HP DNA Transfection reagent (06366244001, Roche). At 24 h post-transfection, HeLa cells were infected with CVB3 at an MOI of 10 for 8 h, fixed with 4% paraformaldehyde and stained with 4', 6-diamidino-2-phenylindole (DAPI). The fluorescence of GFP-LC3 was visualized using a confocal fluorescence microscope.
-
All experiments were performed at least in 3 independent experiments and the results were presented as means ± standard deviation (SD). The Student's t test was used to analyze data. Statistical significance was set at P < 0.05.
Cell and Virus
ER Stress Inhibition
Western Blot Analysis
Semiquantitative RT-PCR
Confocal Microscopy
Statistical Analysis
-
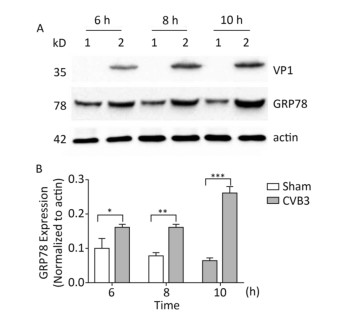
The upregulation of glucose regulated protein 78 kD (GRP78) is an indicator of ER stress in mammalian cells[24, 25]. To determine whether ER stress was activated by CVB3 infection, we first identified the expression of GRP78 in CVB3-infected cells. HeLa cells were infected with CVB3 at an MOI of 10, and the cells were collected at 6, 8, and 10 h postinfection. The expression levels of GRP78 in CVB3-infected HeLa cells were detected by Western blotting. As shown in Figure 1, compared with control cells, GRP78 expression gradually increased and reached a high level at 10 h postinfection. These results confirmed that CVB3 infection triggered ER stress and activated UPR in HeLa cells.
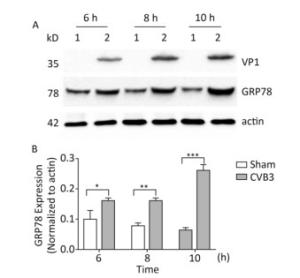
Figure 1. ER stress was induced in CVB3-infected HeLa cells. (A) HeLa cells were infected with CVB3 at MOI of 10. The expression levels of GRP78, VP1 and actin were detected by Western blotting at 6, 8, and 10 h postinfection. 1: HeLa cells, 2: CVB3-infected HeLa cells. (B) Gray scanning analysis of GRP78 to actin. Experiment was repeated three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
-
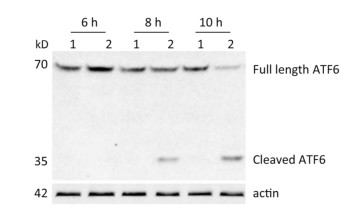
ATF6 is an ER membrane protein with a relative molecular mass of about 90 kD. Under ER stress, ATF6 is translocated to the Golgi apparatus, where it is cleaved to produce an N-terminal–activated form (p50ATF6). As an active transcription factor, p50ATF6 then enters the nucleus and moderates ER stress-associated gene expression[26, 27]. To investigate whether the ATF6 pathway was activated by CVB3 infection, HeLa cells were collected at 6, 8, and 10 h postinfection. Both full-length ATF6 and cleaved ATF6 were detected by Western blotting. Our results showed that full-length ATF6 (p90ATF6) expression decreased with extension of CVB3 infection. Meanwhile, cleaved ATF6 (p50ATF6) could be clearly detected at each time point in CVB3-infected HeLa cells, and the expression of p50ATF6 significantly increased with extension of infection time (Figure 2). Thus, the results suggested that the ATF6 pathway was activated by CVB3 infection.
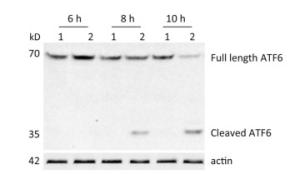
Figure 2. ATF6 pathway was activated in CVB3-infected HeLa cells. After HeLa cells were inoculated with CVB3 at MOI of 10, the expression levels of full-length ATF6, cleaved ATF6 and actin were detected by Western blotting at 6, 8, and 10 h postinfection. 1: HeLa cells, 2: CVB3-infected HeLa cells.
-
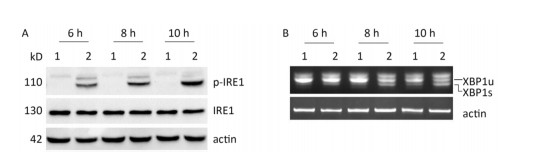
Under ER stress, IRE1 is activated by oligomerization and autophosphorylation and exhibits both endoribonuclease and kinase activity. The IRE1 RNase domain directly splices a 26- nucleotide intron from XBP1 mRNA. Then the spliced XBP1 (XBP1s) mRNA is translated into a potent transcription factor that increases the expression of ER stress–associated proteins[28, 29]. To explore whether CVB3 infection triggered the activation of IRE1, HeLa cells were inoculated with CVB3 and collected at 6, 8, and 10 h postinfection. The expressions of IRE1 and p-IRE1 were detected by Western blotting. Our results showed that CVB3 increased the phosphorylation level of IRE1. The expression levels of p-IRE1 increased gradually and reached a peak at 10 h postinfection (Figure 3A). To investigate whether CVB3 infection activated the IRE1-XBP1 pathway, both the full-length and spliced XBP1 mRNA were amplified by RT-PCR. The results showed that spliced XBP1 mRNA appeared and increased with the extension of the infection time in CVB3-infected HeLa cells (Figure 3B). Taken together, the results indicated that the IRE1-XBP1 pathway was fully activated in CVB3-infected HeLa cells.
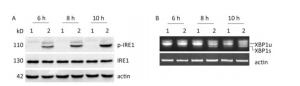
Figure 3. IRE1-XBP1 pathway was activated in CVB3-infected HeLa cells. (A) The expression levels of IRE1, p-IRE1, and actin were detected by Western blotting at 6, 8, and 10 h postinfection. (B) The relative XBP1 mRNA splicing was detected at 6, 8, and 10 h postinfection by RT-PCR. HeLa cells were infected with CVB3 at MOI of 10. 1: HeLa cells, 2: CVB3-infected HeLa cells.
-
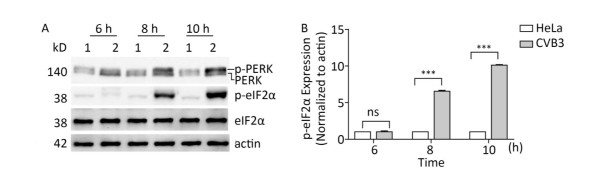
The PERK-eIF2α pathway is an important sensor of ER stress. During ER stress, PERK is activated by dimerization and autophosphorylation. Activated PERK phosphorylates eIF2α and subsequently effectively blocks protein synthesis[26]. To understand whether the PERK-eIF2α pathway was activated by CVB3 infection, the phosphorylation levels of PERK and eIF2α were detected by Western blot. Western blotting showed that more PERK (phosphorylated PERK) migrated at a higher molecular weight in CVB3-infected cells than in mock-infected cells (Figure 4A), which was consistent with previous researches indicating the phosphorylated PERK shifts slower in polyacrylamide gels than the latent form of the kinase[30, 31]. The result showed that CVB3 infection activated PERK. Consistently, phosphorylated eIF2α obviously appeared after CVB3 infection and increased in a time-dependent manner (Figure 4A). These results suggested that CVB3 infection activated the PERK-eIF2α pathway of ER stress.
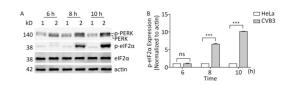
Figure 4. CVB3 infection induced PERK-eIF2α pathway. (A) The expression levels of PERK, p-PERK, eIF2α, p-eIF2α, and actin were detected by Western blotting at 6, 8, and 10 h post infection. HeLa cells were infected with CVB3 at MOI of 10. 1: HeLa cells, 2: HeLa cells infected with CVB3. (B) Gray scanning analysis of p-eIF2α to actin. Experiment was repeated three independent experiments. ***P < 0.001.
-
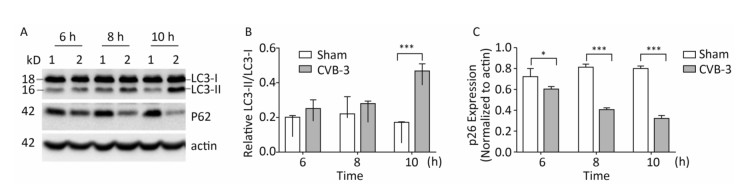
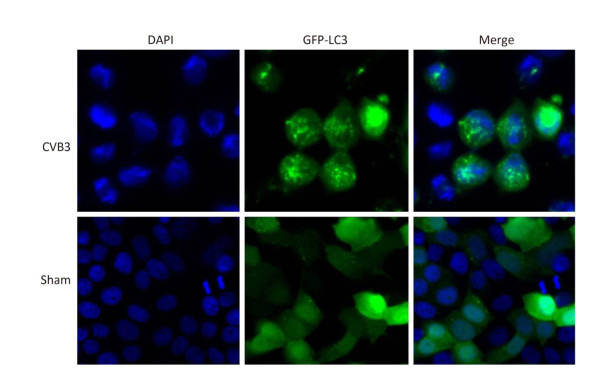
In mammals, LC3 is synthesized as a precursor form and is cleaved at its COOH terminus by the protease ATG4, referred to as LC3-Ⅰ. LC3-Ⅰ is conjugated to phosphatidylethanolamine (PE) to form LC3-Ⅱ, which specifically targets to the autophagosome membrane and participates in the formation of autophagosomes[13, 23]. Therefore the conversion of LC3-Ⅰ to LC3-Ⅱ is a key step in autophagy[23]. To determine whether autophagy was induced by CVB3 infection, we first measured the expression level of LC3-Ⅰ and LC3-Ⅱ by Western blot assay at 6, 8, and 10 h postinfection. As shown in Figure 5A and 5B, CVB3 induced the conversion from LC3-Ⅰ to LC3-Ⅱ, and the expression of LC3-Ⅱ reached its highest level at 10 h postinfection, which demonstrated that CVB3 infection induced autophagy. To further confirm the above results, we examined the distribution of LC3 in CVB3-infected HeLa cells using confocal microscopy. The results showed that CVB3 infection contributed to the obvious accumulation of green fluorescent dots from GFP-LC3 expression (Figure 6), which again showed that CVB3 infection increased formation of autophagosomes.
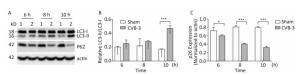
Figure 5. CVB3 infection induced autophagy. (A) The expression levels of LC3-Ⅰ/Ⅱ, P62, and actin were detected by Western blotting at 6, 8, and 10 h postinfection. HeLa cells were infected with CVB3 at MOI of 10. 1: HeLa cells, 2: CVB3-infected HeLa cells. (B, C) Gray scanning analysis of LC3-Ⅱ to LC3-Ⅰ, and P62 to actin. Experiment was repeated three independent experiments. *P < 0.05, ***P < 0.001.
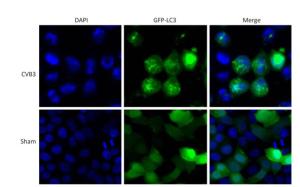
Figure 6. The fluorescence of GFP-LC3 was observed using confocal microscopy. Robust fluorescence punctate (green dots) was presented in CVB3-infected HeLa cells. Diffused fluorescence (green smears) was expressed in normal cells. Cell nuclei were stained with DAPI (×400).
During autophagy, the autophagy receptor SQSTM1/p62 is delivered to lysosomes for degradation. p62 is used as an indicator of autophagic flux or autophagy-mediated protein degration[32]. To determine whether p62 was degraded during CVB3-induced autophagy, HeLa cells were collected at 3 time points postinfection and the expression of p62 was detected using Western blotting. In CVB3-infected cells, p62 expression gradually decreased and reached a minimum level at 10 h postinfection (Figure 5A and 5C). The results demonstrated that CVB3 infection promoted protein degradation by lysosomes.
-
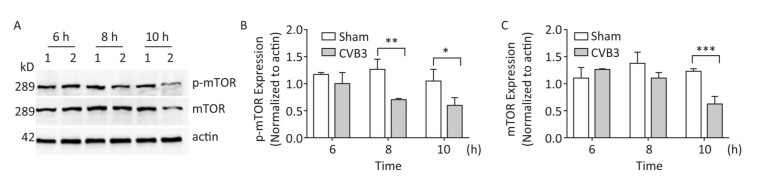
mTOR1 is established as a potent repressor of autophagy in eukaryotes[33]. The PI3K-AKT signaling axis phosphorylates mTOR1 at Ser2448. Phosphorylated mTOR1 exhibits the kinase activity and inhibits autophagy[34]. To understand the molecular basis of CVB3-induced autophagy, we detected the expression of mTOR1 and p-mTOR1. Our results showed that CVB3 infection decreased the expression of total mTOR1 and p-mTOR1 (Ser2448) in a time-dependent manner (Figure 7), which indicated that CVB3 infection may inhibit the kinase activity of mTOR1 to induce autophagy.

Figure 7. CVB3 infection decreased expression of mTOR1 and p-mTOR1. (A) The expression levels of mTOR1, p-mTOR1, and actin were detected by Western blotting at 6, 8, and 10 h postinfection. HeLa cells were infected with CVB3 at MOI of 10. 1: HeLa cells, 2: HeLa cells. (B, C) Gray scanning analysis of p-mTOR1 to actin, and mTOR1 to actin. Experiment was repeated three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
-
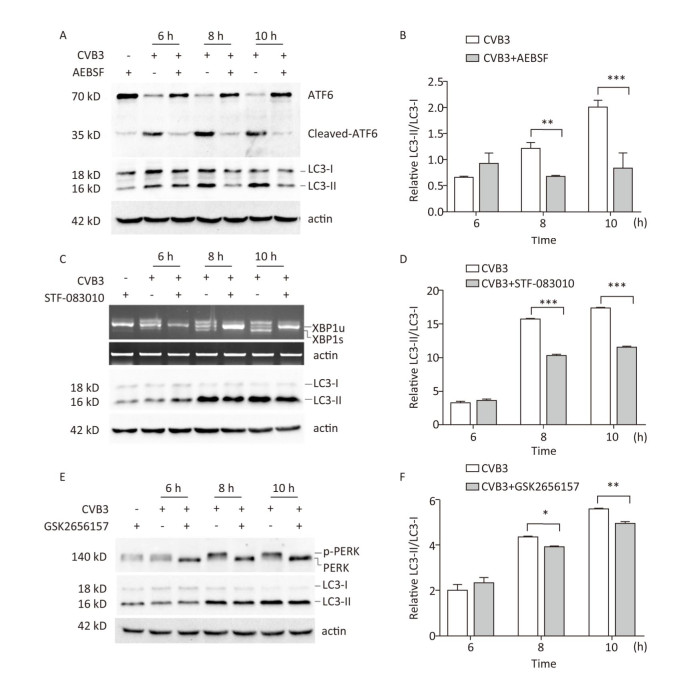
To explore whether CVB3 infection induced autophagy through UPR, ATF6, IRE1, and PERK signal pathway was inhibited by the ATF6 inhibitor AEBSF[35, 36], the IRE1 inhibitor STF-083010[37, 38], and the PERK inhibitor GSK2656157[31, 39]. Our results showed that these inhibitors obviously decreased the formation of cleaved ATF6 protein, spliced XBP1 mRNA, and phosphorylated PERK protein in CVB3-infected cells (Figure 8A, 8C and 8E). Next, the expression levels of LC3-Ⅰ and LC3-Ⅱ were detected using Western blotting. The results showed that the ratio of LC3-Ⅱ to LC3-Ⅰ decreased in the presence of the ATF6 inhibitor (Figure 8A and 8B), IRE1 inhibitor (Figure 8C and 8D), or PERK inhibitor (Figure 8E and 8F) in CVB3-infected cells, which demonstrated that CVB3 infection induced autophagy through the ATF6, IRE1, and PERK signal pathways.
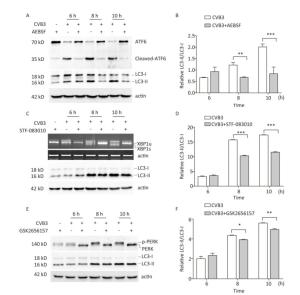
Figure 8. CVB3 infection induced autophagy through ER stress. (A, C, and E) The expression levels of ATF6, p-PERK, LC3-Ⅰ/Ⅱ, and actin were detected by Western blotting, and the relative XBP1 splicing was detected by RT-PCR at 6, 8, and 10 h postinfection. (B, D, and F) Gray scanning analysis of LC3-Ⅱ to LC3-Ⅰ. Experiment was repeated three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
CVB3 Infection Activated ER Stress
CVB3 Infection Induced Activation of the ATF6 Pathway
CVB3 Infection Induced Activation of the IRE1- XBP1 Pathway
CVB3 Infection Induced PERK- eIF2a Pathway
CVB3 Infection Induced Autophagy
CVB3 Infection Decreased the Kinase Activity of mTOR
CVB3 Infection Induced Autophagy through 3 Sensors Pathways of UPR
-
Ongoing studies demonstrate that ER stress regulates autophagy and ATF6, IRE1, and PERK signal pathways of ER stress participate in the activation of autophagy in various ways. Under ER stress, PERK-eIF2α upregulates the expression of transcription factor ATF4, which activates another transcription factor, CHOP. ATF4 and CHOP translocate to the nucleus to regulate autophagy-related gene expression[5]. IRE1 interacts with tumor necrosis factor receptor-associated factor-2 and apoptosis signal-regulating kinase-1 to activate Jun-N-terminal kinase (JNK)[6]. Then, the activated JNK phosphorylates Bcl-2 to free Bcl-2 from the Beclin-1/Bcl-2 complex, which contributes to the nucleation of autophagy. Meanwhile, IRE1-mediated XBP1 splicing also promotes autophagy through transcriptional activation of autophagy-related genes[40, 41]. Proteolytic ATF6, as an activated transcription factor, participates in regulation of the expression of GRP78 and AKT to induce autophagy[42, 43]. In our study, inhibition of IRE1, PERK, or ATF6 signaling pathways of the UPR sensor decreased the ratio of LC3-Ⅱ to LC3-Ⅰ in CVB3-infected HeLa cells, which demonstrated that CVB3 infection triggered autophagy through the IRE1, PERK, and ATF6 signaling pathways.
mTOR is a negative regulator of autophagy. mTOR1 inhibits autophagy through regulation of unc-51-like kinase 1 (ULK1) complex, which composed of ULK1, autophagy-related gene 13 (ATG13), and focal adhesion kinase family- interacting protein of 200 kD (FIP200) in mammals[44, 45]. In our study, CVB3 infection decreased the expression of total mTOR. A possible explanation is that CVB3 2A or 3C protease may cleave mTOR and decrease the expression of mTOR. Because there are many enterovirus protease cleavage sites on mTOR, which were predicted by NetPicoRNA 1.0 Server[46].
Several kinases, including PERK, protein kinase double-stranded RNA-dependent (PKR), general control non-derepressible-2 (GCN2), and heme-regulated inhibitor (HRI) may phosphorylate eIF2α to prevent protein synthesis and regulate stress-related gene expression. These kinase are respectively activated by ER stress, virus infection, uncharged tRNAs, and heme depletion[47]. One report showed that CVB3 infection triggered activation of PKR, not PERK, to phosphorylate eIF2α[9]. However, our results first demonstrated that both PERK and PKR (data not shown) were activated during CVB3 infection.
In conclusion, this study demonstrated that CVB3 infection induced ER stress to initiate autophagy through 3 classical UPR pathways.
the China Mega-project for Infectious Disease 2018ZX10734401
the SKLID Development Grant 2011SKLID104
the China Mega-project for Infectious Disease 2018ZX10734404
the China Mega-project for Infectious Disease 2018ZX10102001
the China Mega-project for Infectious Disease 2018ZX10711001

 Quick Links
Quick Links
 DownLoad:
DownLoad: