
-
Ticks, encompassing a diverse group of arthropods within the class Arachnida (order Ixodida, superfamily Ixodoidea), are classified into three families: Argasidae (soft ticks), Ixodidae (hard ticks), and Nuttalliellidae (nuttalliellid ticks). They are temporary hematophagous ectoparasites capable of infesting a wide range of hosts, including mammals, birds, reptiles, and amphibians. Factors such as bird migration, global climate change, and human activities have expanded tick distribution, thereby increasing the spread of tick-borne diseases. On March 31, 2022, the World Health Organization launched the Global Arbovirus Initiative to heighten global alarm on the potential risk of arbovirus epidemics and pandemics by strengthening surveillance and control of arthropod-borne viral diseases[1]. As obligate hematophagous ectoparasites, ticks can interrupt feeding and switch hosts, facilitating the transmission of pathogens between hosts[2]. They are second only to mosquitoes as vectors of pathogens and have a significant impact on human and animal health[3].
Poxvirus are enveloped, double-stranded DNA virus belonging to the family Poxviridae, order Chitovirales, class Pokkesviricetes, and phylum Nucleocytoviricota[4]. This family comprises two subfamilies: Chordopoxvirinae (CPV), which infects vertebrates, and Entomopoxvirinae (EPV), which infects insects. The genus Orthopoxvirus (OPXV), within the subfamily Chordopoxvirinae, includes 12 viral species, such as camelpox virus (CMLV), cowpox virus (CPXV), mpox virus (MPXV), vaccinia virus (VACV), and variola virus (VARV). MPXV has garnered significant attention due to sporadic reports in Africa, and imported cases in non-endemic countries, including the United Kingdom, Israel, Singapore, and the United States since 2003, with all affected patients receiving prompt treatment[5]. A global mpox outbreak beggining in May 2022 was primarily transmitted through sexual contact among men who have sex with men (MSM). Phylogenetic analyses suggest that MPXV has circulated in humans since 2016, with altered virulence likely resulting from high-frequency mutation in the genome driven by host cytosine deaminases[6]. Beyond MPXV, other orthopoxviral species (CMLV, VARV, VACV, and CPXV), as well as viruses from other genera—such as orf virus, bovine papular stomatitis virus, pseudocowpox virus, grey sealpox virus, red deerpox virus from the genus Parapoxvirus, molluscum contagiosum virus (MCV) from the genus Molluscipoxvirus, yaba-like disease virus, and yaba monkey tumor virus from the genus Yatapoxvirus—can infect humans[7-12]. All poxviruses capable of infecting humans are zoonotic, except for VARV and MCV.
Previous studies have shown that hematophagous arthropods can carry and transmit poxviruses. For example, Avipoxvirus transmission occurs via mosquitoes[13], and Capripoxvirus is transmitted both horizontally and vertically by ticks[14,15]. Leporipoxvirus is carried and transmitted by mosquitoes, fleas, blackflies, mites, and ticks[16,17], whereas Parapoxvirus is mechanically transmitted by flies and ticks[18,19]. Additionally, Suipoxvirus has been shown to undergo mechanical transmission via fleas[20]. Given that ticks typically infest the common hosts of various CPVs (except Salmonpoxvirus), ticks likely serve as vectors for poxvirus transmission between hosts and may facilitate genetic recombination among different poxviruses. However, evidence supporting ticks as carriers of poxviruses remains limited. As DNA viruses, poxviruses detected in tick (meta)transcriptomic data suggests that ticks may act as natural hosts or that poxviruses actively replicate within their blood-feeding hosts. This indicates that ticks could contribute to cross-species transmission, potentially resulting in host jumps and the emergence and spread of novel human zoonotic poxviruses. Therefore, the present study utilized publicly available raw (meta)transcriptomic data from ticks to detect, assemble, and identify poxvirus genomes. The primary aim was to investigate the presence of poxviruses in ticks and assess the potential for gene exchange between poxviruses, their vectors or hosts, and the surrounding environment.
-
Raw tick sequencing data were retrieved from the NCBI Sequence Read Archive (SRA) database on December 25, 2022. Data from miRNA-Seq library strategy and metagenomic library sources were excluded, retaining only (meta)transcriptomic data sequenced using the RNA-Seq library strategy for further analysis.
-
Adapters, low quality sequences, and sequences containing excessive ambiguous nucleotides (“n”) were removed from the raw data using fastp (v0.20.1)[21]. Filtered reads were then compared with the Nucleotide Sequence Database (NT) using bwa (v0.7.17-r1188)[22] to remove host reads and obtain clean data. Species annotation of clean data was performed using Kraken2 (v2.1.2)[23], followed by assembly with Megahit (v1.2.9)[24]. Assembled contigs were expanded using TGI Clustering tools (TGICL) (v2.1)[25], and the extended contigs were species annotated using Kraken2 (v2.1.2)[23].
-
Tick (meta)transcriptomic data were aligned to poxvirus genomes using bwa[22] based on the NCBI for Biotechnology Information Genome and Nucleotide Database, enabling identification of poxvirus species in each sampling pool. The abundance of poxvirus was quantified as reads per million mapped reads.
For visualization, poxvirus distribution across sampling pools was plotted using the "ComplexHeatmap" package (v2.16.0) in R (v4.3.2). Absolute poxvirus abundance in ticks, stratified by species, geographic regions, and host, was visualized using the "ggplot2" package (v3.5.0) in R.
Differences in poxvirus distribution among ticks of varying species, geographic regions, and hosts were evaluated through principal coordinate analysis (PCoA) using the "vegan" package (v2.6.4) in R. The adonis function was used to calculate the intergroup differences among subgroups, and PCoA results visualized using the "ggplot2" package.
-
Assembled poxviral sequences were compared to 45 chordopoxviral reference sequences using BLASTn (v2.2.28+) with an E-value threshold set to 1×10-5. Sequences with matching fragments longer than 45 bp were classified as CPV. Matching fragments were designated as discontinuous sequences, while unmatched fragments were termed unblasted fragments.
-
Whole genome sequences of 52 poxvirus reference sequences were retrieved from NCBI, and the computed nucleotide composition method of the Mega X software was used to calculate the adenine (A), thymine (T), guanine (G), and cytosine (C) occupancy ratios in the continuous sequences and reference sequences. The A+T and G+C percentages were calculated.
-
Amino acid sequences of proteins encoded by the genomes of 52 poxviral reference sequences were retrieved from NCBI. Twenty-five genes encoding conserved poxviral proteins were derived separately, including A2, A3, A7, A18, A22, A23, A24, A28, A32, D1, D5, D6, D11, D12, E1, E9, E10, G5, H2, H4, H6, I8, J3, J6, L1 (according to the gene names of Vaccinia virus Copenhagen)[4]. Each gene’s amino acid sequences were aligned separately using mafft[26] and concatenated by species. The aligned sequences were then imported into MEGA-X and phylogenetic analyses were performed using Maximum Likelihood method with embellishment of the phylogenetic tree by iTol[27].
-
Unblasted fragments were blasted online using the NCBI Nucleotide BLAST function with the expected threshold set to 1×10-5. The blasted results were exported and aligned using mafft[26]. Subsequent phylogenetic analyses and tree embellishments were similar to those of the reference sequences.
-
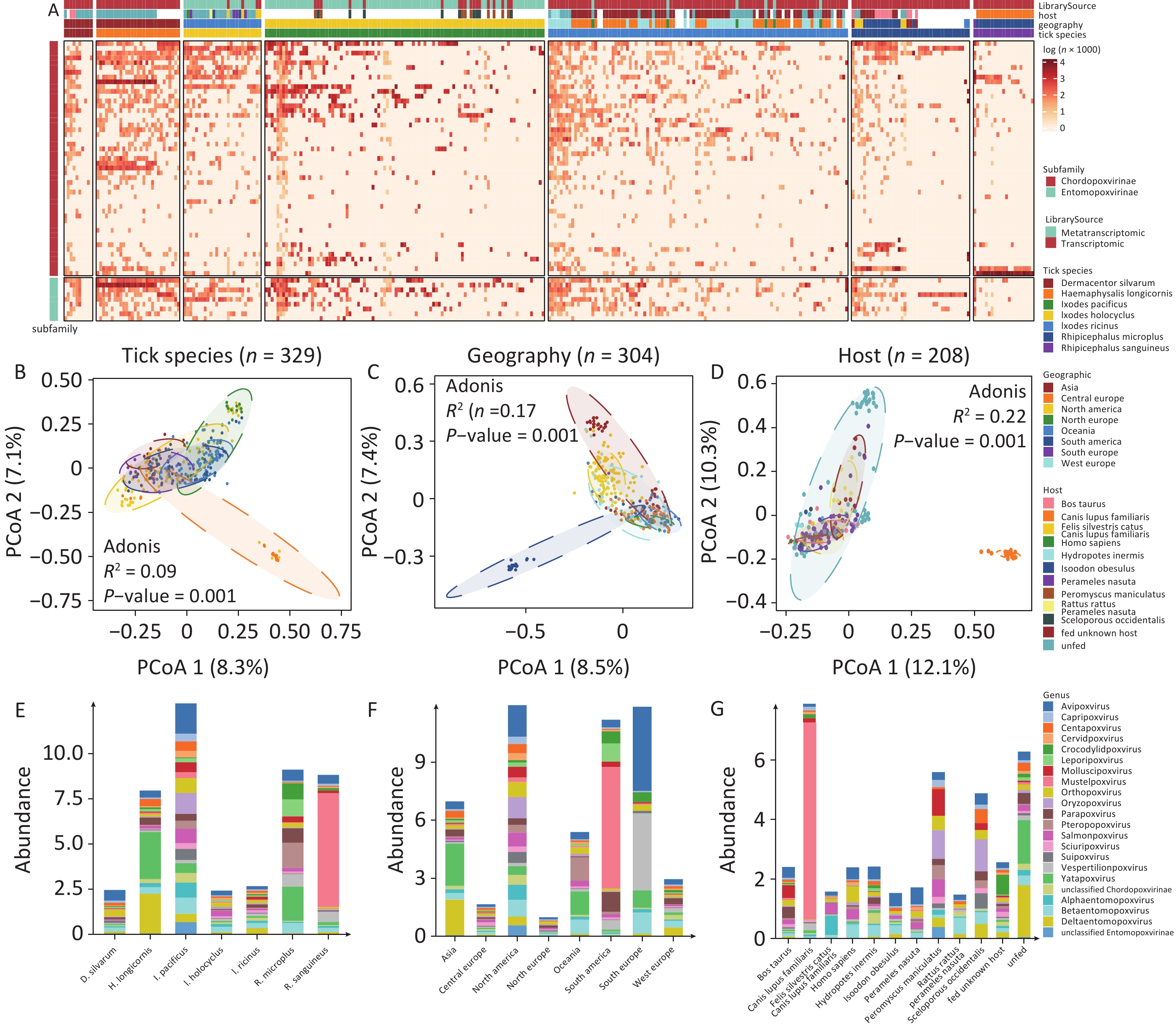
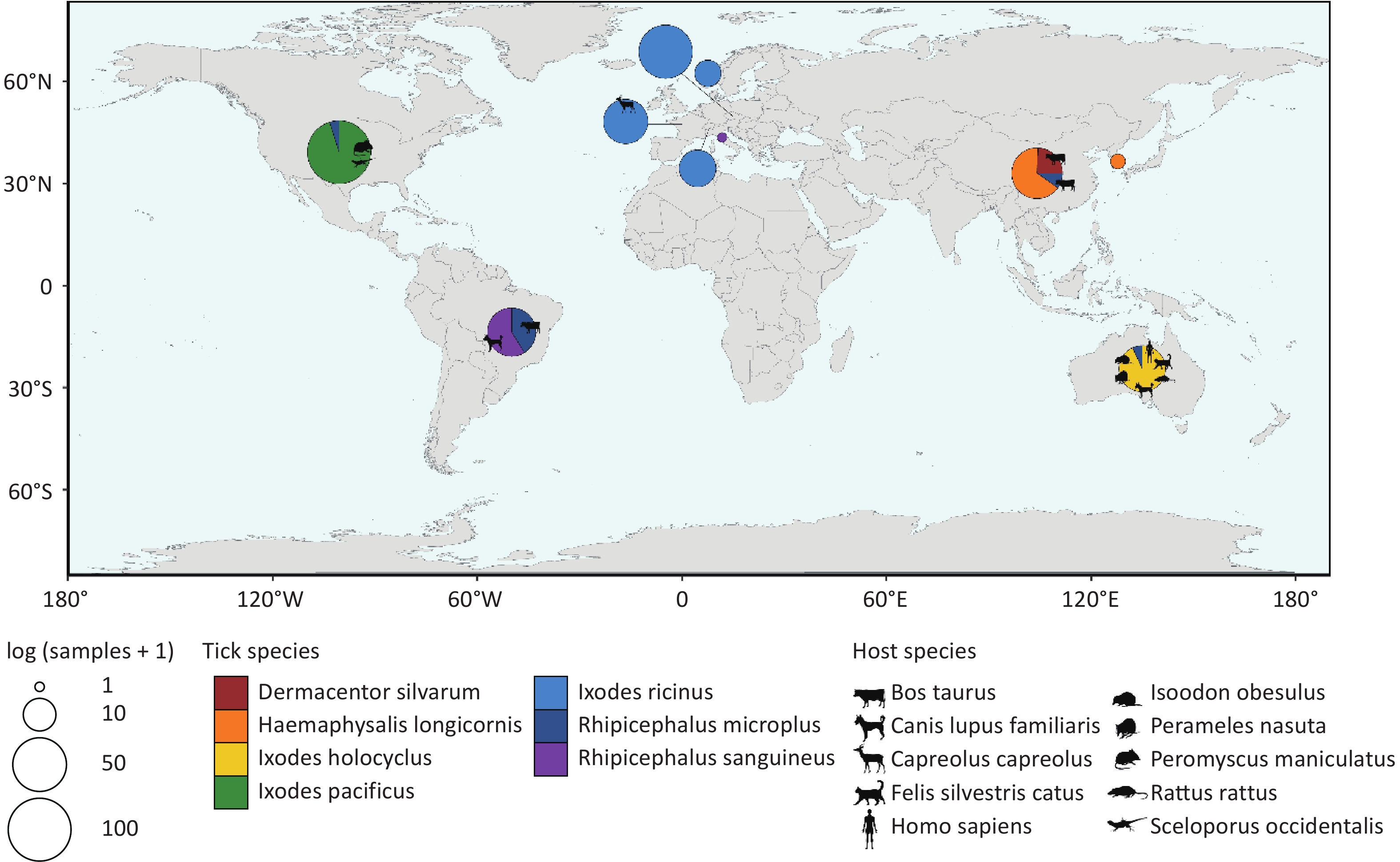
(Meta)transcriptomic raw data from 374 sampling pools, representing 7 tick species, were retrieved after excluding metagenomic and miRNA-Seq data. Basic information of each sample is presented in Supplementary Table S1. The raw data totaled approximately 685 gigabytes, comprising about 1.73 × 1012 base pairs and 8.40 × 109 reads. Following filtering, alignment, annotation, and assembly, the processed data exceeded 8 terabytes. Annotation results were used to identify sampling pools containing sequences assigned to the order Chitovirales, yielding 329 sampling pools from 30 bioprojects. According to database records, these 329 sampling pools used for further analysis originated from seven ticks species: Dermacentor silvarum (D. silvarum) (n = 10), Haemaphysalis longicornis (H. longicornis) (n = 29), Ixodes pacificus (I. pacificus) (n = 97), Ixodes holocyclus (I. holocyclus) (n = 27), Ixodes ricinus (I. ricinus) (n = 104), Rhipicephalus microplus (R. microplus) (n = 41) and Rhipicephalus sanguineus (R. sanguineus) (n = 21). Of these, 304 sampling pools were collected from diverse geographical regions across Europe, Asia, Oceania, South America, and North America, with 59 pools providing explicit host information (Figure 1).
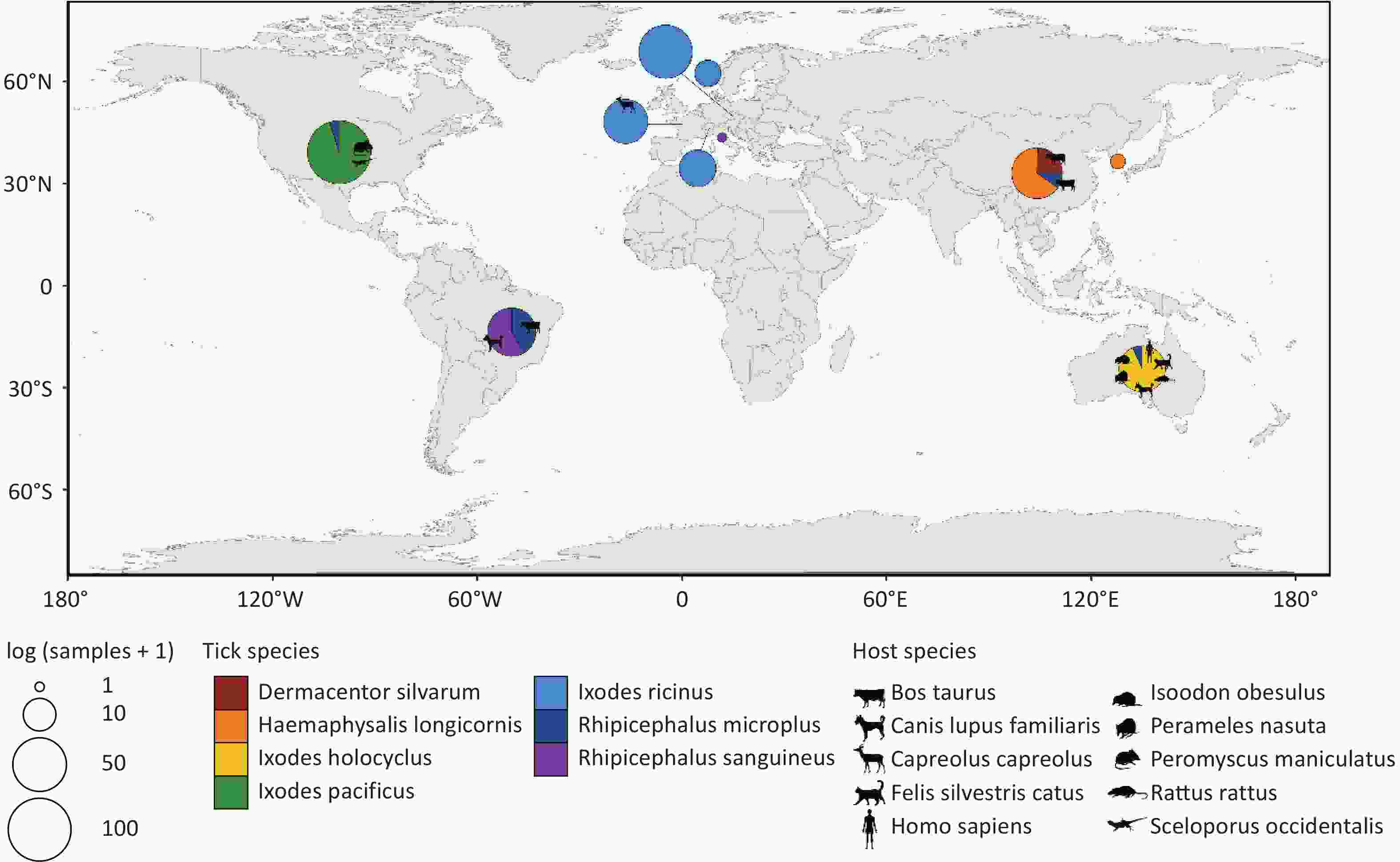
Figure 1. Distribution of sampling pools. Different colors in the pie chart represent different tick species. The size of the pie chart corresponds to the number of sampling pools, and the animal silhouettes indicate the host source of some of the sampling pools. All animal silhouettes were obtained from the Phylopic website (https://www.phylopic.org/images).
-
A total of 49 poxviral species belonging to 17 known genera and 3 unclassified genera of CPV, as well as 9 species of EPVs from 3 known genera and 1 unclassified genus of EPV, were identified by aligning the tick (meta)transcriptomic data with the NT database. The abundance of poxviruses in ticks varied considerably across both species and genera, with marked differences in poxviral species composition and abundance observed among different sampling pools of the same tick species (Figure 2A). The PCoA revealed significant differences in poxvirus diversity among tick species (Adonis, p = 0.001), host origins (Adonis, P = 0.001), and geographical regions (Adonis, P = 0.001) (Figure 2B-D). Analysis of poxvirus composition across subgroups stratified by tick species, host origin, and geographic region revealed that multiple poxviruses were highly abundant in ticks (Figure 2E-G, Supplementary Table S2).
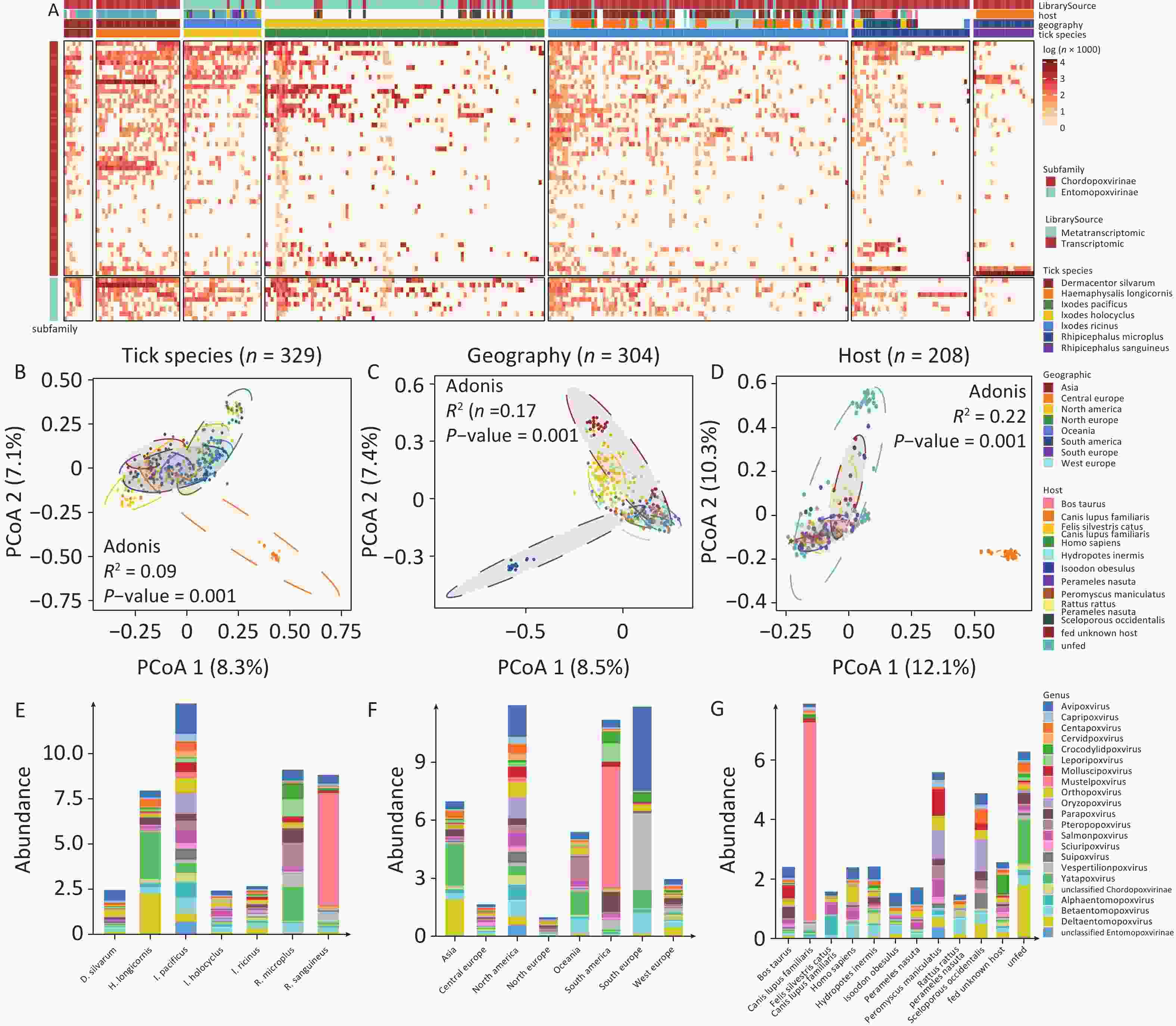
Figure 2. The abundance of poxvirus species in ticks. (A) Poxvirus richness and abundance, calculated as log (reads per million clean reads), with each vertical column representing a sampling pool. (B–D) PCoA analysis of poxvirus composition across tick genera, geographical regions and host origins. (E–G) Absolute abundance of poxviruses in ticks by genera, geographical regions and host origins.
Poxviruses pathogenic to warm-blooded vertebrates were detected at high abundance in various tick species. OPXV, which infects a wide range of warm-blooded animals closely related to humans (e.g., primates, rodents, artiodactyls, and carnivores), was notably abundant in D. silvarum (15.04%), I. holocyclus (8.92%), I. ricinus (6.35%), and I. pacificus (6.44%). MPXV, associated with the 2022 global outbreak, was detected in sampling pools of BioProjects PRJNA693137, PRJNA735703, PRJNA870442, PRJNA494273 and PRJNA311553, submitted before 2022. Other human-specific poxviruses, including Molluscipoxvirus (0.00%–6.04%), Yatapoxvirus (1.92%–32.79%), which can infect primates (including humans), and Parapoxvirus (0.08%–8.71%), which can infect humans, artiodactyls, and carnivores, were also identified in ticks. Rodent-infecting poxviruses, such as Centapoxvirus (0.54%–5.93%), Oryzopoxvirus (0.00%–9.05%), Sciuripoxvirus (0.68%–5.19%), as well as bat-infecting Pteropopoxvirus (0.00%–15.28%) and Vespertilionpoxvirus (1.05%–7.60%), were detected. Mustelpoxvirus, pathogenic to carnivores, was highly abundant in R. sanguineus (up to 71.38%), particularly in samples from Brazilian canines (PRJNA606595). In addition to mammalian poxviruses, the avian-specific Avipoxvirus was also found in ticks, including D. silvarum (24.12%), I. pacificus (13.16%), and I. holocyclus (10.77%).
Reptile- and fish-pathogenic poxviruses were also detected. Crocodylidpoxvirus, pathogenic to crocodiles, was abundant in R. microplus (9.80%). Salmonpoxvirus, affecting fish, showed unexpected richness and abundance in I. holocyclus (12.07%), I. pacificus (6.25%), and I. ricinus (3.66%).
EPV was prevalent in ticks, with more than 15.00% of EPVs in all six tick species except the R. microplus and R. sanguineus. Among these, EPVs constituted 35.67% of poxviruses in H. longicornis, 22.42% in I. pacificus, 23.62 % in I. holocyclus, and 28.63% in I. ricinus. In terms of absolute abundance, Deltaentomopoxvirus was significantly more abundant in H. longicornis and I. pacificus than in the other tick species.
-
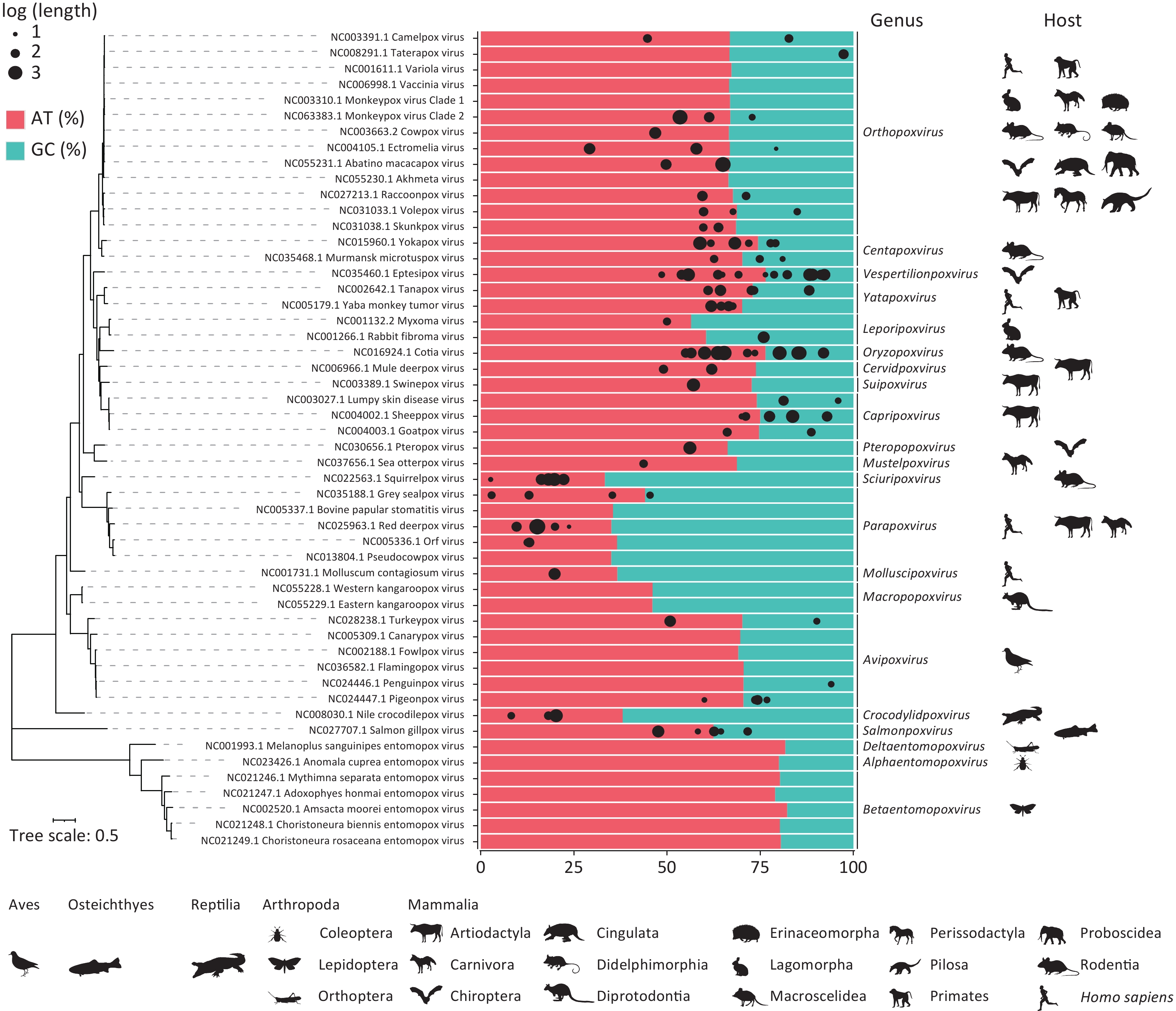
Following the assembly of the matched poxvirus reads, 212 poxvirus sequences longer than 300 bp were assembled, including two sequences exceeding 1,000 bp and 55 sequences exceeding 500 bp (Supplementary Material 1). BLAST analysis revealed that 122 sequences aligned with 45 chordopoxviral reference sequences. The adenine-thymine (AT) nucleotide content of these 122 assembled sequences and 52 reference sequences (Supplementary Table S3) is shown in Figure 3. The AT base ratios in the entomopoxviral genomes (79.00%–82.22%) were higher than those in the CPVs (33.31–76.44%). Genera such as Crocodylidpoxvirus, Parapoxvirus, Macropopoxvirus, Molluscipoxvirus and Sciuripoxvirus had significantly lower AT percentages (33.31%–46.10%) compared to other poxvirus genera. In the homologous OPXVs sequences assembled, the adenine-to-thymine composition ratio was skewed toward adenine, and numerous AT-rich fragments of varying lengths were identified (Supplementary Material 2).
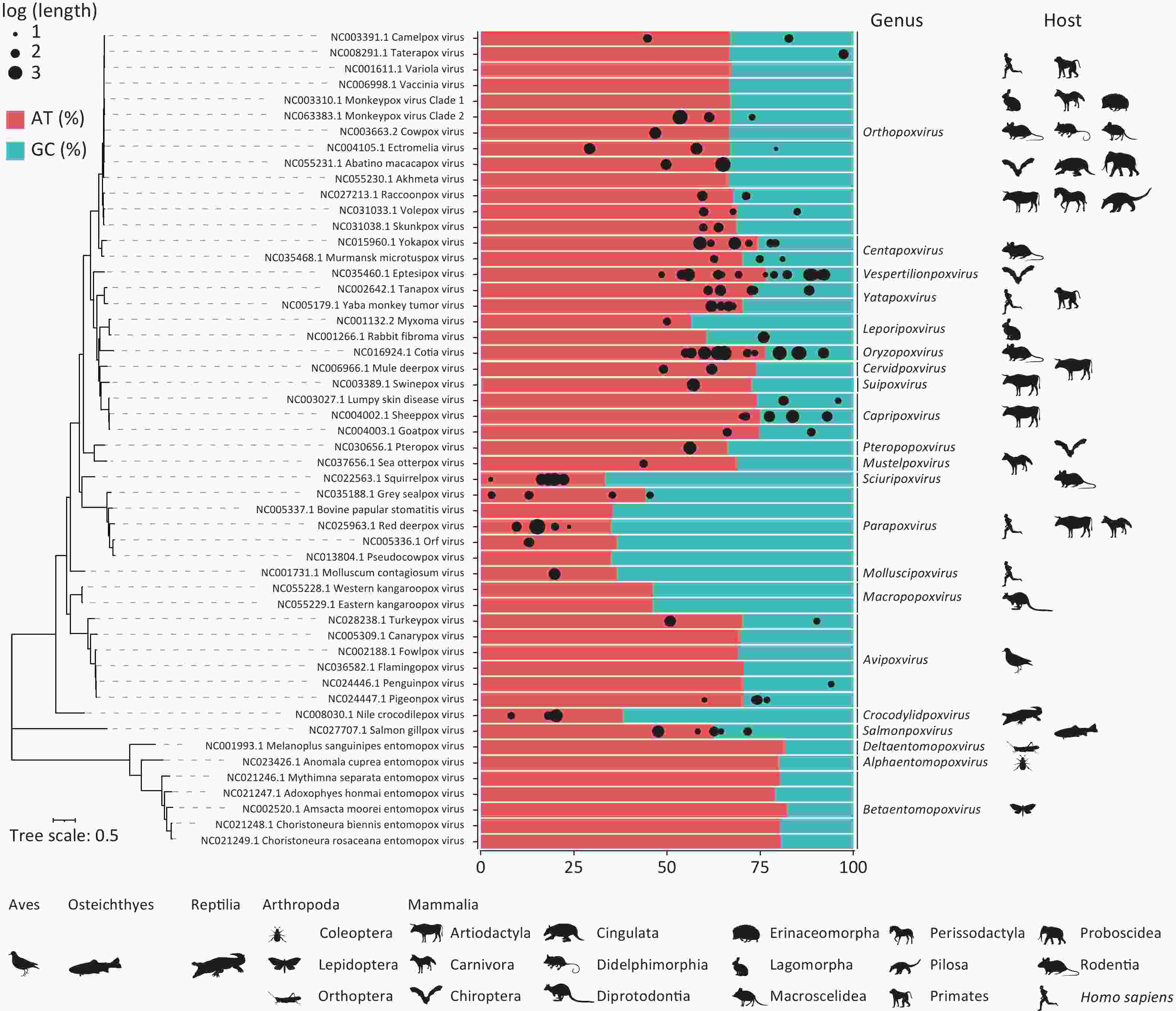
Figure 3. Adenine-thymine nucleotide content based on assembly of poxvirus genomic sequences. The percentages of adenine-thymine (AT) and guanine-cytosine (GC) nucleotides labelled in red and blue were determined by the reference sequences, and the black dots at the top represent the 122 assembled sequences matching the 45 chordopoxviruses, where the size of the dots represents the log (length of the assembled sequences) and the position represents their AT content. Phylogenetic analyses of 52 poxvirus reference sequences were performed using 25 conserved genes, with host status indicated by the animal silhouette below each genus. All hosts of mammalian poxviruses and entomopoxviruses are listed sequentially, with a human silhouette added for genera affecting humans. Host profiles for Avipoxvirus, Crocodylidpoxvirus and Salmonpoxvirus are denoted by their respective classes. All animal silhouettes were sourced from the Phylopic website (https://www.phylopic.org/images).
-
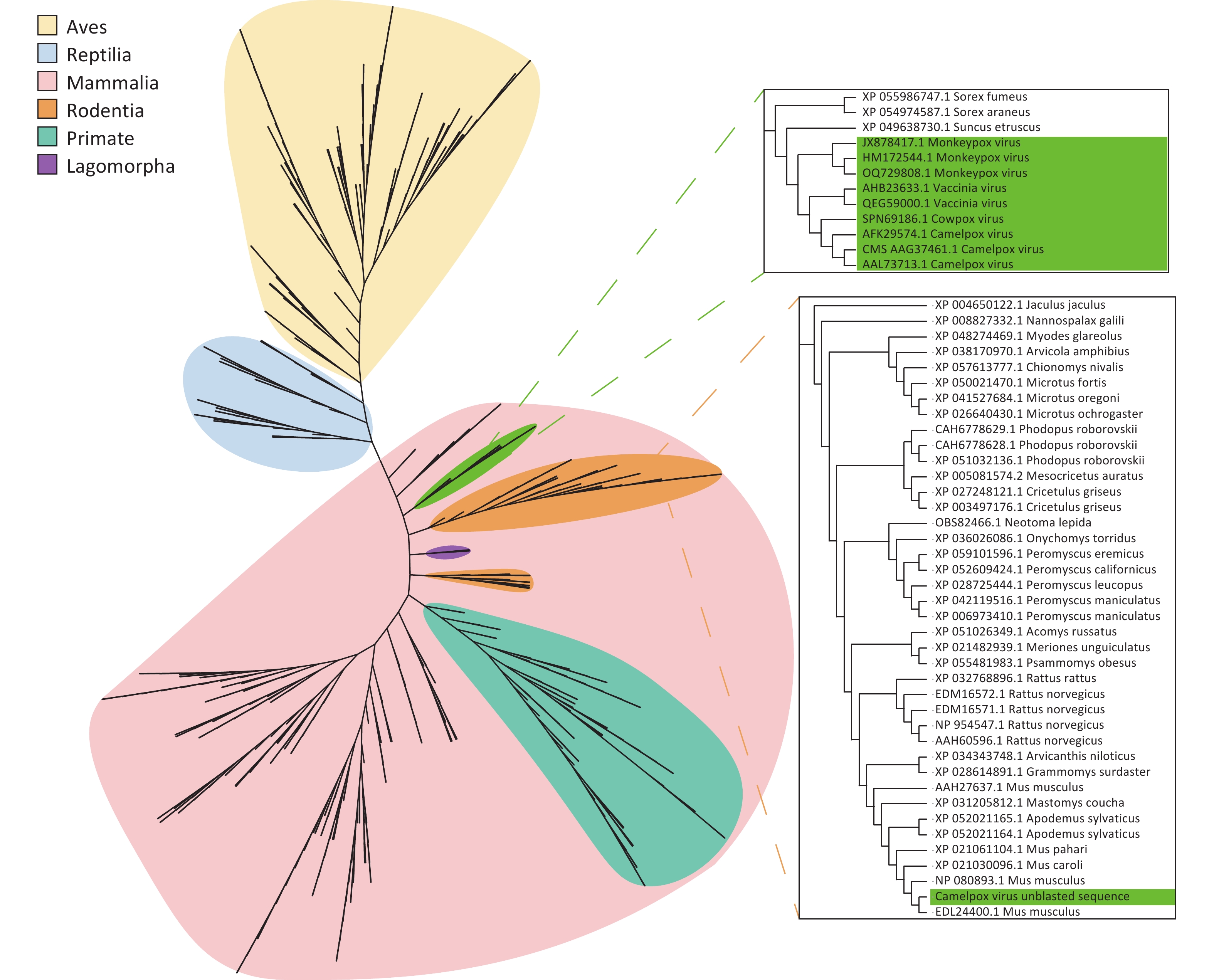
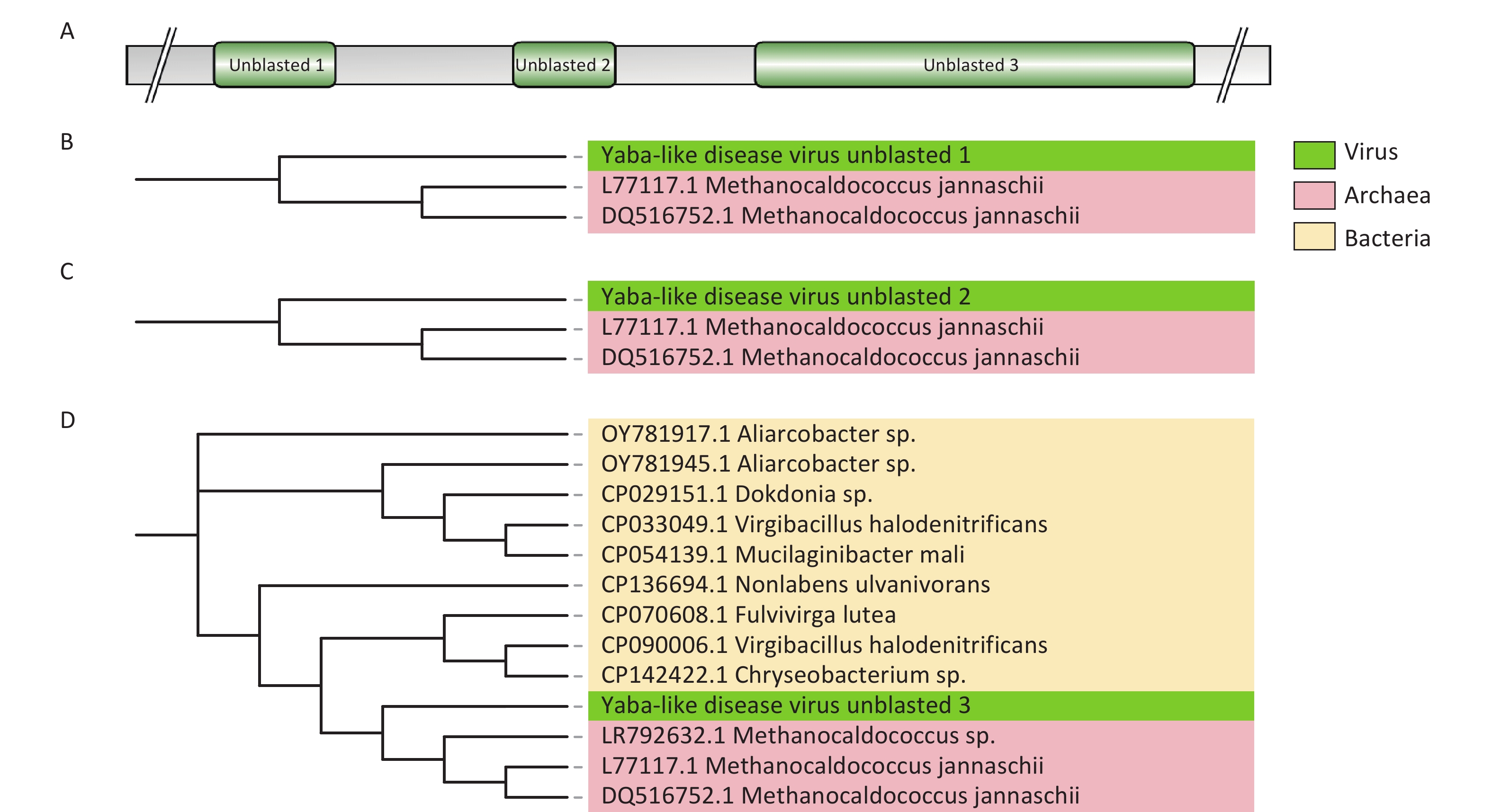
Assembled poxvirus genomic sequences were compared to 45 CPVs reference sequences (Supplementary Table S3) to explore potential evolutionary changes or mutations of poxviruses in ticks. Certain fragments within the poxvirus sequences did not match the corresponding references. We propose that these fragments represent horizontally transferred sequences acquired by poxviruses from their hosts or the environment, and we refer to them as “unblasted sequences”. Upon querying these unblasted sequences in the NCBI nucleotide database website, we found that one unblasted fragment from CMLV closely clustered with the sequence of the rodent-derived transmembrane BAX inhibitor motif-containing 4 (TMBIM4) gene, also known as GAAP, in poxviruses (Figure 4). This observation suggests that the “Camelpox virus unblasted fragment” may have been acquired from rodents. The “Yaba-like disease virus unblasted fragment” was found to align with a variety of bacterial and archaeal sequences, including members of the orders Methanococcales, Flavobacteriales, Bacillales, Sphingobacteriales, Campylobacterales and Cytophagales (Figure 5). Notably, all three unblasted fragments clustered with the genome of Methanococcus, suggesting that poxviruses may acquire genes from the environmental bacteria or archaea. Additionally, a substantial number of unblasted fragments exhibited high similarity to ticks and other arthropods (Figure 6), suggesting possible gene exchange between EPVs and CPVs, or an evolutionary relationship between them.
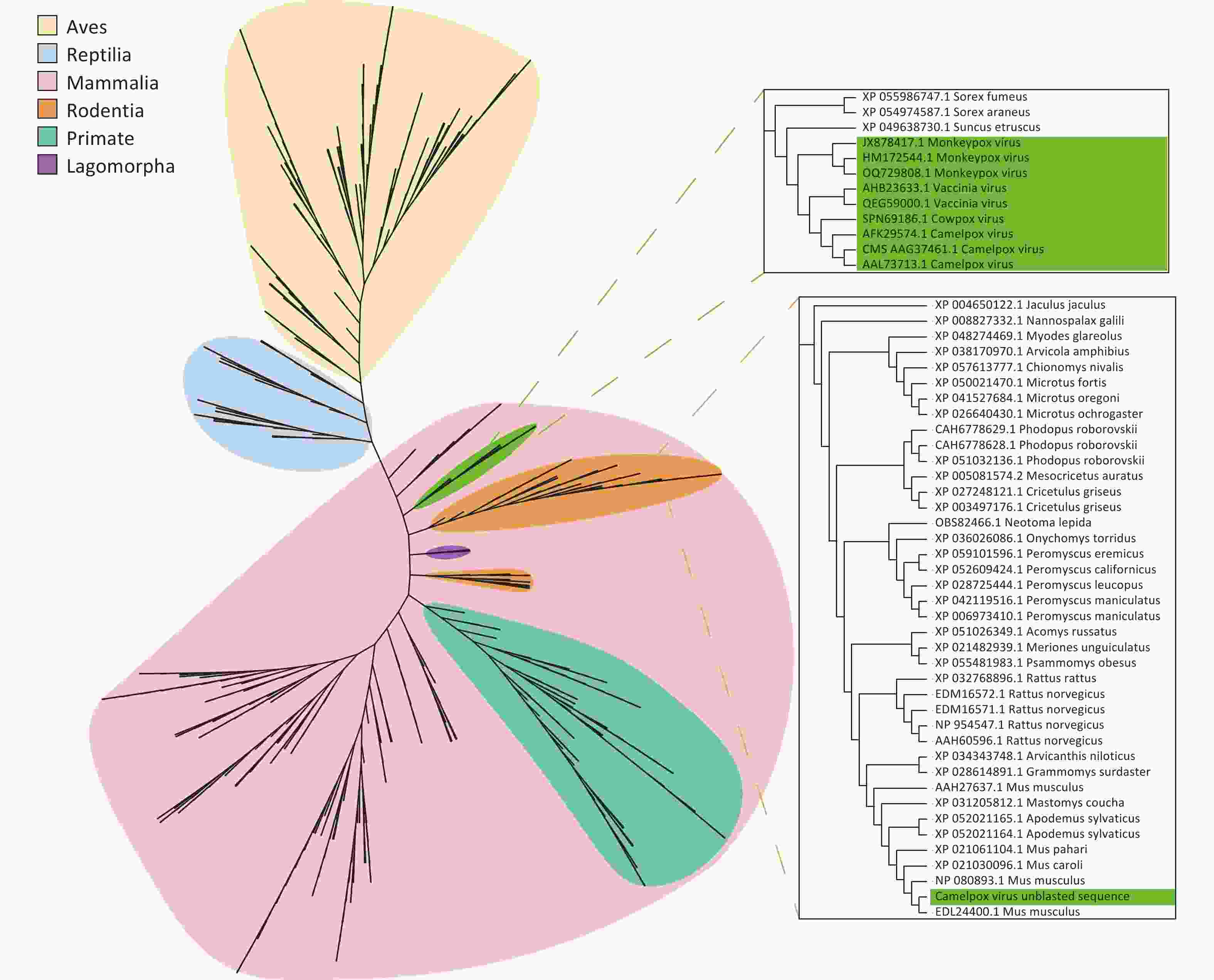
Figure 4. Phylogenetic analysis based on the unblast sequence of the assembled Camelpox virus. Poxviruses are labelled in green, with gene fragments from birds (yellow), reptiles (blue), and mammals (pink). Mammalian genes are further distinguished: rodents (orange), primates (blue-green), and rabbit-like animals (purple).

Figure 5. Phylogenetic analysis based on unblasted fragments of a Yaba-like disease virus. (A) Sequence profile of the Yaba-like disease virus obtained by assembly. (B–D) Phylogenetic trees based on unblasted 1–3 fragments of the yaba-like disease virus.
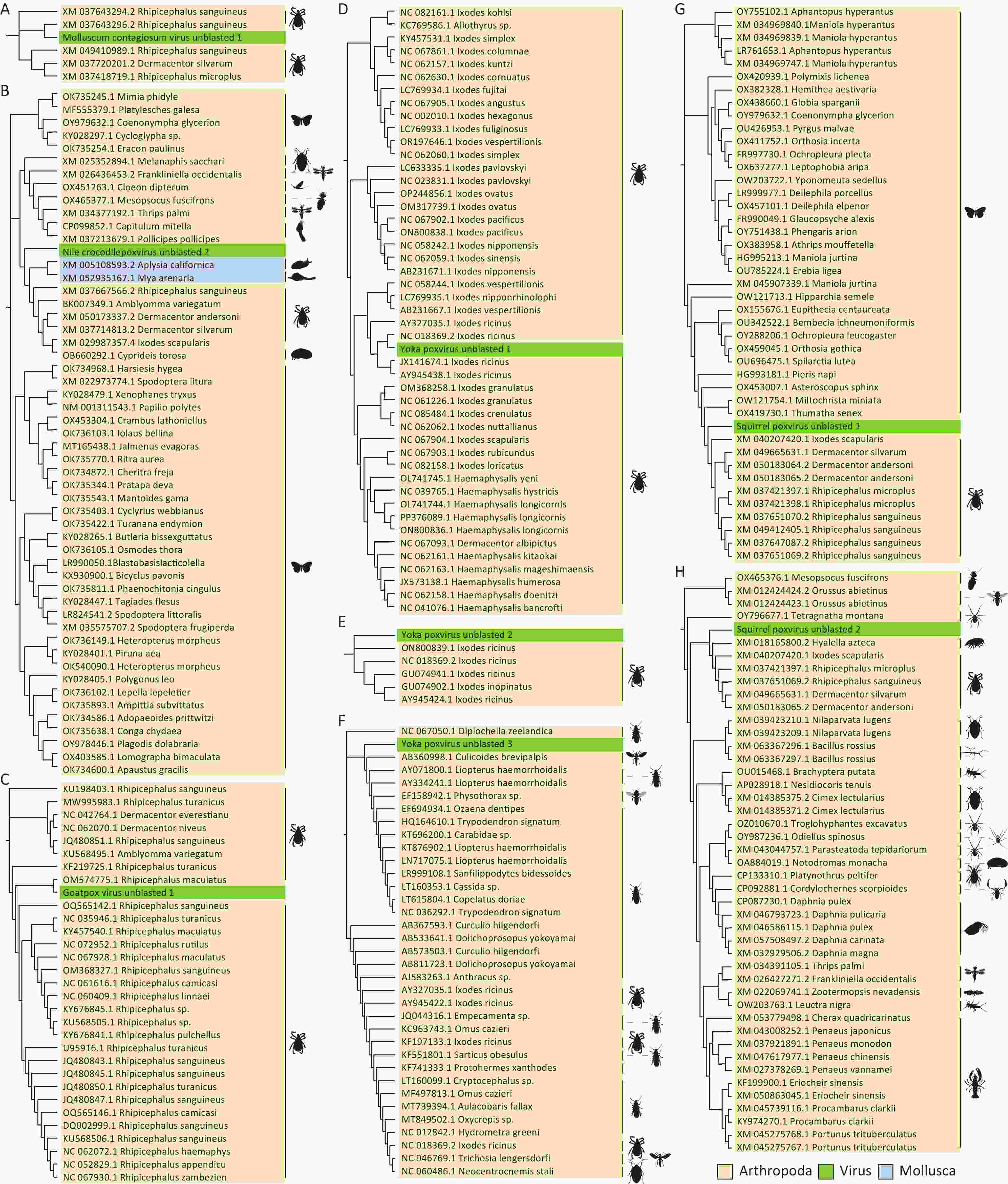
Figure 6. Phylogenetic analysis based on unblasted poxviral sequences that associated with arthropods genes. Poxviruses are labelled in green, the phylum Arthropoda in yellow and the phylum Mollusca in blue, with sequences accompanied by illustrations of their respective animal orders. The animal silhouettes were sourced from the Phylopic website (https://www.phylopic.org/images).
-
This study analyzed raw (meta)transcriptomic data from 329 sampling pools, comprising seven tick species across five continents. From these data, 58 poxviruses species were identified, representing 2 subfamilies and 20 genera, along with 212 assembled poxviral sequences. CPVs and EPVs were abundant in ticks, and the assembled poxviral sequences showed high similarity to those from arthropods, archaea, and mammals. These findings suggest that ticks serve as vectors or vessels for poxvirus transmission and evolution. The potential for gene recombination and horizontal gene transfer within ticks raises concerns, as these processes may enhance poxvirus evolution, alter virulence and adaptability, and contribute to the emergence of novel pathogens.
This study integrated transcriptomic and metatranscriptomic data using two high-throughput sequencing approaches based on RNA sequences derived from ticks in vivo. The key distinction between the two methods lies in their focus: transcriptomics treats ticks as a single species, whereas metatranscriptomics views ticks as complex ecosystems that host diverse microbial communities. Despite this difference, the final sequencing results obtained from both approaches were similar. Transcriptomics can also be applied to studies of host-microbe interactions[28]. By combining both techniques, this study increased the sample size and enhanced the reliability of the findings. Further analysis of the raw data revealed the presence of poxviruses in both transcriptomic and metatranscriptomic datasets, leading to the assembly of 212 poxvirus sequences, 122 of which aligned with CPVs.
The findings indicate that ticks play an important role in poxvirus transmission, likely facilitated by mechanical transport via mouthparts, consistent with previous studies on blood-feeding arthropods as poxviruses vectors[13,15]. This mechanical transmission mode, more efficient than contact transmission, suggests that blood-feeding arthropods, such as ticks, may intensify the poxvirus-host arms race and accelerate viral mutation and evolution[29]. Moreover, ticks may contribute to cross-regional transmission of poxviruses. For instance, Avipoxvirus, which infects birds, was detected in all ticks species examined, suggesting that ticks acquire avian pathogens through blood-feeding. Because ticks can travel across regions while attached to birds, they may facilitate the transregional spread of poxviruses, including those with potential social and public health impacts[30].
All 13 known human pathogenic poxviruses were identified in the tick data. Among these, OPXVs— including VARV—are of particular interest. Although the natural hosts for many OPXVs remain undetermined, rodents are suspected to be principal reservoirs[31,32]. Combining tick-borne poxvirus surveillance with tick host identification could help to confirm the natural reservoirs of these viruses. Unfortunately, due to data limitations, host identification host identification could not be performed in this study. Rodents, the largest mammalian order, are closely related to humans and are known to harbor diverse vertebrate viruses, representing a critical source of zoonotic infections[33,34]. Due to their blood-feeding and host-switching behaviors throughout their life cycle, and the absence of specific cell surface receptors for poxviruses during viral entry into host cells[35], ticks may facilitate host-hopping for chordate poxviruses. Given that highly abundant rodent- and bat-pathogenic poxviruses have been identified in ticks, these arthropods may act as vectors, mediating their transmission among rodents, bats, and other warm-blooded vertebrates. This can promote gene exchange and co-circulation of zoonotic poxviruses—such as OPXV—with viruses in rodents and bats, potentially altering host specificity. Owing to the intense evolutionary arms race between poxviruses and their hosts, poxviruses tend to adapt via gene mutations, recombination, or other horizontal gene transfers (HGT)[6,36,37]. Under the pressure of poxvirus-host-environment interactions, poxviruses co-evolve with their hosts, possibly giving rise to novel zoonotic strains and increasing the public health burden. Therefore, tick-based poxvirus surveillance can provide early warnings of major poxvirus epidemics in non-endemic regions, as well as novel poxviruses that may cause endemic disease.
A tick-based poxvirus surveillance system is essential for monitoring emerging and re-emerging zoonotic poxviruses, such as MPXV. The detection of a substantial abundance of poxviruses in both fed and unfed ticks via (meta)transcriptomics suggests that tick-based surveillance can provide insights into both current and historical poxvirus prevalence in a given region, offering valuable evidence of transmission dynamics and evolutionary patterns. Due to the significant presence of host-derived blood and tissues in fed ticks—and the ability of poxviruses to retain infectivity in starved ticks, with some even being vertically transmitted to offspring[15,38,39]—a higher average poxvirus abundance was observed in unfed ticks than in fed ticks, after adjusting for variations in sample sequencing depth. This suggests that the tick-based poxvirus surveillance system could be a valuable tool regardless of whether the collected ticks are parasitic or free-living. Moreover, this system could help identify the natural hosts of poxviruses, providing a more effective strategy for controlling future epidemics. Tick-based virome analysis also sheds light on the evolution of poxviruses. It has been shown that poxviruses can capture genes from hosts and the environment, and inactivate genes through deletion or premature termination, resulting in nonfunctional or partially functional products[40]. During gene acquisition, poxviruses gain genes for adaptation. In the present study, we found that the assembled CMLV sequence contained a fragment clustered with the rodent TMBIM4 gene, which is homologous to GAAP in the poxviral genome. GAAP is a nonessential virulence gene that inhibits apoptosis[41,42]. The clustering of the “Camelpox virus unblasted fragment” with the rodent TMBIM4 gene—but not with known poxviral GAAP genes—suggests that this gene may have been horizontally transferred from rodents to the poxvirus. This supports the hypothesis that rodents may be the natural hosts of OPXVs. In addition to acquiring genes from animal hosts, poxviruses may also capture genes from bacteria and archaea in the environment. Methanococcales are anaerobic, methanotrophic archaea. Phylogenetic analyses of the assembled “Yaba-like disease virus” sequence revealed a possible gene capture from Methanococcus jannaschii. This type of HGT likely occurs in aquatic environments, where poxviruses can persist for extended periods and encounter mobile genetic elements containing bacterial or archaeal gene fragments. Subsequently, these fragments may enter the poxvirus and be integrated into its genome during replication in the host cell[43,44]. In contrast, mature phages may carry genes from the host bacterium through HGT, genetic recombination, and prophage carriage[45]. Therefore, poxviruses may exchange genes with phages to acquire sequences derived from bacteria or archaea. Previous studies have also provided strong evidence of co-evolution in the replication mechanisms of phages, nucleocytoplasmic large DNA viruses, bacteria, and archaea[46,47]. Based on this, we postulated that the unblasted sequence of “Yaba-like disease virus” was likely obtained from Methanococcus jenneri via extracellular vesicles or phage transduction. Additionally, multiple gene fragments from arthropods were identified in the CPVs assembled in the present study, suggesting that CPVs might organed from EPVs, consistent with previous research[37]. An alternative explanation is that CPVs underwent genetic recombination with EPVs during evolution. Although direct laboratory evidence to support this hypothesis is lacking, such recombination may have occurred in blood-feeding arthropods that parasitize vertebrates. In summary, the incorporation of gene fragments from hosts, co-infecting viruses, and environmental sources into poxviral genomes offers critical insights into the patterns and mechanisms of horizontal gene transfer during viral evolution.
In gene expression and regulation, most poxviruses contain AT-rich fragments, which are primarily driven by poxvirus mutational bias and tend to cause early termination of protein translation[48]. Several studies have demonstrated that these AT-rich fragments may play a role in regulating viral gene expression and contribute to the poxvirus life cycle, as well as interactions with the host[49,50]. The presence of AT-rich fragments within the assembled poxvirus sequences was consistent with our previous observations regarding MPXV virulence genes. Therefore, the present study focused exclusively on the relationship between AT-rich fragments and early stop mutations. Within the B14R gene of MPXV, which encodes an immune-associated protein, AT-rich fragments ranging from 0 to 109 nucleotides were identified between nucleotide positions 590–591 (Refseq, NC_003310) of the coding sequence (Supplementary Material 3). These may act as additional mechanisms to inactivate virulence genes, leading to protein length polymorphisms, premature termination, and ultimately, altered viral virulence[51]. Poxviruses continuously engage in an arm race with hosts during cross-species transmission via direct contact or tick vectors. Throughout this process, poxviruses tend to retain adaptive mutations which may arise from replication errors or be induced by host mechanisms such as the cytosine deaminase apolipoprotein B mRNA editing enzyme catalytic polypeptide-like 3 (APOBEC3)[6,52,53]. Under host and environmental selection pressures, such mutations accumulate and superimpose on the variable terminal regions of the genome, contributing to the formation of AT-rich fragments within host-range genes. These sequences alter the length and function of the host-range proteins, thereby enhancing viral adaptability. This mechanism enables dynamic modulation of virulence and pathogenicity, facilitating a balance with host defenses and ensuring viral persistence[54]. Other viruses—and even host systems—may also regulate protein expression and function via this mechanism. Notably, T and A are highly represented in termination codons (TAA, TGA, and TAG), which appear with a frequency of up to 77.78% in most species and organelles possessing autonomous genetic systems. The modification of accessory genes by poxviruses in response to the host immune system exemplifies an adaptive strategy for host switching, while also reflecting a cost-efficient approach to maximizing exploitation of host cellular resources under stable conditions. These findings underscore the importance of evaluating the structural integrity of proteins encoded by virulence genes in key zoonotic poxviruses.
Analyses based on tick-borne viromes may also offer insights into the discovery of previously unidentified poxviruses. The detection of members from the orders Molluscipoxvirus, Crocodylidpoxvirus, and Salmonpoxvirus, as well as several genera of EPV, in ticks suggests the possibility of novel poxviruses. Molluscipoxvirus is known to infect only humans. Given that only one sample pool (I. holocyclus) was derived from a human host, and considering the increasing public awareness of tick-borne infectious diseases, the detection of Molluscipoxvirus may reflect the misidentification of an unidentified Molluscipoxvirus species as Molluscum contagiosum virus. Crocodylidpoxvirus is a poxvirus that infects crocodiles, whereas Salmonpoxvirus is a poxvirus that infects fish, both of which remain poorly studied. Reptiles are known to carry a substantial tick burden, with ticks capable of remaining attached to semi-aquatic reptiles for extended periods—and even surviving in aquatic environments[55]. Considering the close evolutionary relationship between fish and reptiles, along with the global diversity of uncharacterized poxviruses, the high abundance of Crocodylidpoxvirus and Salmonpoxvirus in ticks likely results from the misclassification of unknown reptilian poxviruses. The presence of EPVs in ticks further suggests that these arthropods may act as natural hosts. EPVs classification is closely tied to the taxonomic order of their hosts, indicating a long coevolutionary history between EPVs and their hosts[56]. Although EPVs have not previously been reported in ticks, and all currently known hosts belong to the class Insecta, our identification of EPVs in seven tick species suggests that ticks may serve as natural hosts for a previously unrecognized group of EPVs.
However, this study has several limitations. A key limitation of this study was the inability to evaluate the effects of temporal and geographical factors on poxvirus evolution and reassortment. Sequencing data were grouped by tick species without distinguishing sampling times or locations, preventing separate analysis of regional and temporal influences on viral diversity and evolution. Future studies incorporating more detailed sampling across time and geographic regions are needed to address these factors and provide a clearer understanding of their roles in poxvirus evolution. Overall, further research is required to elucidate the full role of ticks in the transmission and evolution of poxviruses.
-
In this study, we deep mind 329 (meta)transcriptomic raw datasets from the NCBI database, encompassing seven tick species across five continents. This analysis aimed to examine the diversity and abundance of poxviruses, and phylogenetic characteristics of chordopoxviruses harbored within these ticks. Our findings reveal that the genomic sequences of the assembled viruses include fragments originating from rodents, archaea, and arthropods, suggesting a complex evolutionary process influenced by cross-kingdom interactions. Therefore, we propose that ticks may serve not only as vectors facilitating poxvirus transmission but also as reservoirs fostering poxvirus evolution.
(Meta)transcriptomic Insights into the Role of Ticks in Poxvirus Evolution and Transmission: A Multicontinental Analysis
doi: 10.3967/bes2025.062
- Received Date: 2024-12-04
- Accepted Date: 2025-04-07
-
Key words:
- Poxvirus /
- Tick /
- Evolution /
- Horizontal gene transfer /
- Gene recombination /
- Gene regulation
Abstract:
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
All data for this study were sourced from public databases, and ethical approval was not required.
&These authors contributed equally to this work.
| Citation: | Yuxi Wang, Jingjing Hu, Jingjing Hou, Xiaojie Yuan, Weijie Chen, Yanjiao Li, Qile Gao, Yue Pan, Shuiping Lu, Qi Chen, Siru Hu, Zhongjun Shao, Chenglong Xiong. (Meta)transcriptomic Insights into the Role of Ticks in Poxvirus Evolution and Transmission: A Multicontinental Analysis[J]. Biomedical and Environmental Sciences. doi: 10.3967/bes2025.062

|

 Quick Links
Quick Links
 DownLoad:
DownLoad: