
-
Belonging to the Actinobacterial order Mycobacteriales, Nocardia bacteria exhibit high GC content and are aerobic and Gram-positive, with partially acid-fast and catalase-positive traits[1]. Nocardia species are widely distributed across terrestrial and aquatic environments, and to date, 132 species have been validly published with correct names (https://lpsn.dsmz.de/genus/nocardia), and approximately 95% of known Nocardia taxa are considered to possess pathogenic potential[2]. Notably, some Nocardia species are opportunistic pathogens that can cause severe infections in both immunocompromised individuals and animals, such as N. farcinica, N. cyriacigeorgica, and N. brasiliensis[2-4]. Nocardia is known to synthesize more than 50 bioactive compounds, including non-ribosomal polypeptides (NRPS), polyketides, and terpenes, reflecting both its pharmaceutical potential and its adaptation to diverse ecological or host-associated environments[2,5,6]. Given its importance in both natural and clinical settings, a comprehensive and genome-based reassessment of Nocardia taxonomy, evolution, and functional potential is urgently needed.
Despite their importance, the taxonomy of Nocardia remains problematic. Phenotypic traits and 16S rRNA gene sequences are insufficient for accurate species delineation, leading to frequent misidentifications and ambiguous nomenclature in public databases[7-9]. In recent years, whole-genome sequencing (WGS) has emerged as a powerful tool to refine bacterial systematics by applying values such as average nucleotide identity (ANI) and in silico DNA-DNA hybridization (isDDH) [10]. While previous studies have reclassified subsets of Nocardia genomes, a genus-wide reassessment integrating all publicly available genomes has not been undertaken[2,11,12]. Moreover, the functional potential of Nocardia genomes warrants detailed exploration. Antimicrobial resistance (AMR) and virulence genes are of particular concern in clinical contexts, yet their distribution has not been comprehensively characterized[3]. Equally significant is the biosynthetic capacity of Nocardia. As members of the Actinobacteria, Nocardia species are expected to encode diverse secondary metabolites. Previous reports have identified a handful of compounds with antimicrobial, anticancer, and immunosuppressive activities, but the extent and diversity of their biosynthetic capacity remain largely unknown[2]. Recently, the integration of comparative genomics and metabolite profiling has emerged as a robust analytical paradigm to accurately assess the secondary metabolic potential of diverse microorganisms[13-16].
Therefore, we conducted a comprehensive whole-genome-based study of the Nocardia genus, integrating phylogenomic reconstruction, pan-genome analysis, and functional profiling. Using 751 publicly available genome assemblies, we examined the ecological diversity of the genus, reconstructed a robust phylogenomic tree, and delineated species boundaries based on ANI and isDDH. We further analyzed the Nocardia pan-genome to assess its openness and evolutionary dynamics, profiled AMR and virulence genes to understand clinical relevance, and mined biosynthetic gene clusters (BGCs) to evaluate metabolic potential. This work is designed to establish an updated genomic framework that will facilitate future investigations into Nocardia’s AMR, pathogenic potential, and biosynthetic diversity.
-
From the National Center for Biotechnology Information (NCBI), 813 genome assemblies of Nocardia were obtained as of 15 April 2025. After removing 62 redundant entries resulting from duplicated second-generation sequencing (SGS) and third-generation sequencing (TGS) submissions, 751 non-redundant genomes were retained, representing 116 type strains (Supplementary Table S1) and 635 other strains (Supplementary Table S2) (including 6 informally named species) (Supplementary Table S3), and were used for downstream analyses. Genome assemblies were assessed for quality and completeness using Fastp v0.23.4 (Shifu Chen, Shenzhen, China) [17]. and SPAdes v3.15.5 (Saint Petersburg State University, Saint Petersburg, Russia) [18], and genome annotation was performed with Prokka v1.12 (Torsten Seemann, Melbourne, Australia) [19]. Genome statistics, including assembly size, number of coding sequences (CDSs), and GC content, were estimated using QUAST v5.0.2 (Saint Petersburg State University, Saint Petersburg, Russia) [20] and CheckM v1.1.3 (Australian Centre for Ecogenomics, Brisbane, Australia) [21]. To ensure that functional predictions were not biased by draft genome fragmentation, strict quality control thresholds were applied. Only genome assemblies exhibiting high completeness ( ≥ 95%), low contamination (≤ 5%) as determined by CheckM, and an adequate assembly contiguity (N50 ≥ 50 kb) were retained for downstream comparative functional profiling.
-
Nocardia species were classified into pathogenic and non-pathogenic groups (Supplementary Table S4) based on published literature and the LPSN databases (https://lpsn.dsmz.de/genus/Nocardia), following the criteria proposed by Eripogu et al[2]. Species were categorized as pathogenic if they had been reported in multiple independent human or animal infection cases in peer-reviewed studies[2]. Furthermore, pathogenic species were assigned to biosafety risk groups, with risk group 1 comprising species unlikely to pose significant threats to human health and risk group 2 including species associated with a high risk to human health (Supplementary Table S4).
-
Pangenome and core genome analysis were performed using Roary v3.12.0 (Wellcome Sanger Institute, Cambridge, UK) [22]. For pangenome clustering using Roary, an 80% blastp identity cutoff was selected instead of the default 95% threshold to accommodate inter-species sequence divergence across the genus-wide dataset and prevent the artificial fragmentation of orthologous gene clusters. Single-copy core genes were identified by BLASTn and were constructed using the maximum likelihood method with 1000 bootstrap repeats in iqtree v1.6.11, which was subsequently visualized by iTOL (https://itol.embl.de/). Pairwise ANI values were calculated by Pyani v0.2.12[23] and displayed as a heatmap using the R package pheatmap. In parallel, the isDDH values were calculated through the Genome-to-Genome Distance Calculator 2.1[24].
-
The ARGs were identified using Rgi v6.0.2 (McMaster University, Hamilton, Canada) with a cutoff of 60% identity and 70% coverage against the Comprehensive Antibiotic Resistance Database (https://card.mcmaster.ca/analyze/rgi). Virulence genes were identified using a cutoff of 40% identity, 70% coverage, and an E value of 1e−10 against the Virulence Factor Database (VFDB). Virulence genes and ARGs were visualized using the R package pheatmap.
-
Among the 751 Nocardia genomes analyzed, 273 species were delineated, including both type strains and putative novel taxa. BGCs were predicted from these 273 representative genomes using antiSMASH v6.0 (Technical University of Denmark, Lyngby, Denmark) with relaxed detection settings[25]. To evaluate the association between the total BGCs and genome size, Pearson correlation analysis was performed. The identified BGCs were compared against the MIBiG v3.0 database and categorized as either known or uncharacterized, with the former referring to clusters showing similarity to BGCs or compounds documented in MIBiG, and the latter lacking such matches[26].
-
This genome-scale study encompasses the most complete sampling of Nocardia diversity reported to date. The analyzed strains were derived from diverse environmental sources and from human, animal, and plant hosts. Genomic characteristics, including G+C content, genome size, and CDS counts, are summarized in Supplementary Tables S1 and S2. Briefly, G+C content ranged from 61.5% to 72.5% (mean: 68.33%), genome sizes ranged from 4.75 to 10.95 Mbp (mean: 7.62 Mbp), and coding sequence (CDS) numbers ranged from 4,323 to 10,193, reflecting substantial genomic diversity within the genus.
-
Based on literature and public database evidence, we classified 116 Nocardia species and found that 100 (86.21%) are pathogenic, whereas 16 (13.79%) have no documented pathogenicity (e.g., N. yunnanensis, N. tengchongensis, and N. bovistercoris). Pathogenic species were further categorized into two risk groups. Risk Group 1 comprises 67 species (67%) that predominantly infect immunocompromised hosts or are associated with non-human hosts, such as fish (N. salmonicida) and plants (N. vaccinii). Risk Group 2 includes species capable of causing severe infections in both humans and animals (e.g., nocardiosis), and encompasses major zoonotic pathogens such as N. farcinica, N. cyriacigeorgica, and N. brasiliensis (Supplementary Table S4).
-
The ANI and isDDH values of 751 Nocardia strains were used to evaluate overall genomic similarity. A strain is considered to belong to the same species when it shows ≥ 96% ANI or ≥ 70% isDDH[11]. Among the analyzed members of the genus, ANI values were all above 72.63% and isDDH values above 19.2%. Based on these thresholds, four pairs of synonymous species were identified: N. gamkensis and N. exalbida, N. coubleae and N. ignorata, N. elegans and N. nove, and N. zapadnayensis and N. rhamnosiphila (Supplementary Table S5). In addition, seven strains that had been incorrectly assigned to species were reclassified (Supplementary Table S5). Using ANI and isDDH metrics, we further assigned species status to 32 previously unclassified Nocardia strains (Supplementary Table S6). Moreover, we identified 156 potential novel species represented by 257 Nocardia strains (Supplementary Table S7), as well as five species that had been misclassified as members of Nocardia (Supplementary Table S8), including the type strain N. globerula DSM 44596, which should be reassigned to the genus Rhodococcus[2,11].
-
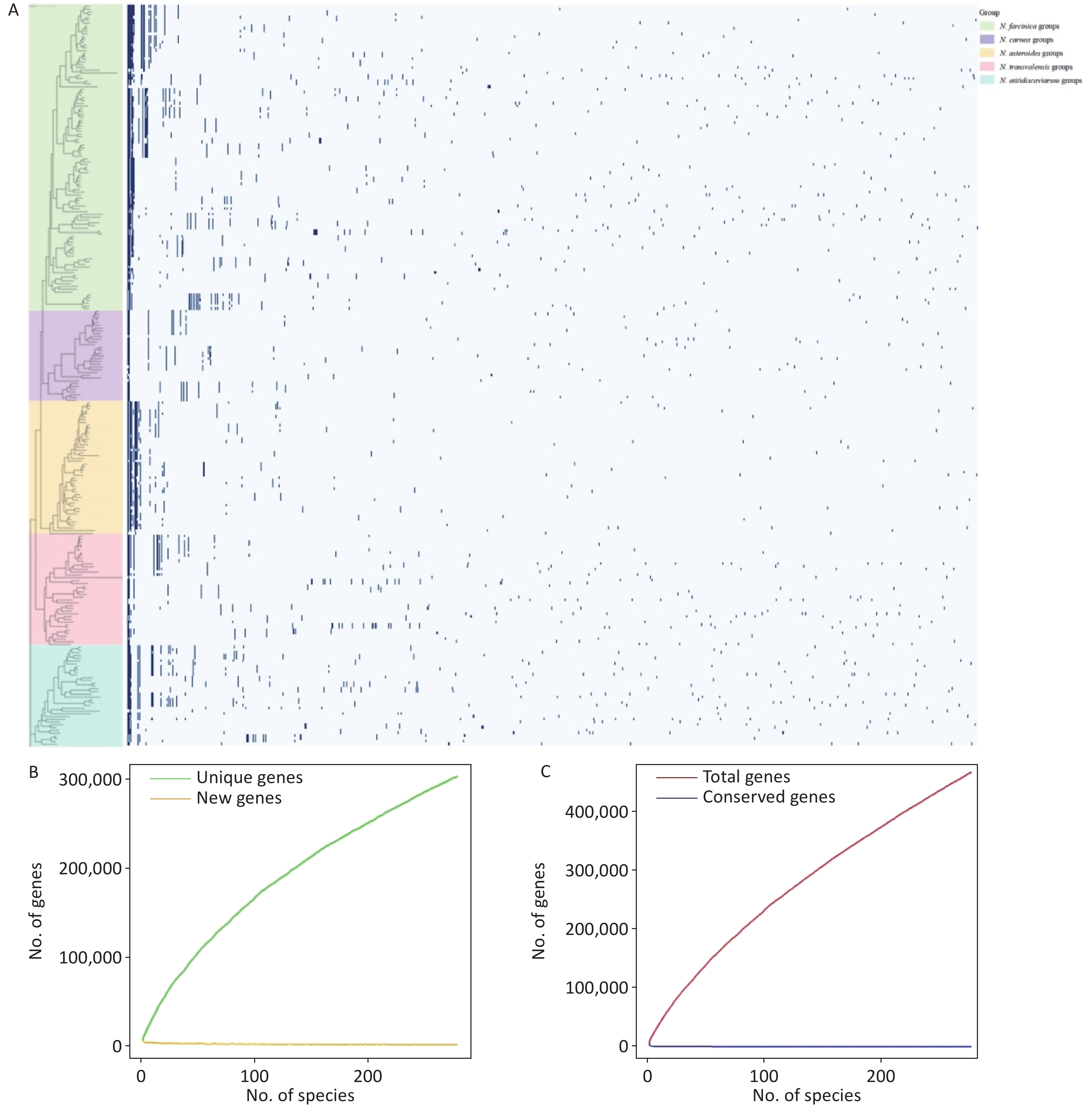
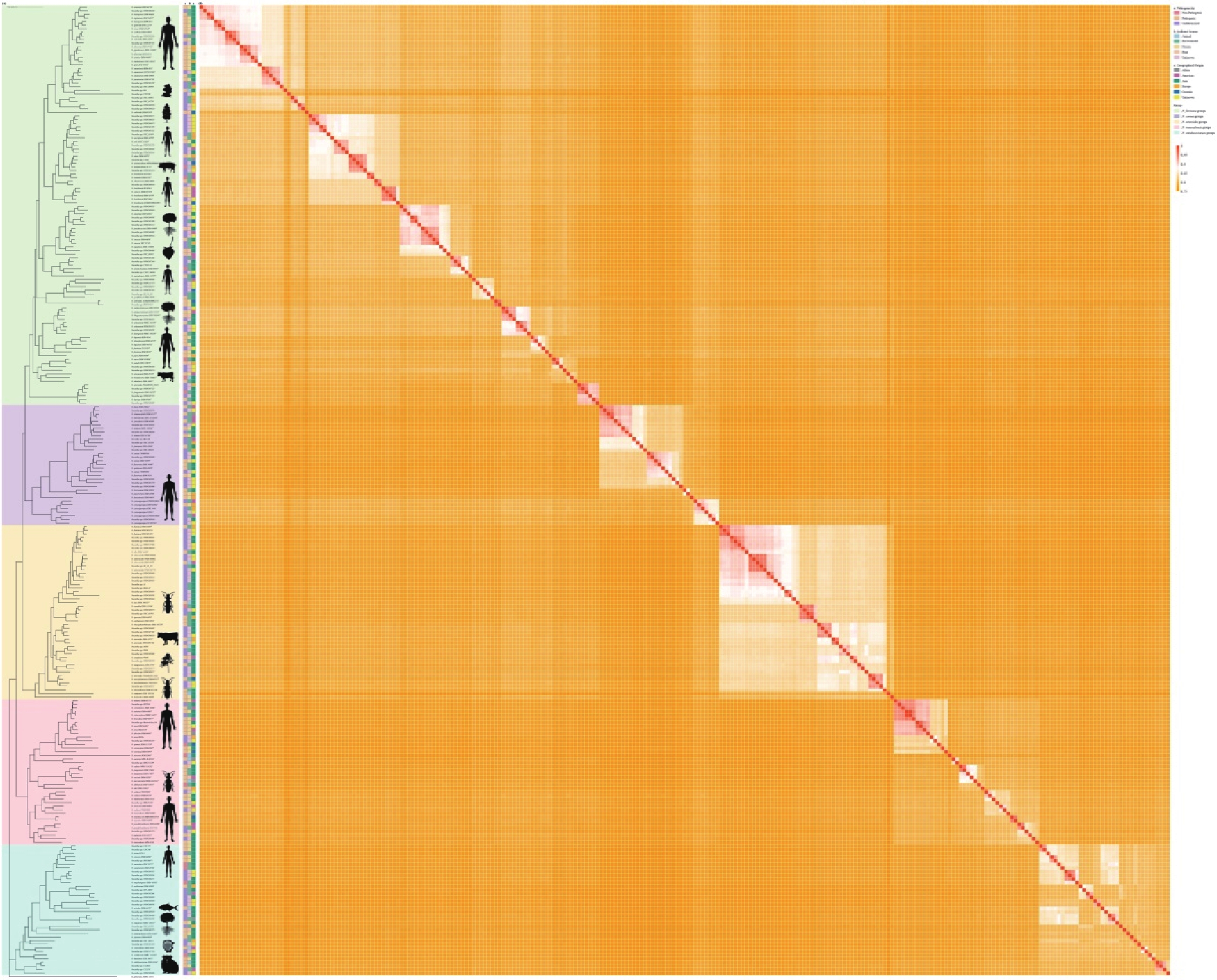
To further explore the evolutionary relationships among these classified species, we first constructed a single-copy core genes–based phylogenetic tree using 746 Nocardia strains, after removing five misclassified non-Nocardia species. Host origins were subsequently visualized, and the resulting topology resolved the strains into five major clades: the N. farcinica, N. carnea, N. asteroides, N. transvalensis, and N. otitidiscaviarum groups (Supplementary Figure S1). We then generated a comprehensive phylogenetic tree and an ANI heatmap based on 111 type strains, six informally named Nocardia strains, and 156 potential novel species, in order to investigate the phylogenetic relationships among 273 Nocardia species (Figure 1). Variations in branch length among species reflect differences in their genetic divergence, which in turn may indicate underlying sequence variability and patterns of phylogenetic clustering. Many strains previously classified as N. cyriacigeorgica clustered within the N. carnea groups and showed a close phylogenetic relationship with N. cyriacigeorgica DSM 44484 T, supporting the taxonomic revision of this species proposed by Xu et al[12].
Figure 1. Whole-genome phylogenetic analysis of 273 Nocardia species (A). Rhodococcus globerulus NBRC 14531 (GCA_001894805.1) as an outgroup. The tree was constructed by the maximum likelihood method with 1000 bootstrap replicates, the division of the genus Nocardia into 5 principal clades was highly supported, with each major clade node achieving a bootstrap value of ≥ 90%. The phylogenetic tree is color-coded to highlight the five major phylogroups. Animal silhouettes represent the main sources of isolation for various groups of Nocardia strains. Heatmap of pairwise average nucleotide identity (ANI) values for 273 genome assemblies of Nocardia (B).
Moreover, the phylogenetic tree provides insights into the clade-specific distribution of Nocardia species, further illuminating their evolutionary dynamics and pathogenic potential. The N. farcinica group, which accounts for approximately 40% of the species, including N. farcinica, N. beijingensis, and N. brasiliensis, is highly pathogenic and associated with severe human infections, likely due to the presence of specific virulence factors that enhance host infectivity[27]. Similarly, species within the N. transvalensis group, such as N. nova, N. africana, and the N. transvalensis complex (including N. transvalensis, N. blacklockiae, and N. wallacei), also exhibit high pathogenic potential. In contrast, the N. carnea, N. asteroides and N. otitidiscaviarum groups exhibit variable virulence, including both low-risk and high-risk pathogenic species.
-
A pangenome analysis of 273 Nocardia species was conducted using Roary with an 80% protein sequence identity cutoff and identified 467,566 gene clusters. The core genome (present in 99% - 100% of species; 297 clusters) represents a set of highly conserved genes that are likely essential for survival. The soft-core genome (present in 95 - < 99% of species; 258 clusters) comprises genes that are nearly ubiquitous and functionally important. Together, these two components reflect a broadly shared functional foundation in Nocardia biology (Figure 2). The accessory genome includes the shell genome (present in 15% - 95% of species; 4,839 clusters) and the cloud genome (present in 2% - 15% of species; 462,172 clusters). The substantial number of accessory genes highlights the high genetic variability within the genus and underscores the potential for functional diversity across species.
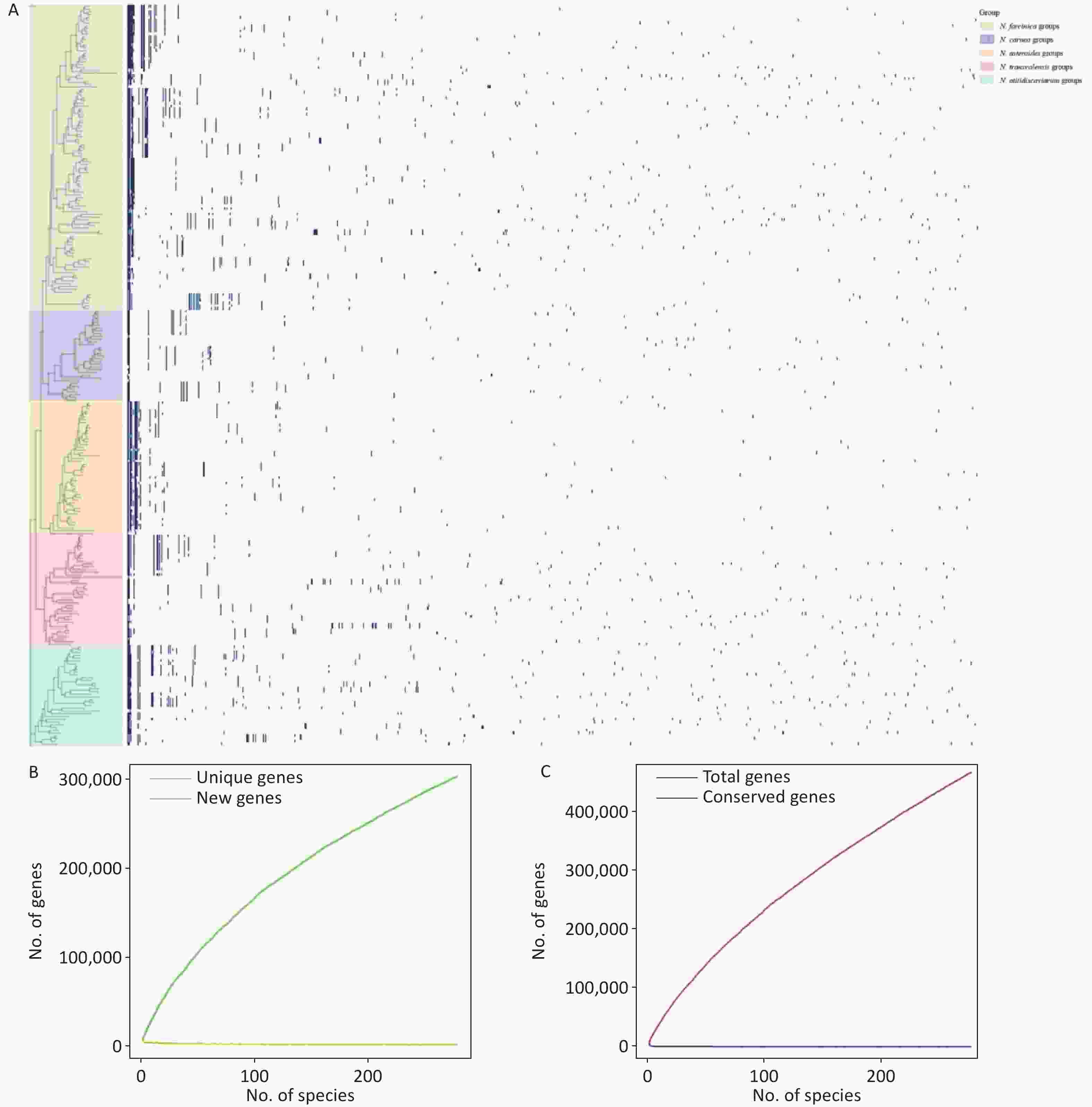
Figure 2. The phylogenetic tree for the 273 species is shown on the left. The gene presence–absence matrix of 467,566 genes is shown on the right. In each row of the matrix, a gene presence is indicated by a blue dot and an absence by a white dot (A). Unique vs new gene accumulation, showing the relationship between the unique genes (green line) and new genes (yellow line) (B). Gene accumulation curve, where the red line represents the total number of genes in the pangenome and the blue line indicates the number of conserved homologous genes (C).
Notably, as additional genomes are incorporated, the number of unique genes continues to rise, reflecting increasing genomic diversity, while the discovery of new genes diminishes, indicating a decreasing likelihood of identifying novel or species-specific genes (Figure 2B). Correspondingly, the total gene count expands with the addition of species, whereas the number of conserved genes remains stable, confirming that Nocardia exhibits an open pangenome (Figure 2C).
-
In this study, 70 ARGs in the pan-genome were identified by BLASTp against the CARD. ARGs showed uneven but clade-associated distribution across the 273 Nocardia species (Supplementary Figure S2). The majority of resistance determinants were linked to β-lactam, aminoglycoside, fluoroquinolone, and macrolide antibiotics, whereas genes associated with rarer antibiotic classes appeared sporadically. The N. farcinica and N. transvalensis groups exhibited the highest resistance gene abundance and diversity, while the N. carnea groups showed notably fewer resistance determinants. Most genes were clade-restricted, although a minority appeared across multiple lineages, suggesting possible horizontal gene transfer. Notably, all Nocardia species naturally carry resistance genes to rifampicin and isoniazid.
-
Core functions such as nutrient acquisition, oxidative stress defense, and basal secretion (e.g., mbt family genes) are conserved in > 95% of Nocardia strains. In contrast, virulence genes including toxins, adhesion factors, and secreted effectors, exhibit strong clade-specific variation. The N. farcinica groups harbor the most diverse and complete virulence repertoire, suggesting the highest pathogenic potential; N. transvalensis and N. otitidiscaviarum groups show intermediate profiles; while N. carnea and N. asteroides groups carry substantially fewer virulence genes, consistent with environmental adaptation or low virulence. Notably, the N. carnea groups lack phospholipase C (plc) and urease (ure) genes, which may contribute to reduced virulence and impaired host colonization (Supplementary Figure S3). Virulence gene content closely mirrors phylogeny, highlighting branch-dependent traits while core pathogenic functions remain evolutionarily conserved.
-
Analysis of BGCs across 273 Nocardia genomes revealed extensive diversity and biosynthetic potential. A total of 10,196 BGCs spanning 46 classes were identified using antiSMASH v6.0, with 19-74 BGCs per genome (mean: 37.35 ± 9.88). Nearly half of the genomes (48.4%, 132/273) contained 25-35 BGCs, while 12.8% (35/273) harbored over 50 (Supplementary Figure S4A). BGC number correlated positively with genome size (PCC=0.559, P<0.001) (Supplementary Figure S4B), indicating that larger genomes tend to encode more biosynthetic pathways; for instance, N. camponoti DSM 100526 (5.21 Mbp) carries 19 BGCs, whereas Nocardia spp. JCM 34519 (9.39 Mbp) harbor 74.
We further classified the identified BGCs into known (6,938 BGCs; 68.05%) and uncharacterized (3,267 BGCs; 31.95%) categories (Supplementary Table S9). The predominance of known BGCs highlights the diverse biosynthetic repertoire of Nocardia. Predominant BGC classes include non-ribosomal polypeptide synthetases (NRPS), terpenes, and type 1 polyketide synthase (T1PKS), universally present across all genomes (Supplementary Figure S4C). Interestingly, several Nocardia genomes were found to encode BGCs for compounds with reported antibacterial and anticancer activities. Specifically, a prodigiosin BGC was detected in N. salmonicida, a phosphonate BGC in N. jiangxiensis, and an Hserlactone BGC in N. stercoris.
Further, the distribution of 46 BGC classes across 273 Nocardia species was examined to understand their evolutionary relationships (Figure 3). NRPS, terpenes, and T1PKS are conserved across all Nocardia species. In contrast, the overall BGC repertoire showed substantial clade-specific variation. Especially, the N. farcinica, N. transvalensis and N. otitidiscaviarum groups exhibit comparatively higher biosynthetic potential, suggesting lineage-specific expansion or retention of secondary metabolite pathways. Further investigation into the correlation between genome size and BGC richness revealed that this expansion is not uniform across all biosynthetic classes. While universally conserved BGCs, such as terpenes, maintain a relatively stable count regardless of genome size, the genome expansion is disproportionately driven by the proliferation of NRPS, T1PKS, and complex hybrid clusters. This disproportionate enrichment is particularly defining for the N. farcinica and N. transvalensis clades, indicating lineage-specific amplifications of these complex secondary metabolite pathways.
-
In this study, we performed a comprehensive genome analysis of 751 Nocardia strains, representing the most complete sampling of the genus to date. Among these, 273 species were characterized, revealing substantial genomic diversity, an open pangenome, and extensive biosynthetic potential, thereby highlighting the evolutionary complexity of the genus. Phylogenetic reconstruction based on single-copy core genes allowed the resolution of five major clades: N. farcinica, N. carnea, N. asteroides, N. transvalensis, and N. otitidiscaviarum groups. Moreover, clade-specific differences in pathogenicity, ARGs, virulence factors, and BGCs suggest that evolutionary pressures have shaped lineage-specific functional diversification. Collectively, these findings provide a genomic framework for understanding the evolutionary dynamics of Nocardia and establish a foundation for linking phylogeny with functional traits, including pathogenic potential and secondary metabolism.
Our phylogenomic analyses further refine the current understanding of Nocardia systematics and reveal the extent of taxonomic inconsistencies within the genus. In agreement with the reclassification proposed by Xu et al. [11,12], several strains previously labeled as N. cyriacigeorgica clustered within the N. carnea groups, underscoring the persistence of misassigned taxa in public databases. These findings further indicate that strains previously labeled as N. cyriacigeorgica should be reclassified and regarded as members of a N. cyriacigeorgica complex. Additionally, we identified four pairs of synonymous species, reclassified seven incorrectly assigned strains, assigned species status to 32 previously unclassified strains, and removed five non-Nocardia species that had been mistakenly placed within the genus, thereby improving taxonomic accuracy and species resolution. Importantly, 156 potential novel species were detected, highlighting the underestimated diversity of the genus[28,29]. These results underscore the value of genome approaches for species delineation and phylogenetic resolution, demonstrating that traditional classification based on limited markers or phenotypic traits may obscure evolutionary relationships and species boundaries. Specifically, the combined use of ANI and isDDH offers a robust framework for species delineation, while the incorporation of phylogenomic data enables a more refined interpretation of evolutionary relationships and host associations[30].
The genomic profiling of ARGs, virulence factors, and BGCs revealed pronounced clade-specific patterns, underscoring the functional diversification of Nocardia. Resistance gene distribution showed clear phylogenetic structuring. The N. farcinica and N. transvalensis groups possessed the greatest abundance and diversity of ARGs, particularly against β-lactams, aminoglycosides, fluoroquinolones, and macrolides, reflecting both clinical relevance and the potential influence of horizontal gene transfer. In contrast, the N. carnea group exhibited comparatively fewer resistance determinants, suggesting a lower risk of multidrug resistance. Notably, all Nocardia species carried intrinsic resistance genes to rifampicin and isoniazid, consistent with the well-documented baseline resistance across the genus[3,31].
Virulence genes exhibited similar clade-dependent patterns. Core functions, nutrient acquisition, oxidative stress defense, and basal secretion, were conserved across all lineages, reflecting their essential role in survival. However, virulence determinants, including toxins, adhesion proteins, and secreted effectors, varied substantially among lineages. The N. farcinica groups retained the most diverse and complete virulence repertoire, congruent with its high pathogenic potential and clinical prevalence[32]. The N. transvalensis and N. otitidiscaviarum groups displayed intermediate virulence profiles, whereas the N. carnea and N. asteroides groups harbored fewer virulence genes. In particular, the absence of plc and ure in the N. carnea group may contribute to diminished host colonization and lower pathogenicity. Mechanistically, plc is crucial for degrading host cell membranes and compromising critical host structures, while ure facilitates intracellular survival by neutralizing the acidic environment of macrophage phagolysosomes[33]. Lacking these key virulence determinants, N. carnea group strains may elicit only a mild, transient host stress response that is readily cleared by the immune system, thereby resulting in the attenuated clinical profile characteristic of this clade[34]. These trends strongly parallel phylogenetic structure, reflecting the coexistence of evolutionarily conserved core functions with lineage-specific adaptations.
Analysis of 273 Nocardia genomes identified 10,196 BGCs across 46 classes, with NRPS, terpene, and T1PKS clusters universally conserved. Larger genomes tended to harbor more BGCs, and the N. farcinica, N. transvalensis, and N. otitidiscaviarum groups exhibited particularly high biosynthetic potential. Importantly, our deeper analysis indicates that this BGC expansion in larger genomes is disproportionately driven by NRPS and T1PKS classes rather than a uniform increase across all BGC types. The pronounced accumulation of these complex, multi-modular gene clusters in clades like N. farcinica and N. transvalensis suggests that genome expansion in these specific lineages is tightly coupled with an enhanced capacity for complex chemical warfare or environmental signaling. A substantial fraction of uncharacterized BGCs indicates the presence of novel pathways and unique chemical entities.2 Notably, several genomes encoded bioactive compounds with antibacterial or anticancer properties, including prodigiosin in N. salmonicida, phosphonate in N. jiangxiensis, and hserlactone in N. stercoris[2,35,36]. These findings highlight the potential of Nocardia as a source of diverse bioactive natural products and suggest avenues for future drug discovery.
Despite the comprehensive scope of this study, some limitations should be acknowledged. First, although our dataset represents the largest genome sampling of Nocardia to date, geographic and environmental biases in genome availability may influence the observed diversity patterns. Second, functional predictions were primarily inferred from genomic data, and experimental validation is required to confirm phenotypic outcomes. Third, while ARG and virulence gene profiles provide insights into potential pathogenicity, host-pathogen interactions and environmental context remain critical for assessing clinical relevance. Future studies should aim to expand genome sampling to mitigate geographic biases. Crucially, to transition these large-scale genomic predictions into a mechanistic understanding, targeted experimental validation represents a key future direction. For instance, the vast reservoir of uncharacterized BGCs offers a ranked target list for bioprospecting, which can be further explored via heterologous expression or targeted gene activation. Importantly, the clade-specific putative virulence factors identified here warrant rigorous functional characterization. Future studies should prioritize the use of in vivo animal infection models (e.g., murine pneumonia models) combined with multi-omics approaches to validate these virulence determinants. Specifically, elucidating how unique virulence genes in highly pathogenic lineages (such as the N. farcinica clade) modulate host immune responses will be essential for validating their precise roles in nocardial pathogenesis.
Overall, this study establishes a framework linking phylogeny, pathogenic potential, antimicrobial resistance, and biosynthetic capacity, providing valuable insights for clinical management and natural product discovery.
HTML
Genome Collection and Annotation
Classification of Pathogenicity
Phylogenetic Tree Construction and ANI Analysis
Antibiotic Resistance Gene (ARG) and Virulence Gene Analysis
BGC Detection
Genomic Features of the Genus Nocardia
Pathogenicity Classification of Nocardia Species
Synonymous Species and Potential Novel Species of Nocardia
Phylogenomic Analysis
Open Pangenome
Clade-related differences of ARGs and phenotypic profiles
Virulence gene Profiles Reflect Clade-Specific Pathogenic Differences
Nocardia’s Rich Biosynthetic Repertoire
Competing Interests The authors declare that they have no competing interests.
Authors’ Contributions Collected the data, processed and drafted the manuscript: Bingqian Du, Ziyu Song and Yuting Duan; Conceived the study: Bingqian Du, Shuai Xu, Yutong Kang and Min Yuan; Critically revised the manuscript: Zhenjun Li, Shuai Xu and Min Yuan; Managed the project: Zhenjun Li; Read and approved the final manuscript: All authors.
&These authors contributed equally to this work.

 Quick Links
Quick Links
 DownLoad:
DownLoad: