

 下载:
下载:

-
The World Organization for Animal Health (OIE) registers African swine fever (ASF) as a reportable infectious disease that is highly contagious in domestic pigs and wild boars. ASF is caused by the African swine fever virus (ASFV), which is the only member of this virus family, Asfarviridae, and the only known DNA arbovirus[1]. The ASFV genome is composed of linear, covalently-closed and double-stranded DNA molecules that vary in length between 170 kbp and 193 kbp and contain 151–167 open reading frames. The length of the genome varies with the length of the variable regions of the different strains of the virus[2]. Currently, 24 genotypes of the ASFV have been identified based on the analyses of the B646L gene, which encodes the capsid protein p72[3]. Eight serogroups of the ASFV have been identified based on the analyses of the EP402R gene, which encodes the serotype-specific protein CD2V[4].
The transmission of ASFV involves direct contact between swine and arthropod vectors carrying the virus, and direct contact with infected pigs or indirect contact with the blood. Moreover, contact with infected feed and excrement or secretions of infected pigs will cause the spread of ASFV. Furthermore, ASFV can be attached to cold transport vehicles and spread over long distances. In addition, studies have shown that ASFV spread through aerosol transmission within a small area[5]. The clinical symptoms exhibited by domestic pigs and wild boars that have been infected with AFS include high fever, lethargy, increased respiratory and nasal secretions, and miscarriages. The mortality rate of infected animals is as high as 100%.
Currently, there is no vaccine or effective treatment for ASF. The spread of ASF can only be prevented through early detection, the implementation of sanitary measures, and strict compliance with traditional disease control methods, which include surveillance, epidemiological investigations, pig tracking, and the elimination of infected individuals. Other measures include the strict quarantine of livestock, biosecurity measures, and the control of animal migration[6]. Passive detection of ASF is the most effective prevention and control method and is used for the early detection of ASF in disease-free areas. After blood samples, nasal secretions, or organs are collected from an animal to be tested, the genome of the virus is detected using polymerase chain reaction (PCR) assays[7], while the antigen of the virus can be detected using ELISA or direct immunofluorescence (DIF)[8]. In addition, virus culture separation technology can be used to detect the ASFV[9]. PCR is considered to be the gold standard test for the early detection of the ASFV because of its high sensitivity, specificity, and stability. A variety of PCR detection methods, including traditional PCR and real-time fluorescent PCR (qPCR), have been developed, verified, and widely used in the detection of different genotypes of ASFV[10-12]. However, it is difficult to apply PCR and qPCR to small-scale pig farms, individual households, and field operations because of instrument limitations. Although researchers have developed a portable battery-powered qPCR instrument for these situations[13], only a small number of samples can be processed simultaneously because of the limitations of the power supply.
Isothermal recombinase-aided amplification (RAA) technology allows the amplification of genes of interest at temperatures between 36 °C and 42 °C; the key component of RAA is the recombinase obtained from E.coli, which can bind tightly to the primers at 36–42 ℃. The recombinase, single-stranded binding protein (SSB), and primer form a complex to scan double-stranded DNA and unwind the double-stranded DNA at the sequence homologous to the primer. SSB prevents single-stranded DNA from renaturation. Then DNA polymerase launches the template synthesis from 3’-terminal of primers for the form of new DNA. RAA has been applied to detect a variety of viral pathogens, including the canine distemper virus[14], infectious bovine rhinotracheitis virus[15], Marek's disease virus[16], and the foot-and-mouth disease virus[17]. In addition, RAA has been applied to detect ASFV, but few studies have reported the detection of ASFV using RAA combined with lateral flow dipstick (LFD) technology. In this work, the work contents of this study were to design specific primers and probes for RAA that targeted the gene sequence of the conserved p72 region of the ASFV, to amplify the target gene using the RAA reaction system, and to perform ASFV detection in combination with an LFD.
-
According to the sequence of the ASFV B646L gene (p72) of the classical African swine fever virus published in the GenBank, The B646L gene was synthesized and cloned into the pMD18-T vector. After the plasmid was amplified in the Luria-Bertani (LB) medium, the plasmid was extracted using a small amount of Plasmid Extraction Kit (Qiagen, Germany). Finally, the plasmid was washed with 50 μL deionized water and stored in a refrigerator at –80 °C.
The nucleic acids were extracted from the inactivated clinical samples using the Nucleic Acid Isolation Kit (magnetic bead method) (GenMagBio, China). The whole extraction process involved cleavage, adsorption, and washing. The DNA was then washed and dissolved in 50 μL RNase-free water. The extracted DNA was stored in a refrigerator at –20 °C.
-
In this study, the conserved sequence of the ASFV B646L gene (p72) isolated from different countries and regions (GenBank accession number: KC835275.2, KF834193.1, KX090924.1, KX151545.1, KY353998.1, LC088171.1, MG596429.1, MK189425.1, MK686054.1, MN199633.1, MN207061.1, MN393476.1, MN537828.1, MN603946.1, MN817977.1, MN886926.1, MN886930.1, MT232750.1) was used as a template to design multiple sets of primers and probes according to the requirements indicated by the guide manual for the RAA Kit (Qitian, China). The 5′ end of the probe was modified with 6-FAM, a base in the middle of the probe was replaced with tetrahydrofuran (THF), the 3′ end was modified with phosphate blocker (P), and the 5′ end of the reverse primer was labeled with biotin. We designed 12 forward primers, 8 reverse primers, and 6 probes for combinatorial screening (Table 1). After the primers and probes were analyzed using basic local alignment search tool (BLAST) contrast, they were synthesized by Sangon Biological Engineering Co., Ltd. (Shanghai, China). Amplification experiments were performed using an RAA assay kit in a total volume of 50 μL, which included 25 μL reaction buffer, 2.1 μL upstream primer (10 μmol/L), 2.1 μL downstream primer (10 μmol/L), 0.6 μL recombinase aided amplification (RAA) probe (10 μmol/L), and 16.7 μL double distilled water (ddH2O) added to the microcentrifuge tube. After 2.5 μL magnesium acetate was slowly dropped onto the lid of the microcentrifuge tube, 1 μL DNA template was added to the microcentrifuge tube, the lid was closed, and the magnesium acetate was briefly centrifuged into the microcentrifuge tube. After centrifugation, the microcentrifuge tube was placed into a constant temperature water bath at 37 °C for 20 min. After the reaction, the amplified products were verified using agarose gel electrophoresis. The amplified products were also verified using LFD. The detection principle of the LFD is that the amplicon ends carry the respective 5′-FAM and 3′-biotin labels, which are subsequently recognized by anti-FAM monoclonal antibodies conjugated to biotin and colloidal gold, respectively. After the amplification reaction, one end of the amplicon will be labeled with FAM, and the other end will be labeled with biotin. Add the amplified product dropwise to the sample pad of the test strip. The FMA labeled on the amplicon will combine with the antibody against FAM modified on the gold nanoparticle. When the amplicon passes through the test line, biotin binds to the anti-biotin on the test strip, thus forming a sandwich structure (Figure 1). The test was positive when one red band and one blue band appeared; the blue band appeared in the quality control area and the red band appeared in the test area.
Table 1. Primers and probes for screening
Primer/probe Sequence (5′→3′) F1 TGTAACGCAGCACAGCTGAACCGTTCTGAAGAAGA F2 AACCGTTCTGAAGAAGAAGAAAGTTAATAG F3 GATACCACAAGATCRGCCGTAGTGATAGACCCCACGT F4 AGCAGTTACGGAAATGTTTTTAATAATAGGTAATGTGATC F5 CGGAAATGTTTTTAATAATAGGTAATGTGATCGGATA F6 ATCGGATACGTAACGGGGCTAATATCAGATA F7 AACGGGGCTAATATCAGATATAGATGAACA F8 CGGGGCTAATATCAGATATAGATGAACATG F9 CGTCTGGAAGAGCTGTATCTCTATCCTGAA F10 GCTGTATCTCTATCCTGAAAGCTTATCTCTG F11 AAGAAGAAAGTTAATAGCAGATGCCGATACCAC F12 CCGATCACATTACCTATTATTAAAAACATTTCC R1 Biotin-CAGAGCAAGAGAATTTTATATTAGTTGGGA R2 Biotin-CGGGAGGAATACCAACCCAGTGGTCATATTAACGT R3 Biotin-TGCTTTGAAGCCACGGGAGGAATACCAACC R4 Biotin-CTGCTCATGGTATCAATCTTATCGATAAATTTCCA R5 Biotin-CCCATTGAATATATGTTTATAGGATTAAAACCTACCTG R6 Biotin-ATGGCCCATTGAATATATGTTTATAGGATTAAAACCTACC R7 Biotin-GCTCTTACATACCCTTCCACTACGGAGGCAATG R8 Biotin-ATAAAATTCTCTTGCTCTGGATACGTTAATATG P1 Fam-ATCGGATACGTAACGGGGCTAATATCAGATA/THF/AGATGAACATGCGTC-P P2 Fam-CGTCTGGAAGAGCTGTATCTCTATCCTGAA/THF/GCTTATCTCTGCGTG-P P3 Fam-GCTTATCTCTGCGTGGTGAGTGGGCTGCATA/THF/TGGCGTTAACAACAT-P P4 Fam-GATGAACATGCGTCTGGAAGAGCTGTATCTCTA/THF/CCTGAAAGCTTATCT-P P5 Fam-TGGGTTGGTATTCCTCCCGTGGCTTCAAAGCAAAG/THF/TAATCATCATCGCAC-P P6 Fam-GGTGCGATGATGATTACCTTTGCTTTGAAG/THF/CACGGGAGGAATACCA 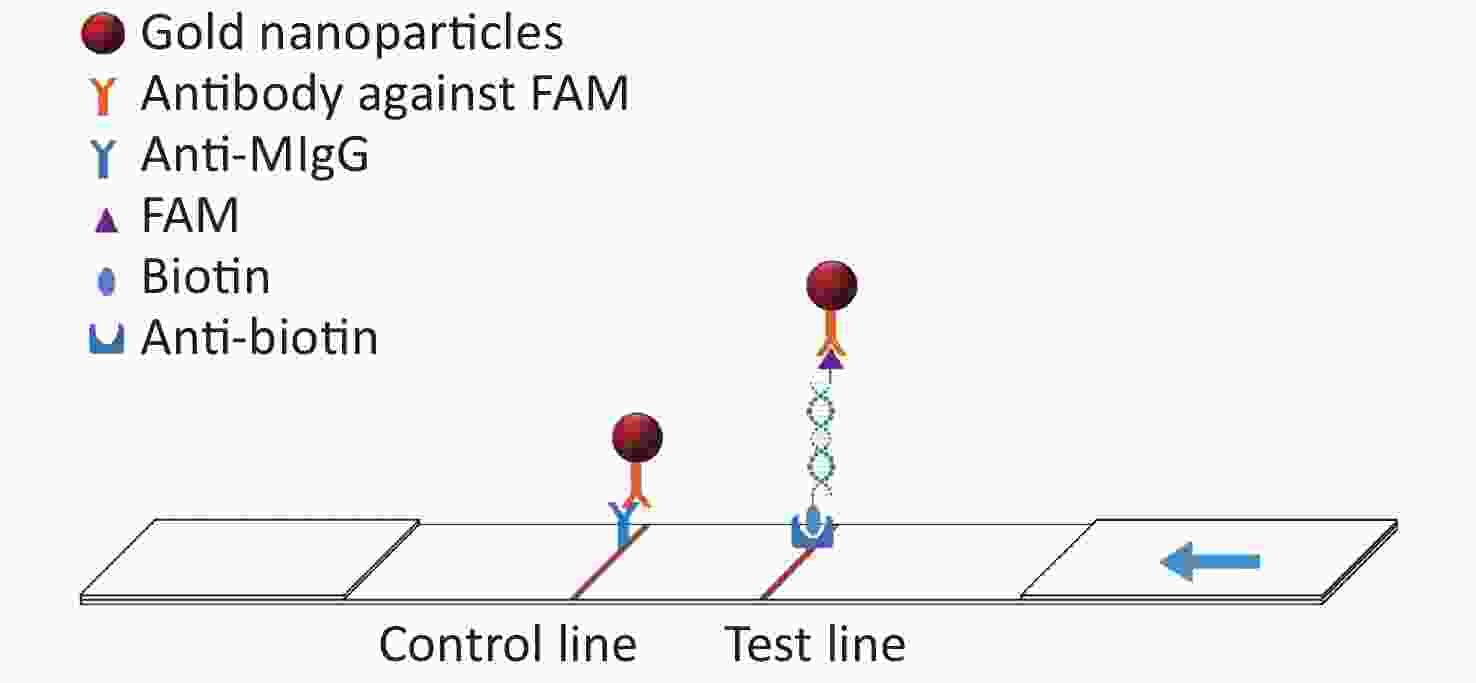
Figure 1. The schematic diagram of the LFD (lateral flow dipstick).
-
In this study, the RAA-LFD assay kit was used for the detection of ASFV. Two experiments were performed. In Experiment 1, the reaction mixture was placed in a constant temperature water bath at 37 °C for 5 min, 10 min, 15 min, 20 min, 25 min, and 30 min. In Experiment 2, the reaction mixture was heated for 15 min at different temperatures: 36 ℃, 37 ℃, 38 ℃, 39 ℃, 40 ℃ and 41 ℃. At the end of the reaction, 2 μL of the amplification product was added to 48 μL of the sample loading buffer, and the 50 μL diluted product was dropped onto the sample pad. The results of the reaction were observed after 5 min.
-
The pMD18t-p72 recombinant plasmid from each reaction was individually diluted to 2 × 107–2 × 101 copies/μL and amplified under optimized experimental conditions. The amplified products were individually examined using RAA LFD after the reaction was completed to determine the lower limits of detection of the RAA-LFD assay. The specificity of the RAA-LFD assay was evaluated using porcine parvovirus (PPV), porcine circovirus type 2 (PCV2), pseudorabies virus (PRV), porcine reproductive and respiratory syndrome virus (PRRSV), and common swine fever virus (CSFV). At least triplicates were used in the experiment.
-
Clinical samples were provided by China Animal Health and Epidemiology Center. The extracted nucleic acids from 30 clinical samples were tested using the RAA-LFD method. Tests of clinical samples were also performed using the qPCR with the 2 × qPCR Mix Kit (Takara Bio Inc., Japan) in combination with the primers and probes recommended by the OIE (Table 2). The PCR reaction system used 10 μL 2 × qPCR mix, 0.4 μL upstream primer, 0.4 μL downstream primer, 0.8 μL probe, and 2 μL template and made up to 20 μL with ddH2O in a PCR reaction tube. The PCR reaction conditions were 95 °C for 30 s, 95 °C for 5 s, and 60 °C for 30 s for a total of 40 cycles. At least triplicates were used in the experiment. Data in this work were presented as mean ± standard deviation.
Table 2. Primers and probe sequences used for qPCR assay
Primer/Probe Sequences(5′→3′) qPCR F CTGCTCATGGTATCAATCTTATCGA qPCR R GATACCACAAGATCRGCCGT qPCR P FAM-CCACGGGAGGAATACCAACCCAGTG-TAMRA -
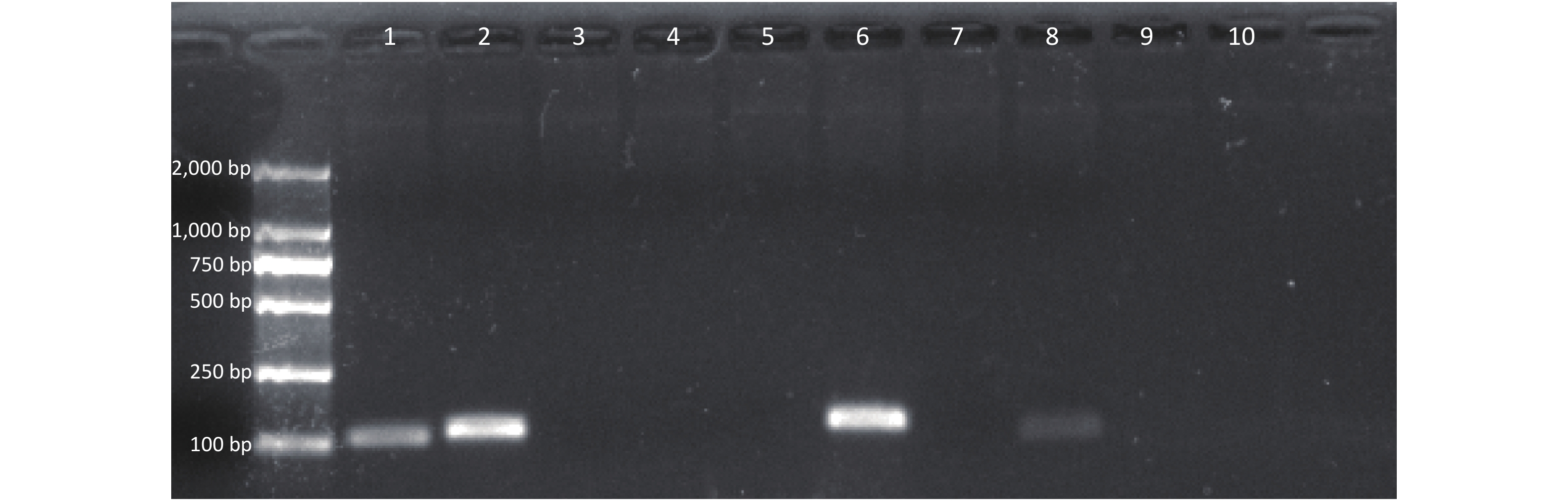
Seven sets of primers and probes were designed and synthesized. After amplification by the RAA Kit, the amplified products were verified using 2% agarose gel electrophoresis (Figure 2). The results showed that a single target band could be amplified in the second and sixth primers and that the amplified bands were clear without obvious primer dimers. The second and sixth products were examined using an RAA-LFD; false positives occurred in products amplified by the second primer, but not the sixth primer. Therefore, primer set 6 was chosen for subsequent experiments. The primer-probe combination is composed of F9, R6, and P3.

Figure 2. Results of agarose gel electrophoresis of RAA amplified products
-
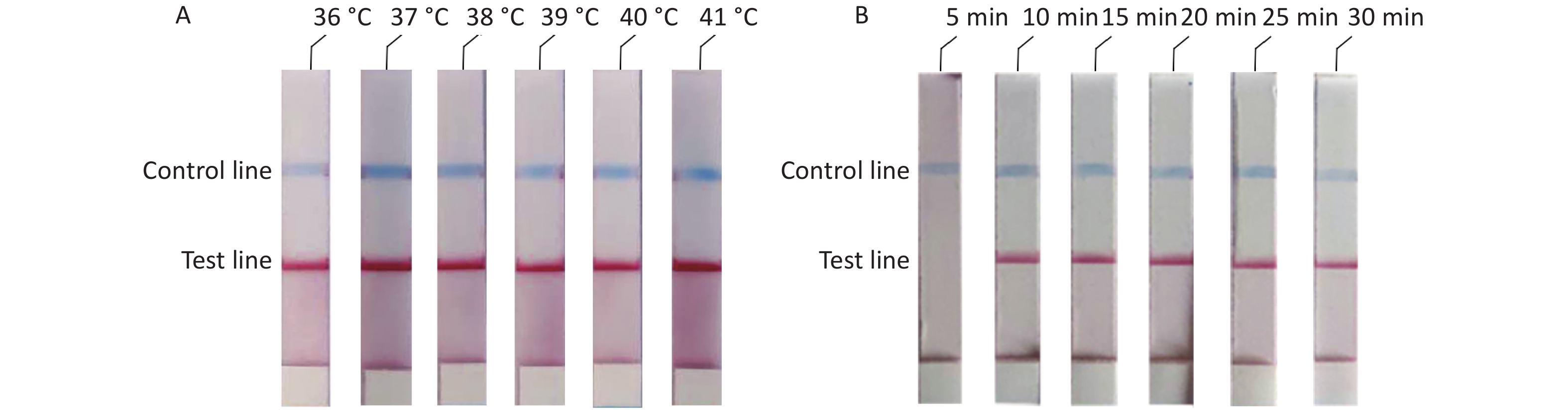
To determine the optimal reaction conditions for the RAA-LFD assay, we performed experiments at different temperatures and reaction times. The experimental results showed that there was no significant difference at 36–41 °C (Figure 3A). Thus, considering the practical application of the rudimentary conditions, we used the reaction at 37 °C in our subsequent experiments. The experiment results to determine the optimal reaction time showed that no amplification products could be detected at a reaction time of 5 min and that there were no obvious differences in the reaction times from 10 min to 30 min (Figure 3B). In summary, we confirmed that the optimal reaction conditions were 10 min at a constant temperature of 37 °C.

Figure 3. Optimal reaction conditions of ASFV-RAA-LFD. (A) Optimal detection temperature of ASFV-RAA-LFD assay. From left to right were the experimental results at 36 °C, 37 °C, 38 °C, 39 °C, 40 °C, and 41 °C; (B) Optimal detection time of ASFV-RAA-LFD assay.
-
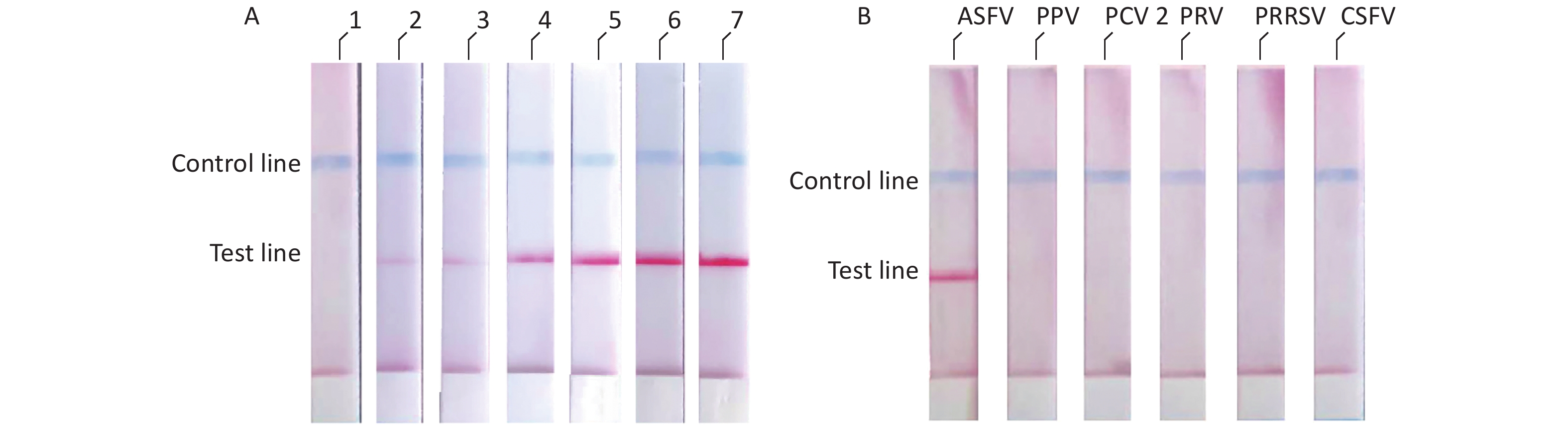
We diluted the pMD18t-p72 recombinant plasmid using a concentration gradient and incubated it at 37 °C for 10 min. The results showed that the ASFV RAA-LFD had a detection limit of 2 × 102 copies/μL of pMD18t-p72 recombinant plasmid per assay (Figure 4A). The specificity of the RAA-LFD assay was verified through the detection of PPV, PCV2, PRV, PRRSV, and CSFV. The experimental results did not show cross-reactivity with other swine viruses (Figure 4B).
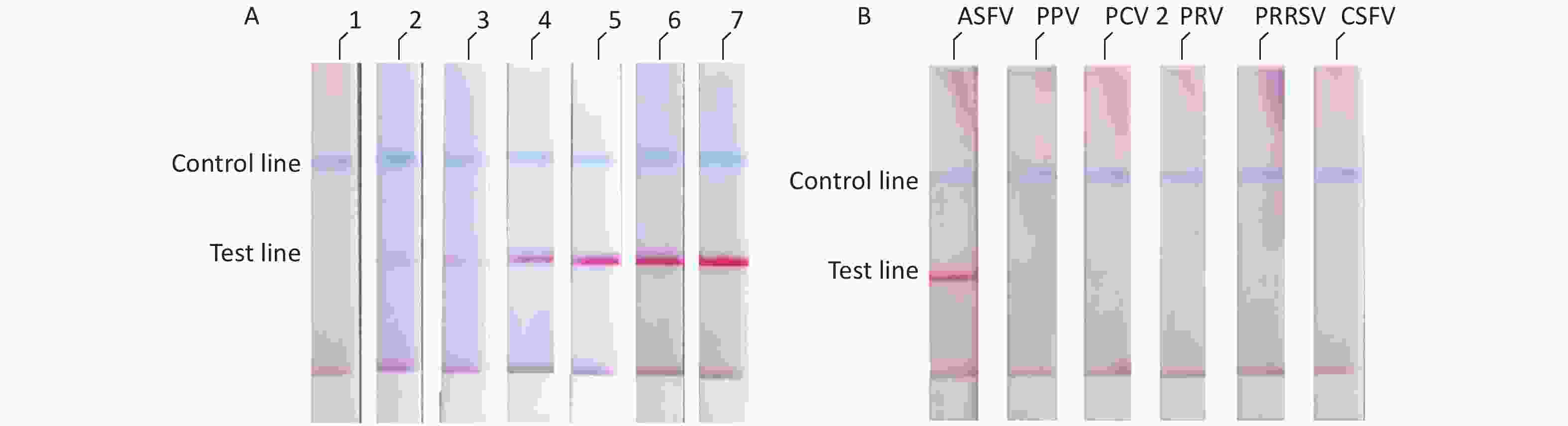
Figure 4. (A) Evaluation of the sensitivity of ASFV-RAA-LFD. Concentration gradient of pMD18t-p72 (copies/μL) were as follow: 1: 2 × 101; 2: 2 × 102; 3: 2 × 103; 4: 2 × 104; 5: 2 × 105; 6: 2 × 106; 7: 2 × 107; (B) Evaluation of the specificity of ASFV-RAA-LFD.
-
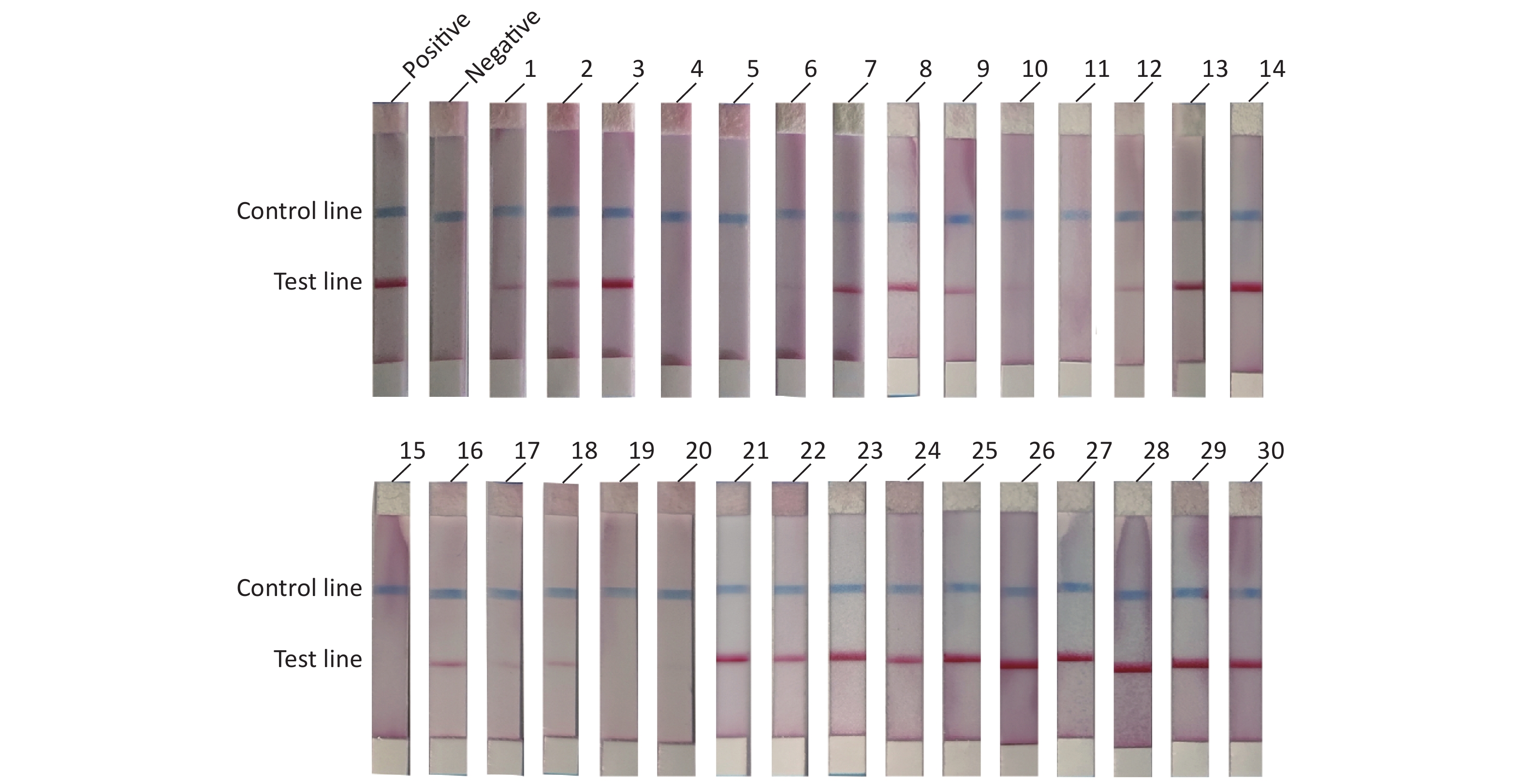
To evaluate the practical application of RAA-LFD, 30 porcine nasopharyngeal swab samples suspected for ASFV were tested. Nucleic acids were extracted from the 30 clinical samples using a magnetic bead-based nucleic acid extraction kit. The samples were tested using qPCR recommended by OIE, and the results showed that among the 30 samples, 23 samples were confirmed to be ASFV positive (Ct value, ranging from 16.23 to 36.44) including 6 weak positives (Ct value > 30). 7 samples were confirmed to be ASFV negative (Ct value, undetermined). The RAA-LFD method was used for nucleic acid amplification and the detection results are shown in Figure 5. The results showed that 21 samples tested positive for ASFV, and 9 samples tested negative for ASFV. ROC analysis based on the detection of clinic samples showed that the AUC value between RAA-LFD and reference qPCR was 0.957 (0.812–0.998, 95% CI). In addition, in comparison to qPCR, the specificity and the sensitivity of RAA-LFD assay for identification of ASFV were 100% (59.0%–100.0%, 95% CI) and 91.30% (72.0%–98.9%, 95% CI) (Table 3).
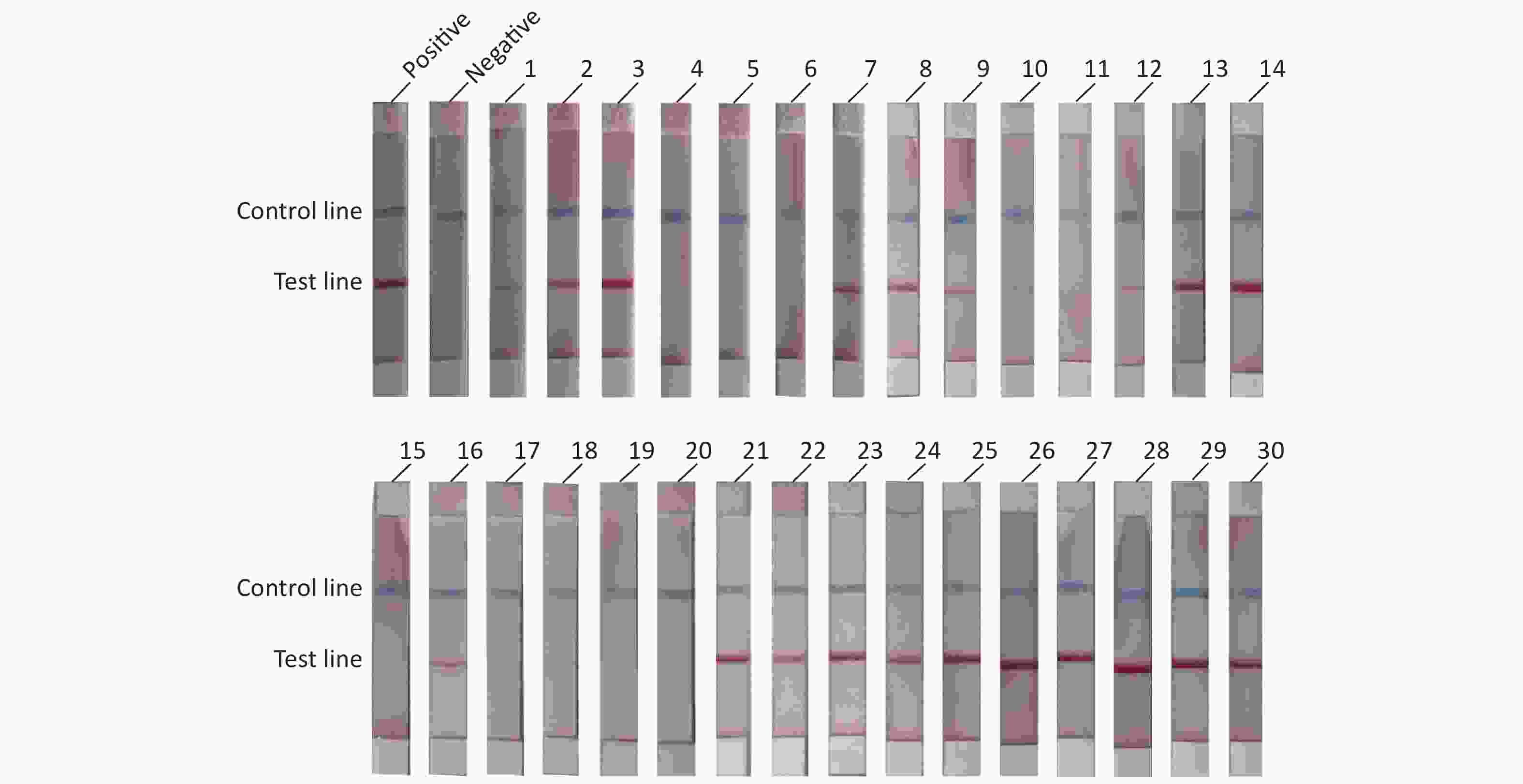
Figure 5. RAA-LFD test results of clinical samples.
Table 3. Diagnostic performance comparison between RAA-LFD and qPCR assays
Variables qPCR Total Performance characteristics (%, 95% CI) Positive Negative Sensitivity Specificity RAA Positive 21 0 21 91.3 (72.0–98.9) 100.0 (59.0–100.0) Negative 2 7 9 Total 23 7 30 -
There are different strains of ASFV that can infect swine, which indicates that the clinical manifestations of ASF can vary. In addition, the manifestations of ASF may be similar to other swine virus infections. Thus, it is difficult to accurately determine whether an animal has been infected with ASF based on its clinical manifestations. Because ASF is able to lurk for 4–20 days in infected animals, the detection of the disease poses a great challenge for the control efforts of ASF in the world. Large-scale pig farms have access to full laboratories that are capable of using qPCR for the rapid detection of ASFV. However, both conventional PCR and qPCR methods require expensive precise instruments and experienced examiners to read the results, which may not be available in rural and remote areas. Farmers who do not have suitable equipment for testing for ASFV in a timely manner because of cost considerations and instrument limitations may miss optimal testing times and, thus, increase the risk of the spread of ASF.
The development of isothermal amplification technology makes it possible to detect and identify viruses in environments without large laboratories. There has been a rapid evolution of isothermal nucleic acid amplification techniques, such as loop-mediated isothermal amplification (LAMP), nucleic acid sequence-based amplification (NASBA), rolling circle amplification (RCA), the helicase-dependent isothermal amplification of DNA (HDA), strand displacement amplification (SDA), and RPA. Compared with LAMP, RAA has a shorter reaction time, lower reaction temperature, and a simpler primer design. The reaction time of RCA is 2–16 h, so compared with RCA, the amplification speed of RAA is faster. NASBA is mainly used for isothermal amplification of RNA. SDA needs a fluorescence spectrophotometer is required to detect the product. HDA needs to react at 60–65 ℃ for 75–90 min. RAA has a lower reaction temperature and shorter reaction time than HDA. Therefore, RAA is a suitable choice for the detection of ASFV.
There are many studies on different assays used for the detection of ASFV. For example, Wang et al. developed a LAMP assay that targeted the p10 gene sequence of the ASFV[18]. Sastre et al. developed a lateral flow assay (LFA) to detect antigens against the VP72 protein of the ASFV[19]. Erickson et al. developed a multiplex reverse transcription PCR and an automated electronic microarray assay and established a multichannel detection system for the detection and discrimination of seven viruses, including ASFV, that affect swine[20]. However, there are few reports on the application of RAA-LFD for the detection of ASFV. Compared with other detection methods, such as the LAMP method[18], single-step multiplex qPCR[21], droplet digital PCR (ddPCR)[22], and polymerase cross-linking spiral reaction (PCLSR)[23], the RAA-LFD method is capable of complete amplification within 10 min under constant temperature conditions and, thus, offers distinct advantages over PCR, which requires complex instruments and is a very time-consuming process.
The experimentally established ASFV assay completed the assay at a constant temperature of 37 °C for 10 min, achieved a lower limit of detection of 2 × 102 copies/μL, and did not cross-react with CSFV, PPV, PRV, PCV2, or PRRSV. The assay is a more convenient and less time-consuming method for detecting the ASFV than the qPCR method that is recommended by the OIE. It also provided technical support for the rapid detection of ASFV in a rudimentary environment.
doi: 10.3967/bes2022.018
Development of a Recombinase-aided Amplification Combined With Lateral Flow Dipstick Assay for the Rapid Detection of the African Swine Fever Virus
-
Abstract:
Objective To establish a sensitive, simple and rapid detection method for African swine fever virus (ASFV) B646L gene. Methods A recombinase-aided amplification-lateral flow dipstick (RAA-LFD) assay was developed in this study. Recombinase-aided amplification (RAA) is used to amplify template DNA, and lateral flow dipstick (LFD) is used to interpret the results after the amplification is completed. The lower limits of detection and specificity of the RAA assay were verified using recombinant plasmid and pathogenic nucleic acid. In addition, 30 clinical samples were tested to evaluate the performance of the RAA assay. Results The RAA-LFD assay was completed within 15 min at 37 °C, including 10 min for nucleic acid amplification and 5 minutes for LFD reading results. The detection limit of this assay was found to be 200 copies per reaction. And there was no cross-reactivity with other swine viruses. Conclusion A highly sensitive, specific, and simple RAA-LFD method was developed for the rapid detection of the ASFV. -
Figure 3. Optimal reaction conditions of ASFV-RAA-LFD. (A) Optimal detection temperature of ASFV-RAA-LFD assay. From left to right were the experimental results at 36 °C, 37 °C, 38 °C, 39 °C, 40 °C, and 41 °C; (B) Optimal detection time of ASFV-RAA-LFD assay.
Figure 4. (A) Evaluation of the sensitivity of ASFV-RAA-LFD. Concentration gradient of pMD18t-p72 (copies/μL) were as follow: 1: 2 × 101; 2: 2 × 102; 3: 2 × 103; 4: 2 × 104; 5: 2 × 105; 6: 2 × 106; 7: 2 × 107; (B) Evaluation of the specificity of ASFV-RAA-LFD.
Table 1. Primers and probes for screening
Primer/probe Sequence (5′→3′) F1 TGTAACGCAGCACAGCTGAACCGTTCTGAAGAAGA F2 AACCGTTCTGAAGAAGAAGAAAGTTAATAG F3 GATACCACAAGATCRGCCGTAGTGATAGACCCCACGT F4 AGCAGTTACGGAAATGTTTTTAATAATAGGTAATGTGATC F5 CGGAAATGTTTTTAATAATAGGTAATGTGATCGGATA F6 ATCGGATACGTAACGGGGCTAATATCAGATA F7 AACGGGGCTAATATCAGATATAGATGAACA F8 CGGGGCTAATATCAGATATAGATGAACATG F9 CGTCTGGAAGAGCTGTATCTCTATCCTGAA F10 GCTGTATCTCTATCCTGAAAGCTTATCTCTG F11 AAGAAGAAAGTTAATAGCAGATGCCGATACCAC F12 CCGATCACATTACCTATTATTAAAAACATTTCC R1 Biotin-CAGAGCAAGAGAATTTTATATTAGTTGGGA R2 Biotin-CGGGAGGAATACCAACCCAGTGGTCATATTAACGT R3 Biotin-TGCTTTGAAGCCACGGGAGGAATACCAACC R4 Biotin-CTGCTCATGGTATCAATCTTATCGATAAATTTCCA R5 Biotin-CCCATTGAATATATGTTTATAGGATTAAAACCTACCTG R6 Biotin-ATGGCCCATTGAATATATGTTTATAGGATTAAAACCTACC R7 Biotin-GCTCTTACATACCCTTCCACTACGGAGGCAATG R8 Biotin-ATAAAATTCTCTTGCTCTGGATACGTTAATATG P1 Fam-ATCGGATACGTAACGGGGCTAATATCAGATA/THF/AGATGAACATGCGTC-P P2 Fam-CGTCTGGAAGAGCTGTATCTCTATCCTGAA/THF/GCTTATCTCTGCGTG-P P3 Fam-GCTTATCTCTGCGTGGTGAGTGGGCTGCATA/THF/TGGCGTTAACAACAT-P P4 Fam-GATGAACATGCGTCTGGAAGAGCTGTATCTCTA/THF/CCTGAAAGCTTATCT-P P5 Fam-TGGGTTGGTATTCCTCCCGTGGCTTCAAAGCAAAG/THF/TAATCATCATCGCAC-P P6 Fam-GGTGCGATGATGATTACCTTTGCTTTGAAG/THF/CACGGGAGGAATACCA  下载: 导出CSV
下载: 导出CSV
Table 2. Primers and probe sequences used for qPCR assay
Primer/Probe Sequences(5′→3′) qPCR F CTGCTCATGGTATCAATCTTATCGA qPCR R GATACCACAAGATCRGCCGT qPCR P FAM-CCACGGGAGGAATACCAACCCAGTG-TAMRA
下载: 导出CSV
Table 3. Diagnostic performance comparison between RAA-LFD and qPCR assays
Variables qPCR Total Performance characteristics (%, 95% CI) Positive Negative Sensitivity Specificity RAA Positive 21 0 21 91.3 (72.0–98.9) 100.0 (59.0–100.0) Negative 2 7 9 Total 23 7 30
下载: 导出CSV
-
[1] Galindo I, Alonso C. African swine fever virus: a review. Viruses, 2017; 9, 103. doi: 10.3390/v9050103 [2] Rojo G, Garcı́a-Beato R, Viñuela E, et al. Replication of African swine fever virus DNA in infected cells. Virology, 1999; 257, 524−36. doi: 10.1006/viro.1999.9704 [3] Bastos ADS, Penrith ML, Crucière C, et al. Genotyping field strains of African swine fever virus by partial p72 gene characterisation. Arch Virol, 2003; 148, 693−706. doi: 10.1007/s00705-002-0946-8 [4] Malogolovkin A, Burmakina G, Titov I, et al. Comparative analysis of African swine fever virus genotypes and serogroups. Emerg Infect Dis, 2015; 21, 312−5. doi: 10.3201/eid2102.140649 [5] Olesen, AS, Lohse L, Boklund A, et al. Transmission of African swine fever virus from infected pigs by direct contact and aerosol routes. Vet Microbiol, 2017; 211, 92−102. doi: 10.1016/j.vetmic.2017.10.004 [6] Gallardo C, Fernández-Pinero J, Arias M. African swine fever (ASF) diagnosis, an essential tool in the epidemiological investigation. Virus Res, 2019; 271, 197676. doi: 10.1016/j.virusres.2019.197676 [7] Shi XJ, Liu XM, Wang Q, et al. A multiplex real-time PCR panel assay for simultaneous detection and differentiation of 12 common swine viruses. J Virol Methods, 2016; 236, 258−65. doi: 10.1016/j.jviromet.2016.08.005 [8] Lokhandwala S, Waghela SD, Bray J, et al. Adenovirus-vectored novel African Swine Fever Virus antigens elicit robust immune responses in swine. PLoS One, 2017; 12, e0177007. doi: 10.1371/journal.pone.0177007 [9] Petrini S, Feliziani F, Casciari C, et al. Survival of African swine fever virus (ASFV) in various traditional Italian dry-cured meat products. Prev Vet Med, 2019; 162, 126−30. doi: 10.1016/j.prevetmed.2018.11.013 [10] Frączyk M, Woźniakowski G, Kowalczyk A, et al. Development of cross-priming amplification for direct detection of the African Swine Fever Virus, in pig and wild boar blood and sera samples. Lett Appl Microbiol, 2016; 62, 386−91. doi: 10.1111/lam.12569 [11] Agüero M, Fernández J, Romero L, et al. Highly sensitive PCR assay for routine diagnosis of African swine fever virus in clinical samples. J Clin Microbiol, 2003; 41, 4431−4. doi: 10.1128/JCM.41.9.4431-4434.2003 [12] Zsak L, Borca MV, Risatti GR, et al. Preclinical diagnosis of African swine fever in contact-exposed swine by a real-time PCR assay. J Clin Microbiol, 2005; 43, 112−9. doi: 10.1128/JCM.43.1.112-119.2005 [13] Liu LH, Atim S, LeBlanc N, et al. Overcoming the challenges of pen-side molecular diagnosis of African swine fever to support outbreak investigations under field conditions. Transbound Emerg Dis, 2019; 66, 908−14. doi: 10.1111/tbed.13103 [14] Wang JC, Wang JF, Li RW, et al. Rapid and sensitive detection of canine distemper virus by real-time reverse transcription recombinase polymerase amplification. BMC Vet Res, 2017; 13, 241. doi: 10.1186/s12917-017-1180-7 [15] Hou PL, Wang HM, Zhao GM, et al. Rapid detection of infectious bovine Rhinotracheitis virus using recombinase polymerase amplification assays. BMC Vet Res, 2017; 13, 386. doi: 10.1186/s12917-017-1284-0 [16] Zeng FW, Wu ML, Ma L, et al. Rapid and sensitive real-time recombinase polymerase amplification for detection of Marek's disease virus. Mol Cell Probes, 2019; 48, 101468. doi: 10.1016/j.mcp.2019.101468 [17] Liu LB, Wang JF, Zhang RX, et al. Visual and equipment-free reverse transcription recombinase polymerase amplification method for rapid detection of foot-and-mouth disease virus. BMC Vet Res, 2018; 14, 263. doi: 10.1186/s12917-018-1594-x [18] Wang DG, Yu JH, Wang YZ, et al. Development of a real-time loop-mediated isothermal amplification (LAMP) assay and visual LAMP assay for detection of African swine fever virus (ASFV). J Virol Methods, 2020; 276, 113775. doi: 10.1016/j.jviromet.2019.113775 [19] Sastre P, Gallardo C, Monedero A, et al. Development of a novel lateral flow assay for detection of African swine fever in blood. BMC Vet Res, 2016; 12, 206. doi: 10.1186/s12917-016-0831-4 [20] Erickson A, Fisher M, Furukawa-Stoffer T, et al. A multiplex reverse transcription PCR and automated electronic microarray assay for detection and differentiation of seven viruses affecting swine. Transbound Emerg Dis, 2018; 65, e272−83. doi: 10.1111/tbed.12749 [21] Haines FJ, Hofmann MA, King DP, et al. Development and validation of a multiplex, real-time RT PCR assay for the simultaneous detection of classical and African swine fever viruses. PLoS One, 2013; 8, e71019. doi: 10.1371/journal.pone.0071019 [22] Wu XL, Xiao L, Lin H, et al. Development and application of a droplet digital polymerase chain reaction (ddPCR) for detection and investigation of African swine fever virus. Can J Vet Res, 2018; 82, 70−4. [23] Woźniakowski G, Frączyk M, Kowalczyk A, et al. Polymerase cross-linking spiral reaction (PCLSR) for detection of African swine fever virus (ASFV) in pigs and wild boars. Sci Rep, 2017; 7, 42903. doi: 10.1038/srep42903 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 2094
- HTML全文浏览量: 899
- PDF下载量: 198
- 被引次数: 0

 Quick Links
Quick Links