




-
Liver transplantation (LT) is the only curative option for simultaneous hepatocellular carcinoma (HCC) and cirrhosis induced by hepatitis B virus (HBV) infection. However, the recurrence rate of HCC after LT is high, and intrahepatic recurrence of the primary tumor and de novo HCC occur frequently in the transplanted liver[1]. The mechanism of HCC occurrence after LT remains unknown. Factors affecting tumor recurrence and outcome in patients with aggressive HCC include tumor size and number, pathological tumor differentiation, vascular invasion, and serum alpha-fetoprotein (AFP) level[2]. Selection criteria for LT recipients, such as the Milan and University of California at San Francisco (UCSF) criteria[3] or Hangzhou criteria[2], consider these factors, and their application has resulted in post-LT 5-year survival rates of 70%–85%, similar to those of patients without HCC[2,3]. Nevertheless, HCC recurs in the transplanted liver within a year after LT in some patients who meet these criteria. Thus, improved understanding of the origin and pathogenesis of HCC recurrence after LT is needed to pursue the development of novel strategies for HCC detection, treatment, and prognosis prediction. Distinguishing between recurrent and de novo HCC after LT would be helpful for treatment selection.
Currently, recurrent HCC of recipient origin and de novo HCC of donor origin cannot be distinguished clearly on the basis of clinical and pathological tumor characteristics. To the best of our knowledge, few methods are available for the identification of tumor origin. Fluorescent in situ hybridization with specific probes for sex chromosomes has been used to determine tumor origin[4], but it is inapplicable in cases of same-sex LT. Based multiple polymerase chain reaction (PCR) amplification of DNA sequences short tandem repeat (STR) analysis (routinely used for paternity and forensic assays) has also been applied to distinguish de novo tumors from donor or recipient origin after LT[5]. However, the analysis is complicated, and its results are difficult to interpret because of high polymorphism. About 85% of gene mutations that induce human disease occur in exon-coding regions[6]. Recent studies, especially those on mutations related to oncogenes, have applied high-throughput whole-exome sequencing to detect genetic variation and single-nucleotide polymorphisms (SNPs) associated with diseases[7]. In the present study, we investigated the origin of HCC after LT [recipient (recurrence) or donor (de novo occurrence)] in one patient by using high-throughput whole-exome sequencing and confirmed the results by highly polymorphic STR analysis. The study provides novel insights into the examination of molecular alterations by whole-exome sequencing to identify the origin of HCC in transplanted livers.
This study involved a 49-year-old male patient who had HBV-induced cirrhosis for 23 years and HCC symptoms for 2 months, with histological confirmation of HBV-positive HCC. The patient met the UCSF criteria for HCC and underwent LT in February 2013. Histological examination of the native liver revealed two cancerous lesions (maximum diameter, 4 cm) in segments VIII and VII, classified as grade G2 in accordance with the Edmondson criteria and without vascular invasion. The transplanted liver was obtained from a male by donation after brain death plus cardiac death due to heart attack. The donor was positive only for anti–hepatitis B core antigen and negative for other markers, including tumor biomolecular markers. The use of organs in this study was in compliance with the Declaration of Helsinki and the Declaration of Istanbul.
During routine follow-up after LT, two HCC lesions (maximum diameter, 3 cm) were found in the transplanted liver without HBV reinfection. The patient’s serum AFP level was 11.5 IU/L (normal range, 0–17 IU/L). The tumor nodules in the transplanted liver were resected at the same hospital in November 2013. The patient died of HCC within a year after resection.
Samples of the recipient’s primary HCC, the secondary HCC in the transplanted liver from the donor, respective adjacent liver tissues, and pre-OLT peripheral blood from the recipient were collected. Fresh tissues were collected from the patient in the operation room, snap frozen in liquid nitrogen, and then stored at –80 ℃ until use. The HCC cases were confirmed histologically, but whether or not the secondary HCC was derived from the recipient (recurrence) or donor (de novo occurrence) could not be determined by pathological analysis. Total genomic DNA was extracted using QIAamp DNA mini kits (Qiagen, Hilden, Germany), and randomly selected 150–200 bp DNA fragments were used to construct a shotgun sequencing library, as described previously[8]. Care was taken during sample handling and processing to prevent human DNA contamination and sample–sample DNA transfer. The analyses were conducted with recognition that the secondary HCC tissue could contain up to 10%–15% of the donor’s liver tissues (e.g., stroma, blood vessels, and extracellular matrix).
The cDNA library was submitted to ligation-mediated PCR amplification and hybridized to the SureSelect Biotinylated RNA Library, followed by washing, elution, and additional amplification. Post-enrichment shotgun libraries were sequenced as 90-bp paired-end reads with an Illumina Genome Analyzer II (HiSeq 2000) using standard sequencing primers in accordance with the manufacturer's instructions. Image analysis and base calling were performed using the Genome Analyzer Pipeline (version 1.7) with default parameters but no quality filtering. The number and length of reads were analyzed statistically. Short reads were aligned to the human genome by using Burrows-Wheeler Aligner software, and mismatched, inappropriate, and duplicate reads were removed with Sequence Alignment/Map tools (SAMtools; http://samtools.sourceforge.net/SAM1.pdf)[4]. The sequencing coverage and depth of coverage per target region were calculated. Only high-quality sequencing datasets were used for subsequent bioinformatic analysis.
SNP analysis of the high-throughput whole-exome sequencing data was performed using SOAPsnp software (BGI Tech Solutions Co., Ltd. Shenzhen, China) as previously described[8]. The SNPs in the tissue and blood samples were confirmed using SAMtools as previously described[8]. In brief, the DNA sequencing data were aligned and compared with normal human reference data from the GenBank database, and the picard tool was used to mark duplicate reads (redundant information produced by PCR). The alignment results from each sample were combined into one BAM-format (compressed SAM) file. The data were compared with the human reference genome (NCBI build 37/HG19) to identify SNPs by using SOAPsnp software. High-confidence single-nucleotide variants (SNVs) were detected using Varscan analysis. Indels were found using SAMtools. Additional filtering was applied to identify SNVs specific to tumor and normal samples by using the dbSNP (version 135) site, which contains common gene polymorphisms without known medical impact. ANNOVAR was then used to annotate and classify these SNVs in accordance with previous research[9]. The numbers of SNPs in the five samples were compared.
For STR analysis, the genomic DNA samples were subjected to PCR amplification using GoldenEye_20ATMkit (Peoplespot Inc., China) with a 3130XL gene analyzer (ABI, USA). The data were then analyzed using the Amelogenin method for STRs. Selected according to locus, the following chromosome markers were examined: D19S433, D5S818, D21S11, D18S51, D6S1043, D3S1358, D13S317, D7S820, D16S539, CSF1PO, Penta D, AMEL, VWA, D8S1179, TPOX, Penta E, TH01, D12S391, D2S1338, and FGA.
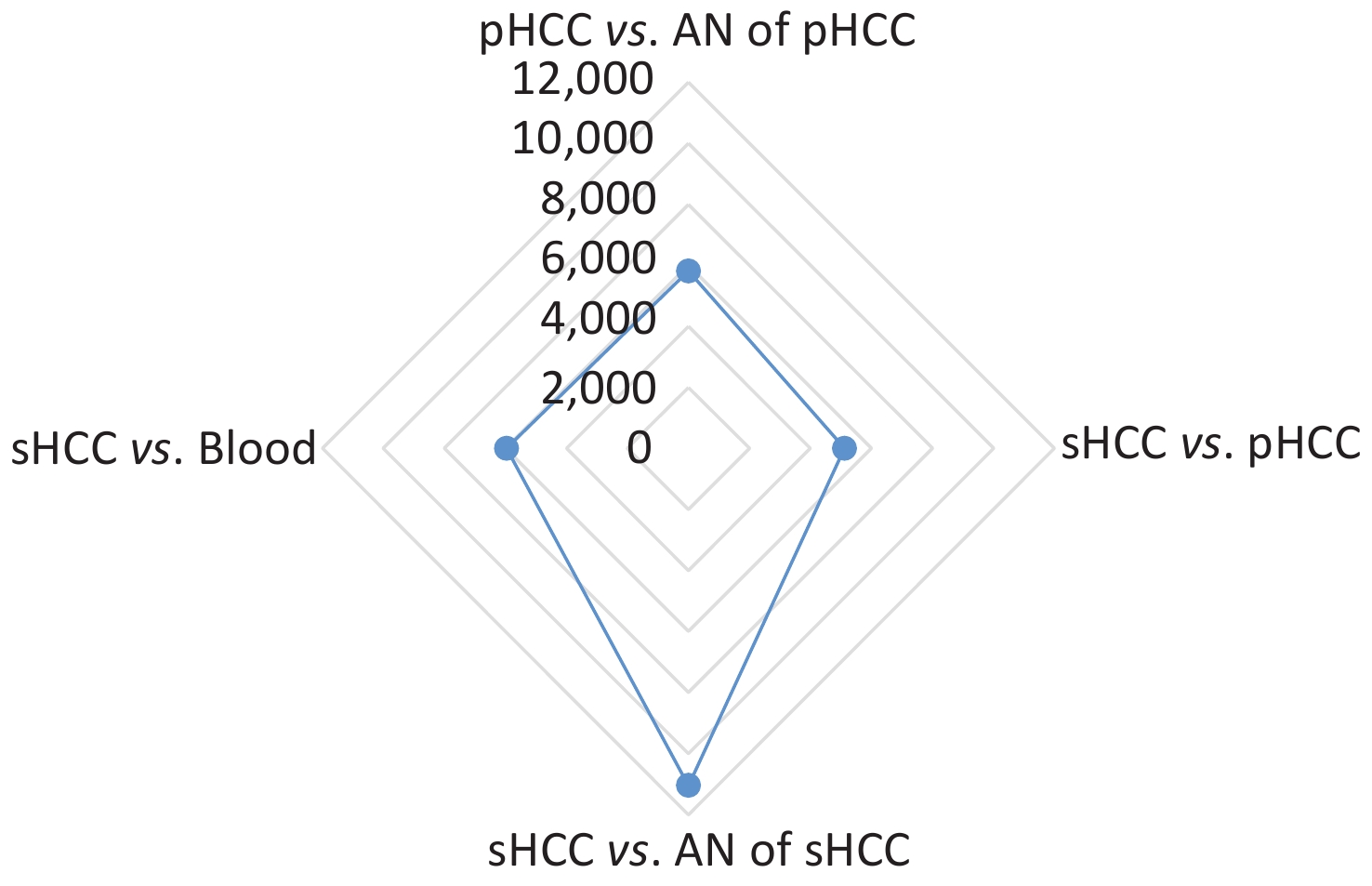
The results of high-throughput whole-exome sequencing and SNP analysis of the five samples are presented in Figure 1. Differences were found in 11,048 SNPs between secondary HCC and adjacent liver tissues, in 5,122 SNPs between primary and secondary HCC tissues, in 5,813 SNPs between primary HCC and adjacent tissues, and in 5,961 SNPs between secondary HCC tissue and blood (Figure 1). These data showed fewer differences in SNPs between the primary and secondary HCCs and more differences in SNPs between the secondary HCC and adjacent liver tissues from the donor (Figure 1). The results indicated that the secondary HCC originated from the recipient’s primary HCC. This patient who met the UCSF criteria without vascular invasion for LT and with negative serum AFP level pre-LT had recurrent HCC a year after LT, but from where and how the primary HCC cells were transmitted to the transplanted liver in such cases remain unknown. Research on HCC confirmed that the molecular mechanism of HCC development and recurrence is complex, involving different genetic and epigenetic alterations[6,7]. Gene mutation may induce physiological signaling disorders, which may contribute to tumor recurrence[6,7,9]. High-throughput whole-exome sequencing is an efficient large-scale technology for finding gene mutations related to protein functional variation. Recently, this technology has been used in the research of cancer genomes, clinical diagnosis, and mechanism of oncogenesis[7,9,10]. In the present study, the origin of HCC in the transplanted liver was identified by whole-exome sequencing SNP analysis.
Figure 1. Comparison of single-nucleotide polymorphisms (SNPs) among the primary HCC (pHCC), secondary HCC (sHCC), adjacent normal (AN) liver tissues of pHCC, and AN of sHCC and pre-transplantation blood samples.
The results of STR analysis for the molecular profiles of 20 loci in the five samples are presented in Figure 2. Genotypes at 18 loci (D5S818, D21S11, D18S51, D6S1043, D3S1358, D13S317, D7S820, D16S539, CSF1PO, Penta D, AMEL, VWA, D8S1179, TPOX, Penta E, TH01, D12S391, and D2S1338) of the primary HCC tissue completely matched with those of its adjacent liver tissue and peripheral blood sample of the recipient, and genotypes at five loci (D18S51, D6S1043, CSF1PO, Penta D, and Penta E) were amplified in the primary HCC, its adjacent liver tissue, secondary HCC tissue, and blood but not in tissue adjacent to the secondary HCC in the transplanted liver. Furthermore, genotypes at FGA loci with 20 alleles of the secondary HCC tissue only matched with that of the primary HCC tissue. STR analysis showed that the main peak of alleles of the secondary HCC was consistent with that of the primary HCC (Figure 2), suggesting that the secondary HCC originated from the recipient’s primary HCC. This result is consistent with that from the whole-exome sequencing analysis.

Figure 2. Short tandem repeat genotyping of 20 loci in the pHCC, sHCC, AN of pHCC, and AN of sHCC and pre-transplantation blood samples (A, B, C, D). Main peaks of alleles of sHCC are consistent with those of pHCC.
STR sequencing is a DNA profiling technology based on PCR analysis loci with high polymorphism of the patients’ genotype for individual identification. Meanwhile, whole-exome sequencing is a genome analysis method based on the target sequence to capture and enrich the DNA sequence of the exon region for identifying disease-related coding variants with accurate SNP detection. Given its high sensitivity, STR analysis can be affected by trace contamination with cells and DNA from other individuals, resulting in mixed genotypes and interpretative difficulties. The limitation of this study is that the tumor and its AN tissue were manually dissected samples but not micro-dissected samples, leading to trace contamination. Consequently, with the secondary HCC tissue containing cells and DNA from the donor’s liver tissues (tumor margin adjacent liver tissue), the result of STR analysis can be disturbed and misunderstood by contamination with cells and DNA from the donor. Thus, micro-dissected samples and avoiding contamination are essential for STR analysis. Another limitation is the few number of cases that underwent detection by whole-exome sequencing. Only one case was subjected to the origin identification of secondary HCC after LT by whole-exome sequencing. In consideration of the individual differences among patients with HCC, more case samples with secondary HCC after LT should be further detected to investigate the difference in origin of HCC recurrence after LT and the effectiveness of whole-exome sequencing analysis.
In conclusion, the current study is, to the best of our knowledge, the first to use high-throughput whole-exome sequencing to determine the primary origin of secondary HCC in a transplanted liver. Whole-exome sequencing analysis may be a feasible method to identify the origin of secondary HCC after LT. The introduction of whole-exome sequencing technologies may open a new area of research, with novel strategies for identifying the origin of secondary HCC after LT and elucidating the mechanism of HCC recurrence after LT.
We thank Dr. HE Ming Hui at the Department of Cancer Research, BGI Tech Solutions Co., Ltd. (Huada Clinical Laboratory Center Co., Ltd.), Shenzhen, China and Dr. DENG Ya Jun at the Beijing Zhongzheng DNA Evidence Institute, Beijing, China for their technical support.
The authors declare that they have no conflict of interest affecting this work.
The datasets generated and analyzed during the present study are available from the corresponding author on reasonable request.
HTML
Reference

 Quick Links
Quick Links
 DownLoad:
DownLoad:
