
-
From 2005 to 2018, 820,588 cases of rotavirus (RV) diarrhea in children under 5 years of age were reported in China. The reported incidence increased from 8.4/100,000 in 2005 to 178.1/100,000 in 2018. The three provinces with the highest incidence rates were Zhejiang (535.2/100,000), Guangdong (334.3/100,000), and Beijing (317.3/100,000). Co-infection with RV and other diarrheal pathogens accounted for 1.8% of cases. The rate of RV infection in children increased rapidly after 6 months of age, and 84.4% were under 2 years of age[1]. Of all the pathogens that cause diarrhea, RV was first identified in the rectum using electron microscopy of monkey swabs and mouse intestinal biopsies in the 1950s and the 1960s, respectively[2]. In humans, RV was first detected in duodenal biopsies of nine children with acute diarrhea, and the name “duo virus” was proposed for the first time[3]. RV, a member of the genus Rotavirus of the family Sedoreoviridae, is an icosahedral-shaped, envelope-free double-stranded RNA (dsRNA) virus with a capsid composed of three concentrically coated structural proteins and a core consisting of a dsRNA genome that encodes 12 viral proteins (VP), including six structural proteins (VP1–VP4, VP6, and VP7) and six nonstructural proteins (NSP1–NSP6)[4]. Membrane-associated NSP4 functions as a viral porin that mediates elevated levels of cytoplasmic Ca2+ released from cellular stores, and functions as an enterotoxin that serves as an important factor in diarrhea[5]. Simultaneously, after interacting with VP6 bilayer particles, NSP4 is transiently encapsulated in a membrane composed of autophagy-tagged and NSP4-hijacked COPII vesicles[6]. VP4 and VP7 possess type-specific antigenic determinants that induce the production of protective neutralizing antibodies in vitro (in a MA-104 monkey kidney cells model) and in vivo in different animal models[7-9]. The middle capsid protein VP6 is a group-specific antigenic determinant and crucial target of the adaptive immune response[10].
Seven groups of RVs (RVA, RVB, RVC, RVD, RVE, RVF, and RVG) have been isolated and characterized. RVA, RVB, and RVC are all capable of causing diarrhea in humans and animals; the most common cause of diarrhea in children under 5 years of age is RVA, which leads to 90% of RV enteritis[11]. According to the different antigenicities of VP7, there are different G genotypes of RVA, the most common of which are G1, G2, G3, G4, G9, and G12. VP4 is a less abundant secondary neutralizing antigen and a protease-sensitive protein with important pathogenicity[12], and depending on its antigenicity, RVA is divided into different P genotypes, the most common P genotypes being P[4], P[6], and P[8]. Among the various G- and P- genotypes, the most common combinations resulting in diarrhea are G1P[8], G2P[4], G3P[8], G4P[8], G9P[8], and G12P[8][13-16]. An RV nomenclature system, Gx-P[x]-Ix-Rx-Cx-Mx-Ax-Nx-Tx-Ex-Hx, based on the calculation of nucleotide sequence percent identity cutoff values for 11 gene fragments of encoded proteins (VP7-VP4-VP6-VP1-VP2-VP3-NSP1-NSP2-NSP3-NSP4-NSP5/NSP6) has been proposed[17]. Furthermore, human RVA strains have been split into three genotype constellations, Wa-like (G1/G3/G4/G9/G12-P[8]-I1-R1-C1-M1-A1-N1-T1-E1-H1), DS-1-like (G2-P[4]-I2-R2-C2-M2-A2-N2-T2-E2-H2), and AU-1-like (G3-P[3]-I3-R3-C3-M3-A3-N3-T3-E3-H3)[15,18].
In this study, we describe the epidemiological characteristics of acute gastroenteritis and RVA in the Pearl River Delta, Guangdong, China, in terms of age distribution and seasonal distribution through a one-year stool sample collection. We investigated the surveillance of the spectrum of RVA infections, explored the genetic evolutionary relationships of RVA strains that were successfully sequenced, and discuss four aspects of gene mutations, including genome constellations and genetic reassortment, selection pressure and codon usage bias analyses, gene polymorphism detection, and protein glycosylation prediction.
-
The study protocol was reviewed and approved by Sun Yat-sen University Zhongshan School of Medicine (approval number: 2017-056). Written informed consent was obtained from all participants or their legal guardians before sample collection. Patient records were coded and de-identified prior to analysis.
-
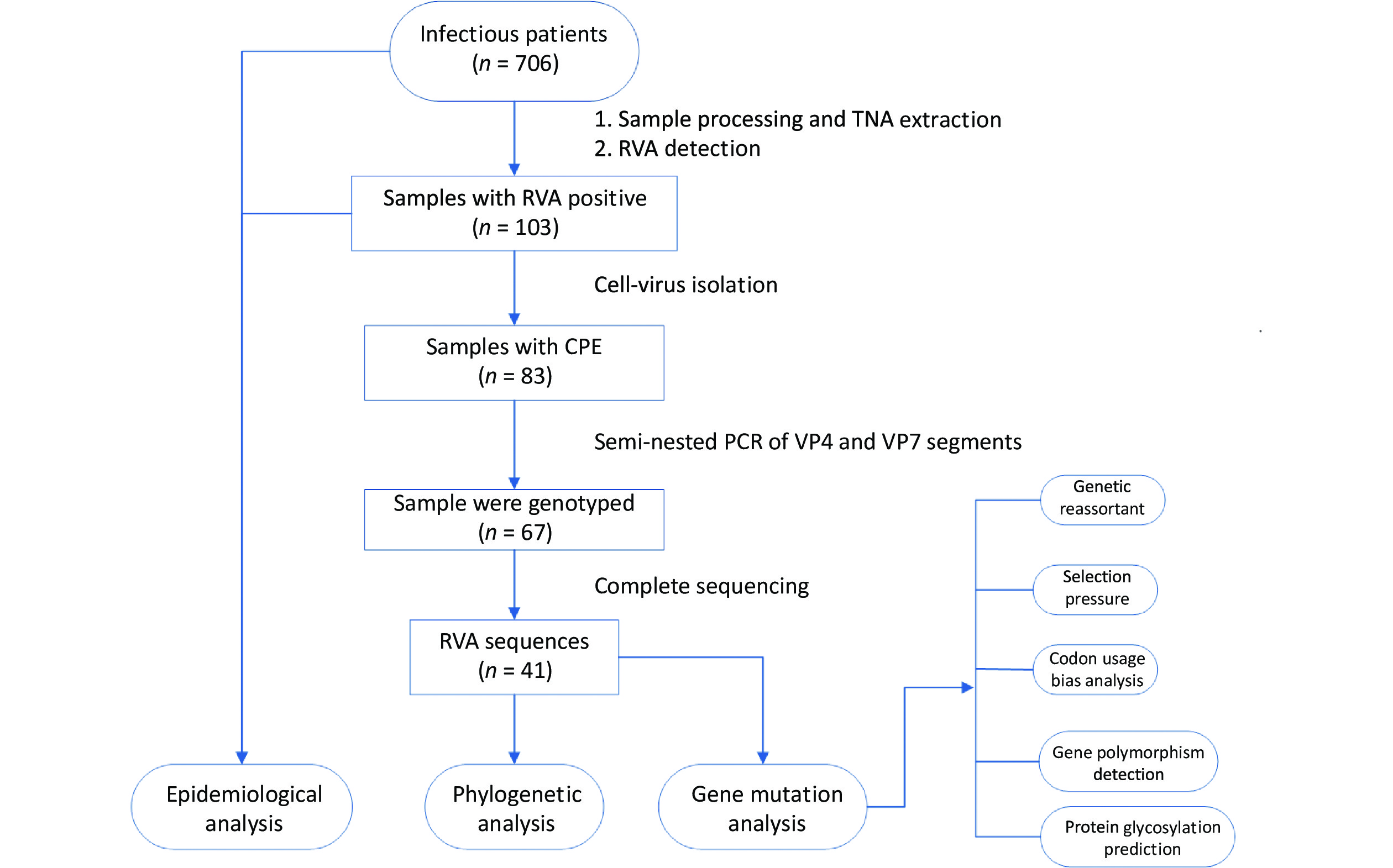
Acute gastroenteritis surveillance was conducted from January 1, 2019, to January 12, 2020, in Guangzhou (n = 241 samples), Dongguan (n = 153 samples), and Jiangmen (n = 312 samples) in Guangdong Province. A total of 706 fecal samples were collected using rectal swabs from patients with acute gastroenteritis, and more than half of the patients were male (59.35%, 419/706). All cases met the diarrhea definition according to the World Health Organization guidelines and were hospitalized with diarrhea as the primary diagnosis. All fecal samples were placed in transport media and stored at –80 °C for further processing. Demographic characteristics and clinical information were collected from electronic medical records. The research design is illustrated in Figure 1.
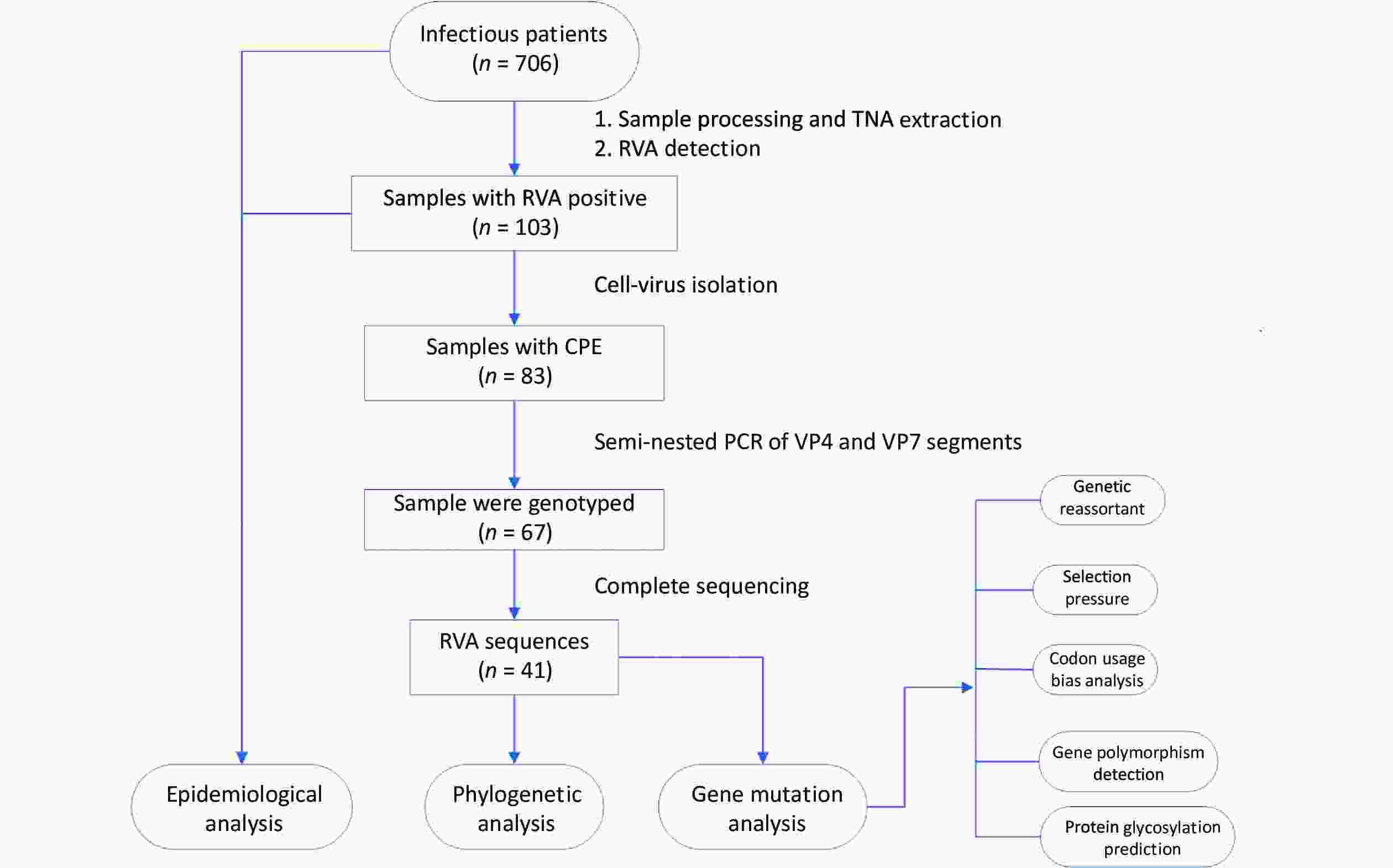
Figure 1. Research design flow chart. Stool samples were collected from patients with acute gastroenteritis in three sentinel hospitals in Guangdong province, China (n = 706). RVA-positive samples were screened through the xTAG Gastrointestinal Pathogen Panel kit (n = 103) and then were inoculated into MA104 cells, yielding 83 isolates. The RVA strains with CPE were amplified by a semi-nested PCR assay (n = 67). Subsequently, the TNAs of RVAs that had been genotyped were sequenced (n = 41). TNA, total nucleic acid; RVA, rotavirus group A; CPE, the appearance of cytopathic effect; PCR, polymerase chain reaction.
-
Fecal samples, at a final concentration of 10% weight/volume, were placed separately in 1 mL phosphate buffer saline supplemented with 10 μL Escherichia coli phage MS2 (internal control), 100 μL chloroform, and 2 g sterile glass beads, and centrifuged at 3,000 rpm for 30 minutes. Total nucleic acid (TNA) was extracted from all suspensions using a QIAamp MinElute Virus Spin Kit (Qiagen, Germany) according to the manufacturer’s protocol.
-
The xTAG Gastrointestinal Pathogen Panel kit (Luminex, USA) is based on multiplex amplification PCR and bead hybridization for simultaneous qualitative detection and identification of viral, bacterial, and parasitic nucleic acids in human fecal samples from patients with signs and symptoms of acute gastroenteritis. The RVA-positive stool suspensions were first screened, and the pathogenic spectrum was analyzed using the above-mentioned kit with an automated Luminex 200x platform (Luminex, USA) according to the manufacturer’s instructions. The data were analyzed using xTAG Data Analysis Software version 2.0 (TDAS 2.0). Positive RVA samples were isolated from MA-104 cells, and second-generation RVA samples, which showed cytopathic effect (CPE), were analyzed by fluorescent quantitative PCR. To ensure that the RVA sample had a sufficient viral load for further testing, RVA isolation was performed. MA-104 monkey kidney cells obtained from the Chinese Center for Disease Control and Prevention were used for live viral culture assays. RVA-positive sample suspensions were added at 200 μL per tube to inclined culture tubes containing MA-104 cell monolayers at a count of 2.0 x 105, and then 2 mL of maintenance medium (MM) was supplemented for blind passaging for three generations. The MM was composed of Roswell Park Memorial Institute 1640 (GIBCO, USA), 1% 1M HEPES (GIBCO, USA), 1% 100 IU/mL penicillin-streptomycin (PS; GIBCO, USA), and 4% EDTA-free trypsin solution (GIBCO, USA). Subsequently, the tubes were incubated at 37 °C in a 5% CO2 incubator for 2–7 days and then observed for CPE by an inverted microscope.
-
The TNAs of the RVA suspensions with CPE were further investigated using a semi-nested PCR test of the VP7 and VP4 segments for the RVA subtypes. Target RVA segments were amplified using a TaKaRa One-Step RT-PCR Kit (TaKaRa, China). Primers for the target segments were based on a previous study[19]. The PCR products were determined and verified with the high-performance capillary electrophoresis (HPCE) assay technique; the specific genotype of RVA can be determined by combining electrophoresis bands with the product length of G/P typing.
-
TNAs were genotyped using a semi-nested assay and sent to MicroFuture Technology Co., Ltd (Beijing, China) for complete sequencing. Because of nucleic acid degradation during sample sequencing, the sequences of 11 segments of RVA strains in our study were imported into BLAST [National Center for Biotechnology Information (NCBI)] and the RotaC genotyping tool (https://www.bv-brc.org/, accessed on April 11, 2021) for verification by the nucleotide sequence percentage identity cutoff value[17]. Entropy analysis was performed using a BioEdit Entropy [H(X)] plot to identify the aligned amino acid sequences of eleven segments of the virus. All sequences obtained in this study were deposited in the SRA Public Genomic Database under the BioProject accession number PRJNA910682.
The complete reference sequences were retrieved from NCBI (https://www.ncbi.nlm.nih.gov/) and the reference sequence numbers are presented in Table 1. A total of 14 G1P[8] sequences (VP7, VP4, VP6, VP1–VP3, NSP1–NSP5 = 14), 128 G2P[4] sequences (VP7 = 128, VP4 = 128, VP1–VP3 = 93, VP6 = 107, NSP1 = 93, NSP2 = 94, NSP3 = 93, NSP4 = 101, NSP5 = 93), 139 G3P[8] sequences (VP7 = 139, VP4 = 139, VP1–VP3 = 94, VP6 = 136, NSP1–NSP5 = 94), and 105 G9P[8] sequences (VP7 = 105, VP4 = 105, VP1–VP3 = 81, VP6 = 90, NSP1 = 81, NSP2 = 82, NSP3–NSP5 = 81) were downloaded and analyzed. Mafft v7.037 was used for sequence alignment with default settings[20]. Alignment was manually checked and corrected using BioEdit v7.0.9.0[21]. The maximum-likelihood (ML) phylogenetic tree was constructed using IQ-Tree v1.6.12, with the GTR+F+G4 nucleotide substitution model as the best-predicted model[22]. One thousand bootstrap replicates were used to deduce branch support. The phylogenetic tree was visualized using FigTree v1.4.2[23]. R v4.1.2 (R Foundation for Statistical Computing, Vienna, Austria) was used to calculate the genetic distance of sequences, obtain homology, and to generate plots.
Clinical data RVA-negative RVA-positive RVA alone RVA co-infection Other pathogens positive P value P value n = 603 n = 103 n = 62 n = 41 n = 201 RVA-negative vs.
RVA-positiveRVA alone vs.
RVA co-infectionSex, n (%) Male 358 (59.37) 61 (59.22) 38 (61.29) 23 (56.10) 124 (55.72) 0.978 0.600 Female 245 (40.36) 42 (40.78) 24 (38.71) 18 (43.90) 77 (44.28) Age (years), median (P25, P75) 1.25 (0.89, 5.00) 1.33 (1.00, 12.00) 1.54 (1.00, 13.50) 1.00 (0.96, 8.00) 1.00 (0.83, 7.00) 0.211 0.289 Clinical symptom s, n (%) Fever (≥ 37.3 °C) 142 (23.55) 27 (26.21) 11 (17.74) 16 (39.02) 53 (26.37) 0.558 0.016 Vomitus 245 (40.63) 35 (33.98) 17 (27.42) 18 (43.90) 79 (39.30) 0.202 0.084 Antibiotic use 163 (27.03) 27 (26.21) 17 (27.42) 10 (24.39) 53 (26.37) 0.863 0.732 Note. RVA, rotavirus group A. Table 1. Demographic characteristics and clinical symptoms of patients
-
SimPlot v3.5.1 was used to plot the similarity to identify putative recombination breakpoint cutoffs by displaying the genetic distance comparisons between chosen sequences and comparison sequences when the lines intersected the predicted site of recombination[24]. A sliding window of 200 nucleotides was used, moving in 20 nucleotide steps, and Kimura’s two-parameter method was applied. Mutation rates were identified for RVA strains using gene selection pressure, which was estimated using the codon-based Z-test of selection (Nei-Gojobori method) in MEGA v6.06 software[25]. Positive selection (dN > dS; where dS and dN are the numbers of synonymous and non-synonymous substitutions per site, respectively) and purifying selection (dN < dS) were considered when the P-value was less than 0.05. Codon usage bias was analyzed, and the relative synonymous codon usage (RSCU) values of each codon for all coding sequences (CDSs) of 11 segments of each genotype of the RVA strains were calculated using MEGA v6.06[25]. In the absence of codon bias, the RSCU value was 1. For the preferred codon, the RSCU was greater than 1. For rare codons, the RSCU was less than 1. RSCU values < 0.6 or > 1.6 were considered to be underrepresented or overrepresented codons, respectively[26]. Entropy analysis of the gene polymorphisms was performed using the BioEdit Entropy [H(X)] plot (BioEdit v7.0.9.0) to identify the aligned amino acid sequences of eleven segments of the virus[21]. For easily mutated amino acid sites, the entropy value was ≥ 0.6. For potential high mutation amino acids sites, the entropy threshold was ≥ 1. Bioinformatics Aider v1.423[27] was used to analyze the mutational signatures of the gene polymorphisms and obtain base substitutions. The YinOYang-1.2 server and NetNGlyc-1.0 server were used to predict O-(beta)-GlcNAc glycosylation and N-linked glycosylation sites, respectively[28].
-
The database was established using EpiData v3.1 (http://www.epidata.dk/; accessed on January 2, 2019). Statistical figures were drawn using R v4.1.2. Continuous variables were presented as the median (interquartile range), and categorical variables as ratio or percentage. The Chi-square test or t-test were used to analyze demographic traits, epidemiological characteristics, and clinical symptom data. Two-tailed tests were performed for statistical significance. Statistical significance was established at an α level of 0.05.
-
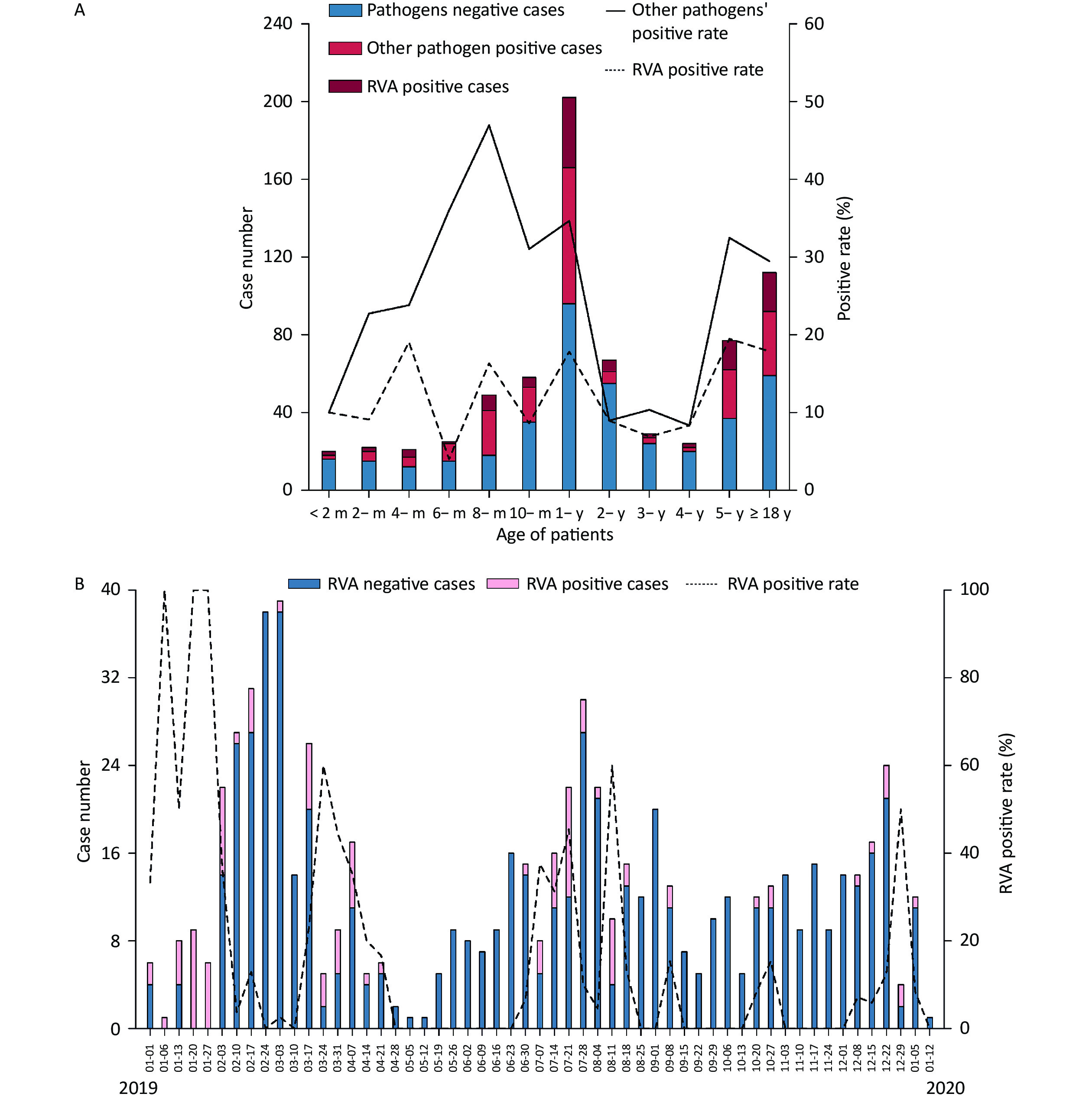
The included individuals were aged 28 days–85 years. The study population consisted of eight age groups: 0 to 6 months (9.92%, 70/706), 6 months to 1 year (36.26%, 256/706), 1 to 2 years (15.72%, 111/706), 2 to 5 years (14.31%, 101/706), 5 to 18 years (8.64%, 61/706), 18 to 30 years (4.53%, 32/706), 30 to 50 years (4.25%, 30/706) and > 50 years (6.37%, 45/706). A total of 103 RVA-positive samples were identified, yielding 83 isolates. The median age was 1.25 years in all patients with RVA-negative acute gastroenteritis, 1.33 years in RVA-positive patients, and 1.00 years in those with other pathogens. RVA was detected in different age ranges (1 month to 85 years) (Table 1 and Figure 2A). A total of 46.17% (326/706) of patients were aged 0–1 years, of whom 43.69% (45/103) were RVA-positive. The positivity rate for other pathogens varied in the opposite direction to that for RVA, with the latter showing four peaks at 4 months, 8 months, 1-year, and 5 years of age and the lowest at the age of 6 months (Figure 2A). Furthermore, a significant difference was observed in the RVA positivity rate in patients younger than 2 years of age, which was higher than that in aged 2 to 5 years (14.42% vs. 7.93%, P < 0.05).
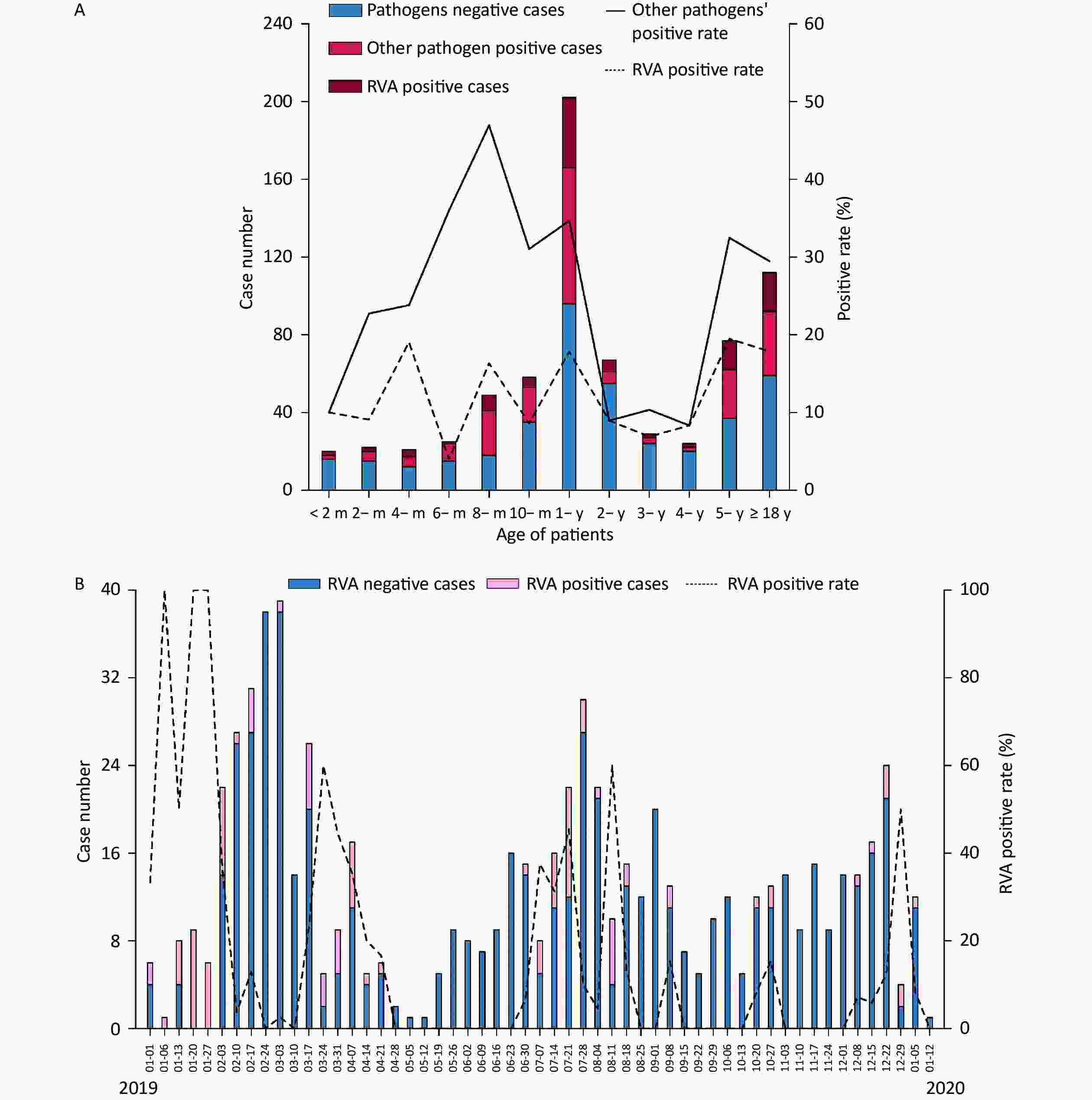
Figure 2. Distribution of acute gastroenteritis in the Pearl River Delta region during the study period (from January 1, 2019 to January 12, 2020). (A) Age distribution of patients suffering from acute gastroenteritis. Pathogen-negative cases (steel blue bars); other pathogen positive cases (fluorescent pink bars); RVA-positive cases (dark red bars); other pathogens-positive rate (%) (straight line); RVA-positive rate (dotted line). (B) Weekly time trend distribution of patients with diarrhea infection. RVA-negative cases (steel blue bars); RVA-positive cases (pink bars); RVA-positive rate (%) (dotted line). RVA: rotavirus group A.
-
Two peaks in the seasonality of acute gastroenteritis were observed: from February to March 2019 (29.32%, 207/706) and from July to August 2019 (21.10%, 149/706). As illustrated in Figure 2B, a weekly variation in occurrence of acute gastroenteritis was observed, with relatively occurrence in weeks 8–10 (February 17 to March 3; 15.3%, 108/706), week 31 (July 28; 4.25%, 30/706), week 7 (February 10; 3.82%, 27/706), and week 12 (March 17; 3.68%, 26/706). A small peak was observed around the 52nd week (December 22; 3.40%, 24/706). Furthermore, RVA was detected in all seasons throughout the study period, and was mainly endemic from January to April 2019 (55.34%, 57/103). Moreover, there was no statistically significant difference in the monthly RVA positivity rate (P = 0.335). The weekly distribution of RVA showed multiple peaks around early spring (January 6–13, January 20–27, January 27-February 3, March 17–24), late summer (July 7–14, July 21–28), and autumn–winter (August 11–18, September 8–15, October 27–November 3, and December 29–January 5, 2020).
-
Clinical symptoms including fever, vomiting, and antibiotic application are shown in Table 1. Of the 103 RVA-positive patients, 27 had fever, 35 had vomiting, and 27 had used antibiotics before fecal sample collection. No significant differences were found between RVA-negative and RVA-positive individuals in terms of fever, vomiting, or antibiotic use. No significant differences were found between individuals with RVA alone and those with RVA and a co-infection, in terms of vomitus, or antibiotic application. Subjects with RVA and a co-infection had a significantly higher positive rate of fever than those with RVA alone (39.02% vs. 17.74%, P < 0.05). As shown in Table 2, of the 103 RVA-positive cases, 39.81% (41/103) were co-infected with other pathogens. Of these, 22 were infected with viral pathogens (adenovirus, norovirus GII, norovirus GI, and astrovirus), bacteria (Salmonella, Clostridium difficile, and Campylobacter), and parasites (Cryptosporidium and Giardia), 7 were infected with three pathogens, and 12 were infected with more than three pathogens.
Type of detection Pathogens No. of samples (%) Single infection RVA 62 (60.19) 2 types RVA Salmonella 4 (3.88) RVA C. difficile 4 (3.88) RVA Adenovirus 4 (3.88) RVA Giardia 3 (2.91) RVA Norovirus GII 2 (1.94) RVA Cryptosporidium 2 (1.94) RVA Norovirus GI 1 (0.97) RVA Campylobacter 1 (0.97) RVA Astrovirus 1 (0.97) 3 types RVA Cryptosporidium Vibrio cholerae 2 (1.94) RVA Adenovirus C. Difficile 1 (0.97) Co-infection RVA Adenovirus Norovirus GI 1 (0.97) RVA Adenovirus Astrovirus 1 (0.97) RVA C. difficile Campylobacter 1 (0.97) RVA Campylobacter Norovirus GII 1 (0.97) 4 types RVA Giardia C. difficile ETEC 2 (1.94) RVA Giardia C. difficile Norovirus GI 1 (0.97) RVA Campylobacter C. difficile Norovirus GII 1 (0.97) RVA Cryptosporidium Salmonella ETEC 1 (0.97) RVA Cryptosporidium Campylobacter Norovirus GI 1 (0.97) 5 types RVA Entamoeba histolytica Vibrio cholerae Norovirus GII Giardia 1 (0.97) 6 types RVA Salmonella C. difficile Norovirus GI Giardia ETEC 2 (1.94) 8 types RVA Entamoeba histolytica Norovirus GII Astrovirus ETEC STEC C. difficile 1 (0.97) 9 types RVA Entamoeba histolytica Cryptosporidium Norovirus GI Norovirus GII ETEC C. difficile Giardia 2 (1.94) Total 103 (100.00) Note. RVA, rotavirus group A; C. difficile, Clostridium difficile; ETEC, enterotoxigenic E. coli; STEC, Shiga toxigenic E. coli. Table 2. Distribution of infection types in patients with diarrheal disease
-
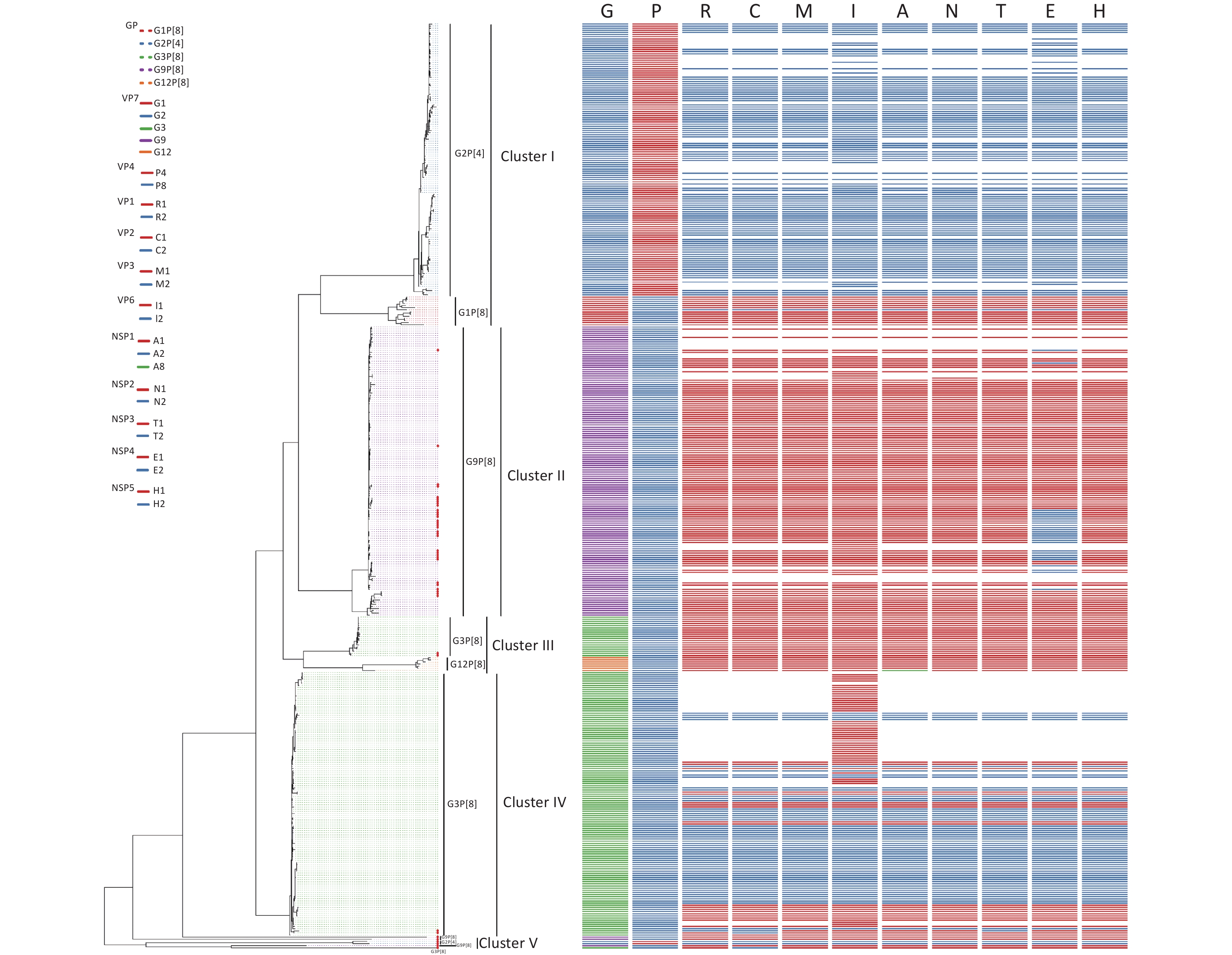
Sixty-seven RVA samples were genotyped successfully, including sixty G9P[8], five G3P[8], and two G2P[4] strains, and 45 strains were successfully sequenced. Of 45 RVA strains, there were two G2P[4], five G3P[8], 34 G9P[8], one G2P[x], and three GxP[8] genotypes. To estimate the homology and relationship of the RVA strains in our study, phylogenetic analysis was performed using the ML method in the IQ-Tree to compare the 41 complete sequences obtained with those in GenBank (Figure 3). Information on all sequences used for the phylogenetic analysis is available in Table 1. The evolutionary tree indicated that 425 different RVA sequences formed four clusters and presented five genotypes. The 41 RVA-positive viral sequences in this study were scattered in four groups (cluster II, cluster III, cluster IV, and cluster V) and were identified as belonging to the three genotypes, which was consistent with the distribution in the phylogenetic dendrogram (Figure 3). As shown in Figure 3, four of the five G3P[8] belonged to the corresponding genotypes of clusters III and IV, with 80.68%–97.93% nucleotide identity. Most of the G9P[8] sequences were gathered in cluster II, with 91.77%–99.49% nucleotide identity. In particular, two RVA sequences belonged to the G2P[4] genotype in cluster V, which was not in the same branch as the G2P[4] reference sequences, and shared a nucleotide identity of 100%.
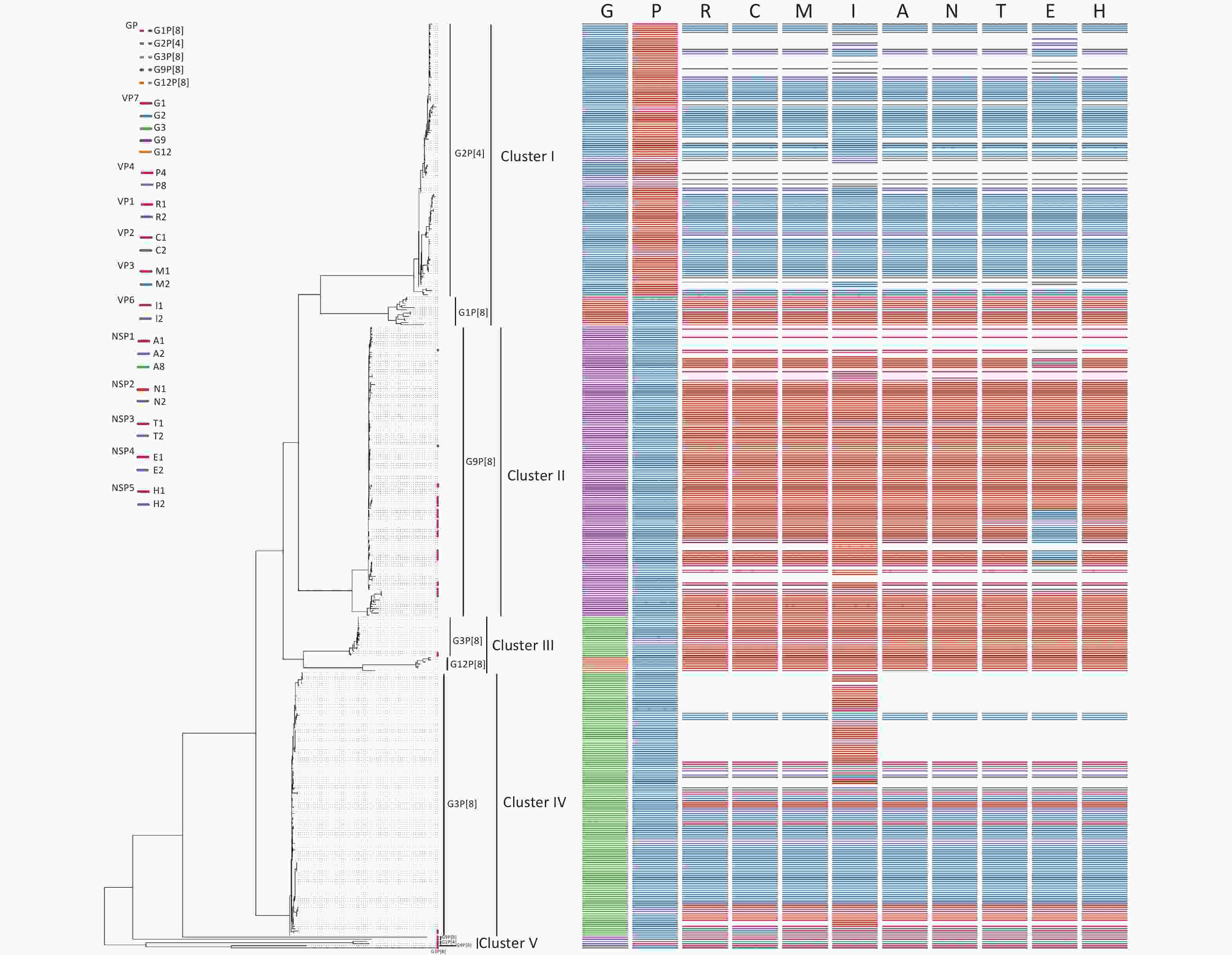
Figure 3. Phylogenetic analysis of the rotavirus strains collected in the Pearl River Region Guangdong, China, from January 2019 to January 2020, compared with reference sequences. The phylogenetic tree was constructed using the ML method based on the GTR+F+G4 model in the IQ-Tree program and tested using a 1,000 bootstrap value. Colors in the key distinguish different genes, including G, P, R, C, M, I, A, N, T, E, and H. Our viral strains are labeled with red squares. Abbreviations of eleven segments are shown on the top of the gene constellation bars. VP: viral protein; NSP: nonstructural protein; G, glycosylated gene; P, protease-sensitive gene; R, RNA-dependent RNA polymerase gene; C, core protein gene; M, methyltransferase gene; I, inner capsid gene; A, interferon antagonist gene; N, NTPase gene; T, translation enhancer gene; E, Enterotoxin gene; H, phosphoprotein gene. All branch lengths are scaled according to the number of substitutions per site.
-
According to the whole genome-based genotyping system, the analyzed RVA strains were divided into Wa-like and DS-1-like, and a reassortment was generated between the two genome constellation strains[17,29]. The comparison results revealed the existence of recombination events in the R, C, and T genome segments of one of the five G3P[8] strains and the E genome segments of eighteen of thirty-four G9P[8] isolates. To confirm these results, reassortment analysis was performed based on G3P[8] and G9P[8] using SimPlot software. Two potential recombinant strains are G3P[8] and G9P[8]. Three reassortment events were found in D8/G3P[8] (Figure 4A) and one reassortment event was found in D18/G9P[8], D23/G9P[8], D39/G9P[8], D42/G9P[8], D44/G9P[8], J241/G9P[8], S8/G9P[8], S10/G9P[8], S14/G9P[8], S15/G9P[8], S16/G9P[8], S24/G9P[8], S26/G9P[8], S35/G9P[8], S51/G9P[8], J258/G9P[8], S30/G9P[8], and S54/G9P[8] (Figure 4B). As illustrated in Figure 4A1–4A3, the parents of the D8/G3P[8]/R, D8/G3P[8]/C, and D8/G3P[8]/T segments were G2P[4]/R2 (accession number: KC571499.1), G2P[4]/C2 (accession number: KC571493.1), and G2P[4]/T2 (accession number: KC571494.1). As depicted in Figure 4B1–4B15, the parent of the G9P[8]/E recombination segments in our study was G2P[4]/E2 (accession number: KC152930.1). It is necessary to emphasize that ‘parent’ here does not allude to genuine progenitors of the reassortment strains, but to those members of the populations whose genetic sequences were most closely similar to that of the reassortment strains. These structural and nonstructural protein genotype recombination events occurred, and the recombinant regions were primarily in NSP3, VP1, and VP2 of G3P[8] and NSP4 of G9P[8]. Therefore, in this study, two G2P[4] strains both had DS-1-like genomes (G2-P[4]-I2-R2-C2-M2-A2-N2-T2-E2-H2), one G3P[8] strain possessed Wa-like constellations with the exception of R2 VP1 genotype, C2 VP2 genotype, and T2 NSP3 genotype, respectively (G3-P[8]-I1-R2-C2-M1-A1-N1-T2-E1-H1), and eighteen isolates had mono-genome reassortment in the NSP4 segment (G9-P[8]-I1-R1-C1-M1-A1-N1-T1-E2-H1) as shown on the bold genotypes. No AU-1-like genomes were observed (Table 3).
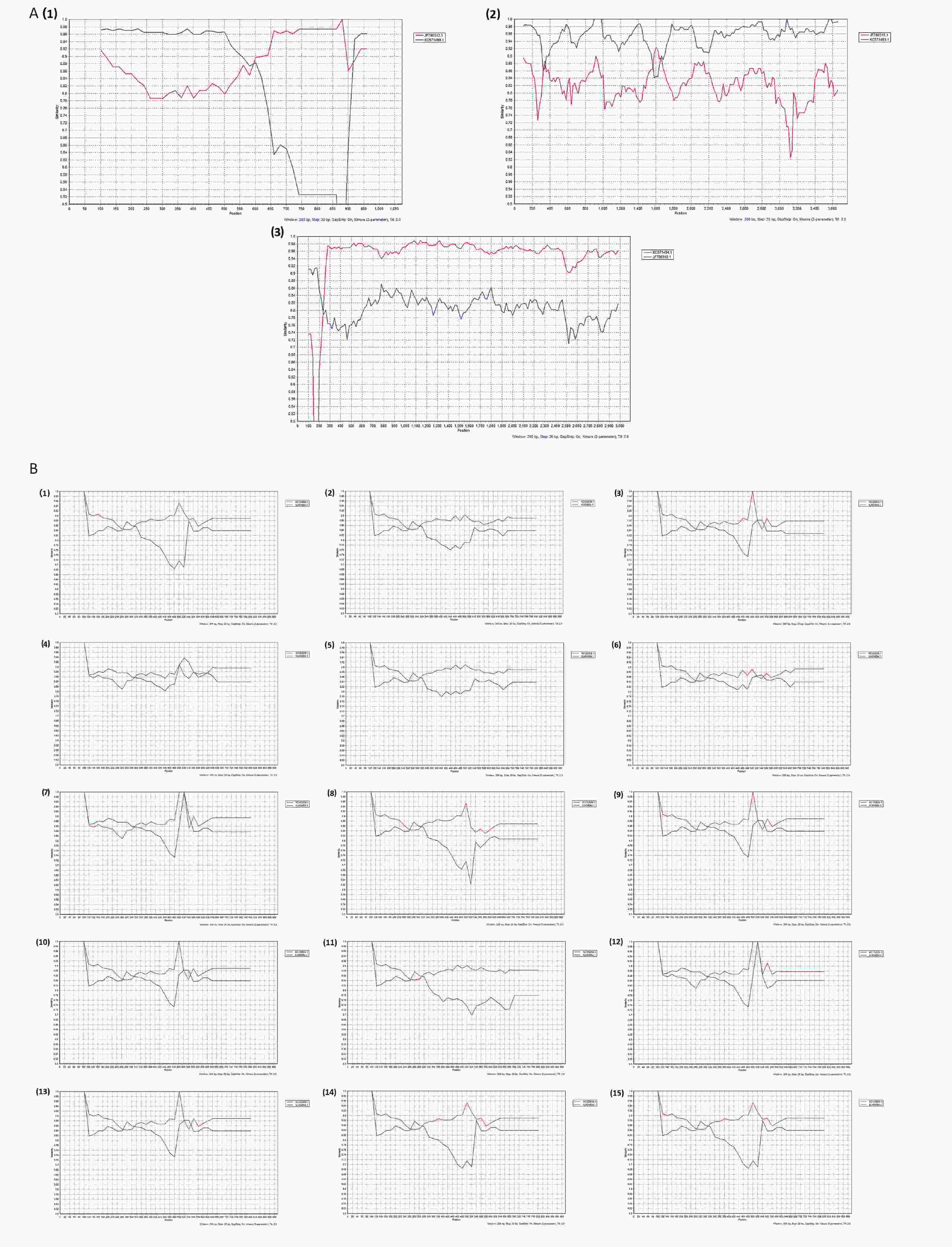
Figure 4. Plots of similarity (generated by SimPlot) of the set of reference sequences from the GenBank to the one G3P[8] and fifteen G9P[8] genomes. Each curve was a comparison between the genome in our study and a reference genome in the GenBank. (A) Recombination analysis of D8/G3P[8]; (A1) D8/G3P[8]-NSP3. D8/G3P[8]-NSP3 was the query sequence, KC571499.1 and JF790313.1 were the G2P[4]-NSP3 (T2) and G3P[8]-NSP3 (T1), respectively. D8/G3P[8]-NSP3 was the potential recombination region; T, Translation enhancer gene; (A2) D8/G3P[8]-VP1. D8/G3P[8]-VP1 was the query sequence, KC571493.1 and JF790315.1 were the G2P[4]-VP1 (R2) and G3P[8]-VP1 (R1), respectively. D8/G3P[8]-VP1 was the potential recombination region; R, RNA-dependent RNA polymerase gene; (a3) D8/G3P[8]-VP2. D8/G3P[8]-VP2 was the query sequence, KC571494.1 and JF790319.1 were the G2P[4]-VP2 (C2) and G3P[8]-VP2 (C1), respectively. D8/G3P[8]-VP2 was the potential recombination region; C, core protein gene; (B) Recombination analysis of G9P[8]-NSP4; KC454664.1 and KC152930.1 were the G2P[4]-NSP4 (E2) and G9P[8]-NSP4 (E1), respectively; E, enterotoxin gene; (B1) D18/G9P[8]-NSP4; (B2) D23/G9P[8]-NSP4; (B3) D39/G9P[8]-NSP4; (B4) D42/G9P[8]-NSP4; (B5) D44/G9P[8]-NSP4; (B6) J241/G9P[8]-NSP4; (B7) S8/G9P[8]-NSP4; (B8) S10/G9P[8]-NSP4; (B9) S14/G9P[8]-NSP4; (B10) S15/G9P[8]-NSP4; (B11) S16/G9P[8]-NSP4; (B12) S24/G9P[8]-NSP4; (B13) S26/G9P[8]-NSP4; (B14) S35/G9P[8]-NSP4; (B15) S51/G9P[8]-NSP4.
Strains Gene segments VP7 VP4 VP1 VP2 VP3 VP6 NSP1 NSP2 NSP3 NSP4 NSP5 D12 G2 P[4] R2 C2 M2 I2 A2 N2 T2 E2 H2 S45 G2 P[4] R2 C2 M2 I2 A2 N2 T2 E2 H2 D8 G3 P[8] R2 C2 M1 I1 A1 N1 T2 E1 H1 J233 G3 P[8] R1 C1 M1 I1 A1 N1 T1 E1 H1 J274 G3 P[8] R1 C1 M1 I1 A1 N1 T1 E1 H1 S19 G3 P[8] R1 C1 M1 I1 A1 N1 T1 E1 H1 S29 G3 P[8] R1 C1 M1 I1 A1 N1 T1 E1 H1 D17 G9 P[8] R1 C1 M1 I1 A1 N1 T1 E1 H1 D18 G9 P[8] R1 C1 M1 I1 A1 N1 T1 E2 H1 D23 G9 P[8] R1 C1 M1 I1 A1 N1 T1 E2 H1 D24 G9 P[8] R1 C1 M1 I1 A1 N1 T1 E1 H1 D38 G9 P[8] R1 C1 M1 I1 A1 N1 T1 E1 H1 D39 G9 P[8] R1 C1 M1 I1 A1 N1 T1 E2 H1 D42 G9 P[8] R1 C1 M1 I1 A1 N1 T1 E2 H1 D44 G9 P[8] R1 C1 M1 I1 A1 N1 T1 E2 H1 D45 G9 P[8] R1 C1 M1 I1 A1 N1 T1 E1 H1 J225 G9 P[8] R1 C1 M1 I1 A1 N1 T1 E1 H1 J241 G9 P[8] R1 C1 M1 I1 A1 N1 T1 E2 H1 J258 G9 P[8] R1 C1 M1 I1 A1 N1 T1 E2 H1 J288 G9 P[8] R1 C1 M1 I1 A1 N1 T1 E1 H1 S19 G9 P[8] R1 C1 M1 I1 A1 N1 T1 E1 H1 S3 G9 P[8] R1 C1 M1 I1 A1 N1 T1 E1 H1 S8 G9 P[8] R1 C1 M1 I1 A1 N1 T1 E2 H1 S10 G9 P[8] R1 C1 M1 I1 A1 N1 T1 E2 H1 S14 G9 P[8] R1 C1 M1 I1 A1 N1 T1 E2 H1 S15 G9 P[8] R1 C1 M1 I1 A1 N1 T1 E2 H1 S16 G9 P[8] R1 C1 M1 I1 A1 N1 T1 E2 H1 S18 G9 P[8] R1 C1 M1 I1 A1 N1 T1 E1 H1 S21 G9 P[8] R1 C1 M1 I1 A1 N1 T1 E1 H1 S22 G9 P[8] R1 C1 M1 I1 A1 N1 T1 E1 H1 S23 G9 P[8] R1 C1 M1 I1 A1 N1 T1 E1 H1 S24 G9 P[8] R1 C1 M1 I1 A1 N1 T1 E2 H1 S25 G9 P[8] R1 C1 M1 I1 A1 N1 T1 E1 H1 S26 G9 P[8] R1 C1 M1 I1 A1 N1 T1 E2 H1 S27 G9 P[8] R1 C1 M1 I1 A1 N1 T1 E1 H1 S30 G9 P[8] R1 C1 M1 I1 A1 N1 T1 E2 H1 S35 G9 P[8] R1 C1 M1 I1 A1 N1 T1 E2 H1 S43 G9 P[8] R1 C1 M1 I1 A1 N1 T1 E1 H1 S50 G9 P[8] R1 C1 M1 I1 A1 N1 T1 E1 H1 S51 G9 P[8] R1 C1 M1 I1 A1 N1 T1 E2 H1 S54 G9 P[8] R1 C1 M1 I1 A1 N1 T1 E2 H1 Note. The Wa-like genotype constellation is represented in red, and DS-1-like in green. RVA: rotavirus group A; VP: viral protein; NSP: nonstructural protein; G: glycosylated gene; P: protease-sensitive gene; I: inner capsid gene; R: RNA-dependent RNA polymerase gene; C: core protein gene; M: methyltransferase gene; A: interferon antagonist gene; N: NTPase gene; T: translation enhancer gene; E: enterotoxin gene; H: phosphoprotein gene; numbers represent the type of gene. Table 3. Whole-genome typing characteristics of RVA epidemic strains
-
Mutation rates were identified for RVA strains using gene selection pressure. The results indicated that the NSP3 and VP6 segments of G2P[4], NSP1, NSP3, NSP4, and VP3 of G3P[8], and VP3 of G9P[8] were under positive selection, as indicated by significant P values of 0.003, 0.036, 0.005, < 0.001, < 0.001, < 0.001, and < 0.001, respectively. However, the NSP5, VP1, and VP4 segments of three genotype strains, VP3 of G2P[4], VP6 and VP7 of G3P[8], and VP7 of G9P[8], were observed to be under purifying selection (Table 4).
Genotypes Segments positive selection/purifying selection NSP1 NSP2 NSP3 NSP4 NSP5 VP1 VP2 VP3 VP4 VP6 VP7 G2P[4] −/− −/− 0.003/− −/− −/0.004 −/0.032 −/− −/0.020 −/0.001 0.036/− −/− G3P[8] 0.005/− −/− 0.000/− 0.000/− −/0.041 −/0.000 −/0.000 0.000/− −/0.027 −/0.000 −/0.005 G9P[8] −/− −/− −/− −/− −/0.002 −/0.000 −/− 0.000/− −/0.016 −/− −/0.012 Note. P value greater than 0.05 is indicated by −. VP: viral protein; NSP: nonstructural protein. Table 4. Putative glycosylation sites of RVA strains
To further probe the evolutionary characteristics of RVA and clarify whether selection pressure affected codon usage, codon usage bias was analyzed, and the RSCU values of each codon for all CDSs of 11 segments of the three RVA strain genotypes were calculated using MEGA v6.06. The RSCU values were considered as 61 components, excluding three stop codons (UAA, UAG, and UGA). As shown in Table 2, the overall RSCU values ranged from 0.01 (codon GGC and GGG) to 3.44 (codon CCA) in the NSP1 segments, from 0.06 to 3.3 in NSP2, 0.03 to 4 in NSP3, 0.04 to 4 in NSP4, 0.01 to 3.16 in NSP5, 0.04 to 2.98 in VP1, 0.01 to 3.86 in VP2, 0.01 to 3.2 in VP3, 0.09 to 3.08 in VP4, 0.04 to 2.82 in VP6, and 0.06 to 3.67 in VP7. A/U-ending codons were preferred for synonymous codon usage in the CDS of RVA strains. The results revealed that the codons were clustered into three branches (positive codon usage bias branch, no codon usage bias branch, and negative codon usage bias branch), and some codons differed significantly in codon usage bias, such as GCG (A), GAC (D), and GGG (G) preference in the NSP4 segments, but CCU (P) preference in VP6.
-
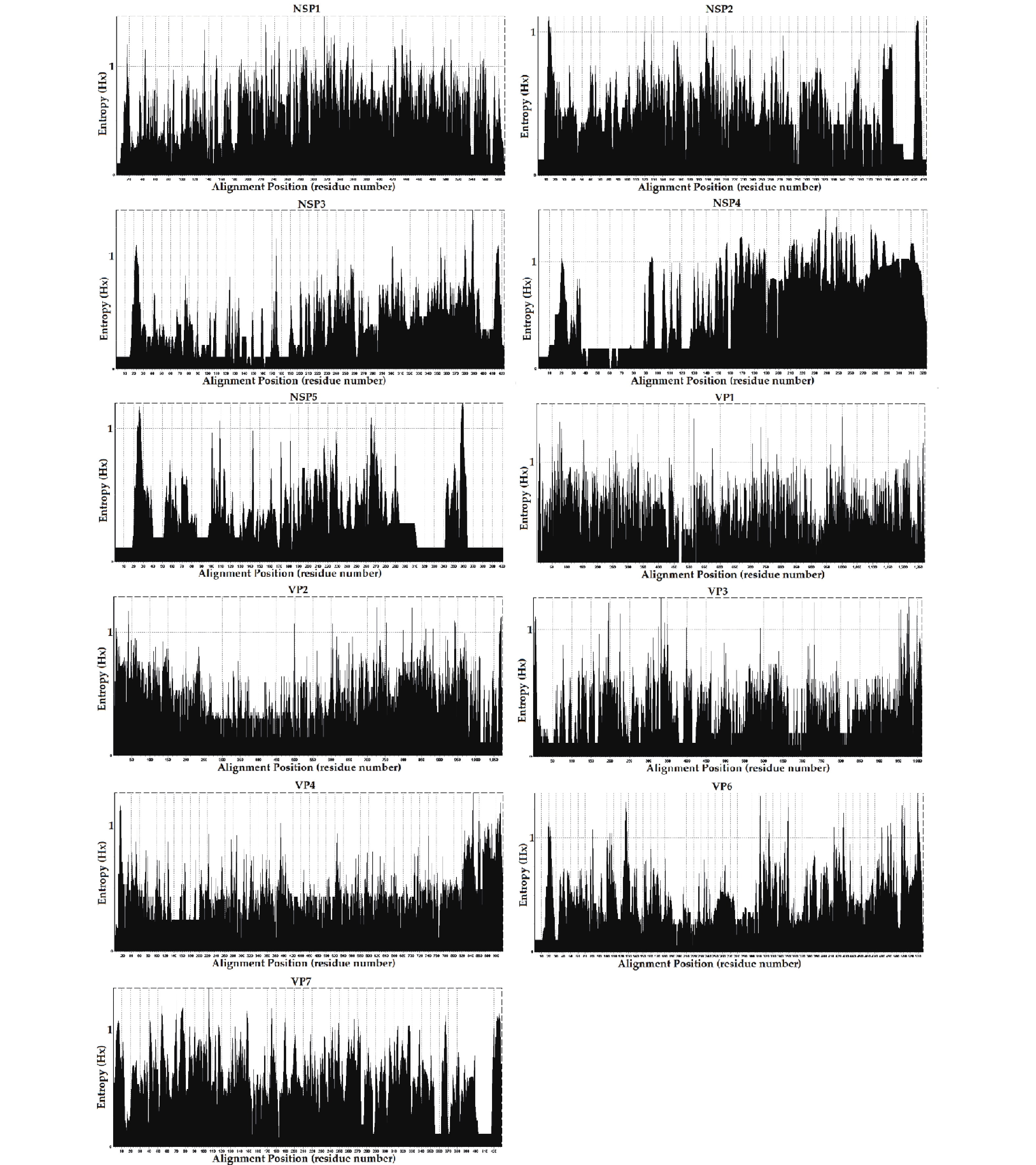
The entropy value (threshold ≥ 0.6, easily mutated amino acid sites) percentages were as follows; NSP1: 66.10% (390/590), NSP4: 57.89% (187/323), VP7: 51.28% (220/429), VP1: 43.82% (557/1,271), VP2: 30.94% (332/1,073), NSP2: 27.94% (121/433), VP6: 27.56% (148/537), NSP3: 21.04% (9/423), VP3: 18.40% (186/1,011), NSP5: 16.96% (68/401), and VP4: 16.38% (150/916). The frequencies of potential high mutation amino acids sites (threshold ≥ 1) were; NSP4: 2.07% (67/323), NSP1: 0.93% (55/590), VP7: 0.82% (35/429), VP6: 0.39%5 (21/537), VP1: 0.35% (44/1,271), NSP5: 0.30% (12/ 401), NSP3: 0.26% (11/423), VP2: 0.20% (21/1,073), VP3: 0.19% (19/1,011), VP4: 0.19% (17/916), and NSP2: 0.16% (7/433) (Figure 5). However, further research in conjunction with site-directed mutagenesis is needed to determine the relationships between the identified mutations and their corresponding functional changes. A detailed understanding of the mechanisms underlying these strains is required to verify their pathogenicity and zoonotic potential[30]. We also explored mutational signatures from 11 segments of two G2P[4], five G3P[8], and thirty-four G9P[8] sequences, using the corresponding reference strains as reference sequences (the reference sequence numbers are summarized in Table 2. Following the principle of complementary base pairing, four kinds of bases formed twelve types of base substitutions in the three genotypes of the RVA strains: G>A, A>G, T>C, C>T, A>T, T>A, A>C, G>T, C>A, T>G, G>C, and C>G. Base substitutions refer to base transversions and transitions. The results showed some variations in signatures, including single-base substitution, double-base substitution, clustered-base substitution, and small insertion-and-deletion signatures (Table 3).
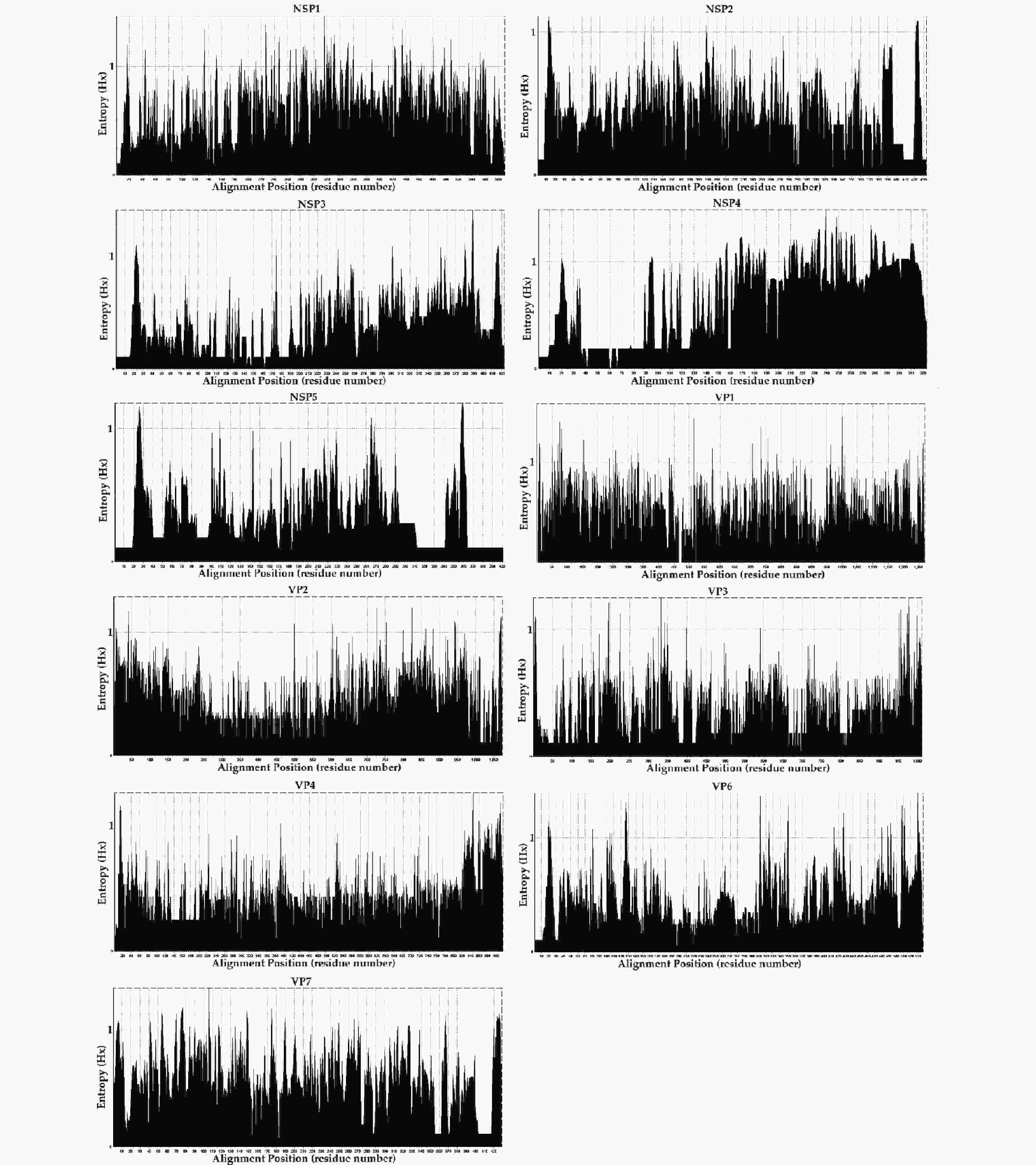
Figure 5. Entropy [H(X)] plot of amino acid residues of eleven segments of RVA strains. The entropy value ≥ 0.6 and ≥ 1 represented easily mutated amino acid sites and high mutation amino acid sites, respectively. The names of eleven segments are shown above each entropy graph.
-
O-glycosylation is a post-translational modification of proteins that is of particular importance to the molecular mechanisms of health and disease, and N-glycosylation profoundly affects receptor binding and antigenicity of proteins[31,32]. To explore the roles of protein glycosylation in host-pathogen interactions, O-ß-GlcNAc attachment sites and N-glycosylation sites of 11 encoding proteins of the RVA strains were predicted from sequence information. Information on all the O-ß-GlcNAc and N-glycosylation sites used for protein glycosylation prediction is shown in Tables 4–1 to 4–22. The prediction results showed that there were 53, 34, 13, 2, 13, 95, 76, 38, 90, 32, and 25 types of N-glycosylation sites and 46, 16, 10, 27, 9, 102, 134, 32, 69, 46, and 26 types of O-ß-GlcNAc sites in the NSP1, NSP2, NSP3, NSP4, NSP5, VP1, VP2, VP3, VP4, VP6, and VP7 segments, respectively. It is worth noting that not all putative glycosylation sites may functional, and the actual glycosylation sites need to be further determined.
-
In this study, we investigated the epidemiology of RVA in patients with diarrheal disease in the Pearl River Delta region of Guangdong, China, from January 1, 2019 to January 12, 2020. We detected 103 RVA-positive samples from the 706 stool samples (14.59%). The differences in detection rates between studies can be explained in part by differences in the scope of the studies, objects, confirmation methods, focus pathogens, and epidemic intensity[33]. Based on the age distribution of RVA infection, the RVA-positive rate of diarrhea cases ≤ 2 year of age was higher than for patients aged 2 to 5 year of age (P < 0.05). This reinforced previous reports in which researchers found that RVA mainly affected infants at different peak ages including 6-10 months, 7-12 months, 9-14 months, 6-24 months, and 12-24 months[34-37]. Globally, RVA infections, with an epidemic peak in the cold season, show seasonal and regional characteristics of incomplete synchronization[34,38-41]. In our study, RVA was detectable in all seasons throughout the year. Nevertheless, RVA infection peaked from January to April but also in mid-July. The reasons for this may be as follows: (a) the study region had a subtropical monsoon climate with a long summer duration, and water and food were susceptible to contamination; (b) in 2019, Guangdong was affected by typhoons that appeared in early to mid-July. It has been reported that the occurrence of the typhoon was a risk factor for the incidence of acute gastroenteritis, mainly affecting those aged ≤ 5 years old[42]. In the 103 positive cases infected with RVA, identified by testing with the Luminex xTAG Gastrointestinal Pathogen Panel, clinical features included fever (27/103, 26.21%), vomiting (35/103, 33.98%), and antibiotic use (27/103, 26.21%). Interestingly, we discovered no statistically significant differences between the groups in terms of fever, vomiting, or antibiotic application. Inconsistent results have been described in previous research, where it was reported that 5.4% of RV diarrhea cases had fever[43]. We found that 41 (39.81%) RVA-positive patients were co-infected with other pathogens, including Salmonella, Clostridium difficile, adenovirus, and Giardia. We discovered that fever (39.02% vs. 17.74%, P < 0.05) was more frequent in RVA co-infection patients than in RVA alone ones which is inconsistent with the findings of Chen et al.[44].
The predominant genotype was G9P[8], which accounted for 58.25% (60/103) of all RVA strains, according to key segment genotyping of stool samples. The trends in the prevalence of the RVA genotypes in the Pearl River Delta region of Guangdong have been described previously in other regions of the world, including France[45], Spain[46], Iran[47], Wuhan[48] and Hangzhou[49]. The molecular characteristics of RVA were further explored by complete sequencing of RVA isolates, which showed that the forty-one viral sequences were scattered in four different clusters and represented three genotypes, out of the four genotypes commonly reported in China[50-53] and suggesting the prevalence of RVA genetic diversity in this region. Genome rearrangements, can provide important information with respect to intraspecific and interspecific gene recombination and cross-species transmission from animals to humans, and may also contribute to RV diversity[18,54]. RVA have a high mutation rate, and gene mutations and reassortment are the principle drivers of RVA evolution[14,55,56]. Preliminary analysis of the 11 segments of RVA sequences suggested the existence of potential reassortment events. Using SimPlot software analysis, three recombination events were found in one G3P[8] strain and one recombination event was discovered in 18 G9P[8] strains, with the recombinations affecting coding regions for structural and nonstructural proteins. Reassortment was detected between genotypes, including between G2P[4] and G3P[8], and between G2P[4] and G9P[8]; the reassortments were in VP1, VP2, NSP3, and NSP4. These findings are consistent with those of previous studies reporting that VP1, VP2, VP6, NSP1, NSP2, NSP3, and NSP4 are involved in recombination events[57-59]. It should be noted that the NSP4 reassortment region is a crucial segment of the virus that plays a role in morphogenesis, viral assembly, and enterotoxin production, and is considered to be involved in the virulence of RV[13,60-63]. Further research is needed on the functional effects of these various genetic recombinations.
Entropy is a useful method for quantifying amino acid sequence diversity. Structurally or functionally important amino acid residues may be related to entropy values[64,65]. Amino acid diversity, including potential easily mutated and highly mutated amino acid sites, was analyzed by entropy analysis. The gene reassortment in the NSP3 segments of G3P[8] might have been under strong positive selection, but whether it enabled efficient and effective functional changes requires further site-by-site analysis. The flexibility afforded by the positive selection sites allowed for a more successful recombination of the NSP3 segments of the G3P[8] genotype. The prevalence of non-synonymous mutations with high entropy values corresponding to positive selection sites in the segments may indicate the high variability of these segments[30], like NSP1 and NSP4. Previous studies have reported the decisive role of section pressure in codon usage bias[66-68]. Analyses of selection pressure and codon usage bias suggested that the evolution of genotype segments was chiefly driven by positive or negative selection, whereas other segments or genotypes tended to be conserved. We found that the segments experienced positive selection, indicating that these segments were confronted with greater external selection pressure and were likely to evolve in the direction of mutation. A previous study reported that recombinant forms of residues 112-175 in NSP4 are capable of causing diarrhea in mice[69]. One N-glycosylation site was predicted at amino acid residues 63 and 80 of NSP4 in S18/G9P[8] and S27/G9P[8], respectively, and none of these sites affected NSP4 function. Furthermore, protein glycosylation prediction results revealed the existence of multiple potential N-glycosylation sites and O-ß-GlcNAc sites, and these post-translational modifications may involve structural changes to increase spatial or temporal regulation, including the modulation of protein folding, protein stability, enzyme activity, subcellular localization, and inter-reactions to modulate protein function and subsequently affect viral replication and pathogenesis[70]. The occurrence of these events is a potential factor capable of increasing genetic diversity and viral evolution, altering clinical disease manifestations, and may be associated with increased virulence[71].
In summary, we provide evidence for the prevalence of RVA in Guangdong Province and present the coexistence of multiple RVA genotypes and their corresponding genetic diversity. Moreover, we present evidence regarding the genetic evolution of the virus and provide a basis for further understanding of its evolutionary traits.
Epidemiological Surveillance: Genetic Diversity of Rotavirus Group A in the Pearl River Delta, Guangdong, China in 2019
doi: 10.3967/bes2024.031
Funds:
This study was funded by the grant National Key R&D Program of China (2017ZX10103011-004 and 2018YFC1603804) and the Science and Technology Program of Guangdong Province (2018B020207013 and 2019B030316013).
- Received Date: 2023-05-08
- Accepted Date: 2023-10-24
-
Key words:
- Infectious diarrheal disease /
- Rotavirus /
- Phylogenetic analysis /
- Gene rearrangement /
- Codon usage bias /
- Genetic diversity
Abstract:
The authors declare no conflicts of interest.
&These authors contributed equally to this work.
| Citation: | JIANG Jie Ying, LIANG Dan, WANG Li, XIAO Yun, LIANG Yu Feng, KE Bi Xia, SU Juan, XIAO Hong, WANG Tao, ZOU Min, LI Hong Jian, KE Chang Wen. Epidemiological Surveillance: Genetic Diversity of Rotavirus Group A in the Pearl River Delta, Guangdong, China in 2019[J]. Biomedical and Environmental Sciences, 2024, 37(3): 278-293. doi: 10.3967/bes2024.031

|

 Quick Links
Quick Links
 DownLoad:
DownLoad: