

 下载:
下载:



-
Proteus mirabilis causes complex urinary tract infections, which may lead to sepsis or systemic inflammatory response syndrome, with a fatality rate of 20%–50% [1]. P. mirabilis, the most common pathogen of the genus Proteus, is a member of the Enterobacteriaceae that causes urinary system infections, second only to Escherichia coli. P. mirabilis accounts for 3% of nosocomial infections [2]. With the widespread irregular use of antibiotics, P. mirabilis antibiotic resistance has increased. Multidrug-resistant P. mirabilis producing broad-spectrum β-lactamase was reported for the first time in Italy [3]. Subsequently, Poland, China, Japan, and other countries and regions have reported multidrug-resistant P. mirabilis [4-8], showing global spread. Due to the continuous increase in antibiotic resistance, the prevention and control of P. mirabilis infections is challenging. Therefore, high-resolution P. mirabilis typing is needed.
High-resolution sequence typing is important to assess the epidemiological links and identify possible sources of transmission during outbreaks. Currently, multilocus sequence typing (MLST) [9, 10] and pulsed-field gel electrophoresis (PFGE) [11-13] are the two most commonly used typing methods for outbreak investigations. However, their main disadvantages are the low resolution in MLST [14, 15] and difficulties in standardization of PFGE [16]. To the best of our knowledge, there are few studies on MLST of P. mirabilis, and most studies have focused on MLST of carbapenem-resistant Gram-negative clinical isolates containing P. mirabilis [9, 10].
In recent years, with the continuous development of genomic epidemiology, the gene-by-gene (GbG) method has been advocated as an extension of MLST [17]. This method has the advantages of portability, scalability, and independence. It is regarded by PulseNet International as an important method for the identification of bacterial strains by high-throughput sequencing. The open source chewBBACA algorithm [18] provides a simple bioinformatics pipeline to construct cgMLST schemes of target strains. To type P. mirabilis globally and monitor the clonal groups (CGs) of potentially high-risk P. mirabilis, we used chewBBACA as a bioinformatics pipeline for GbG analysis to construct and verify the cgMLST scheme of P. mirabilis. Our results support the prevention and control of P. mirabilis infections.
-
Genomic sequences available as of September 10, 2021, for P. mirabilis (Enterobacteria) were downloaded from the National Center for Biotechnology Information (NCBI) genome sequence repository (
http://www.ncbi.nlm.nih.gov/genome/ ). The downloaded sequences included 74 complete genomes and 679 whole genome shotgun sequences available as scaffolds (116), chromosomes (6), or contigs (557). After removing low-quality assemblies (gene assembly rate < 95% or contamination rate > 5%), all remaining genomes were included in the preliminary analysis. -
In the present study, a chewBBACA suite was designed to create and evaluate the core genome GbG typing scheme, followed by allele calling in available P. mirabilis strains. ChewBBACA performs scheme creation and allele calls for complete or draft genomes generated by de novo assembly. Before starting, we created a training file in Prodigal v2.6.3 [19] using the reference genome HI4320 of P. mirabilis. The genome sequence of P. mirabilis HI4320 (RefSeq assembly accession: GCF_000069965.1) was used only as a reference genome to predict the whole genome MLST (wgMLST) loci and was then removed from further analysis.
A total of 72 complete genome sequences were annotated in the first step. In this step, the algorithm defined the coding sequences (CDSs) of each genome, and then all CDSs in the genome were compared in a pairwise manner to generate a single FASTA file containing the selected CDSs. Then, a two-step evaluation process was conducted to identify all CDSs in the genomes. First, CDSs that have the same sequence but are smaller than other CDSs were removed, and the larger CDSs were retained. Second, the remaining CDSs were gathered in unique loci by performing an all-against-all BLASTP search and calculating the blast score ratio (BSR) [20]. CDSs with a pairwise BSR of > 0.6 were considered to be alleles at the same locus, and the larger allele (in bp) was retained. The remaining wgMLST loci were used to define the cgMLST scheme. We selected candidate loci from 100% (threshold) of the available complete genomes (72 genomes) to define our cgMLST scheme. The second step was validation of candidate loci based on a total of 635 unfinished P. mirabilis genomes. We reduced stringency and maintained candidate loci shared by 99% of the isolates. This cutoff was chosen based on the quality of the tested genomes, since the sequences used in this step were incomplete genomes. ChewBBACA is available at
https://github.com/B-UMMI/chewBBACA . -
To define CGs in a non-arbitrary manner, we analyzed the distribution of the number of allelic mismatches (loci with different sequences) among the pairs of genomes. We performed a single-link algorithm designed to classify isolates from cgMLST data. Similar to the classification of classical MLST data, the single-link algorithm was also used for cgMLST data to accurately classify isolates [21].
-
A minimum spanning tree (MST) was constructed using the MSTree method in GrapeTree (version 2.1) for the allelic profiles obtained for each isolate by the cgMLST scheme.
-
Based on our cgMLST scheme, MSTs for different geographical locations were built to investigate possible evolutionary relationships. We focused on the distribution of CGs in different countries or regions and examined the shared and unique CGs between them.
-
We used the ABRicate pipeline (
https://github.com/tseemann/abricate ) to annotate the antibiotic resistance genes (ARGs) and virulence genes in the P. mirabilis genome using the CARD (https://card.mcmaster.ca/ ) and VFDB (http://www.mgc.ac.cn/VFs/main.htm ) databases. The annotation parameters for ARGs and virulence genes were gene coverage > 60% and identity > 70%. -
As of September 10, 2021, 753 genome sequences were available at the NCBI genome sequence repository (
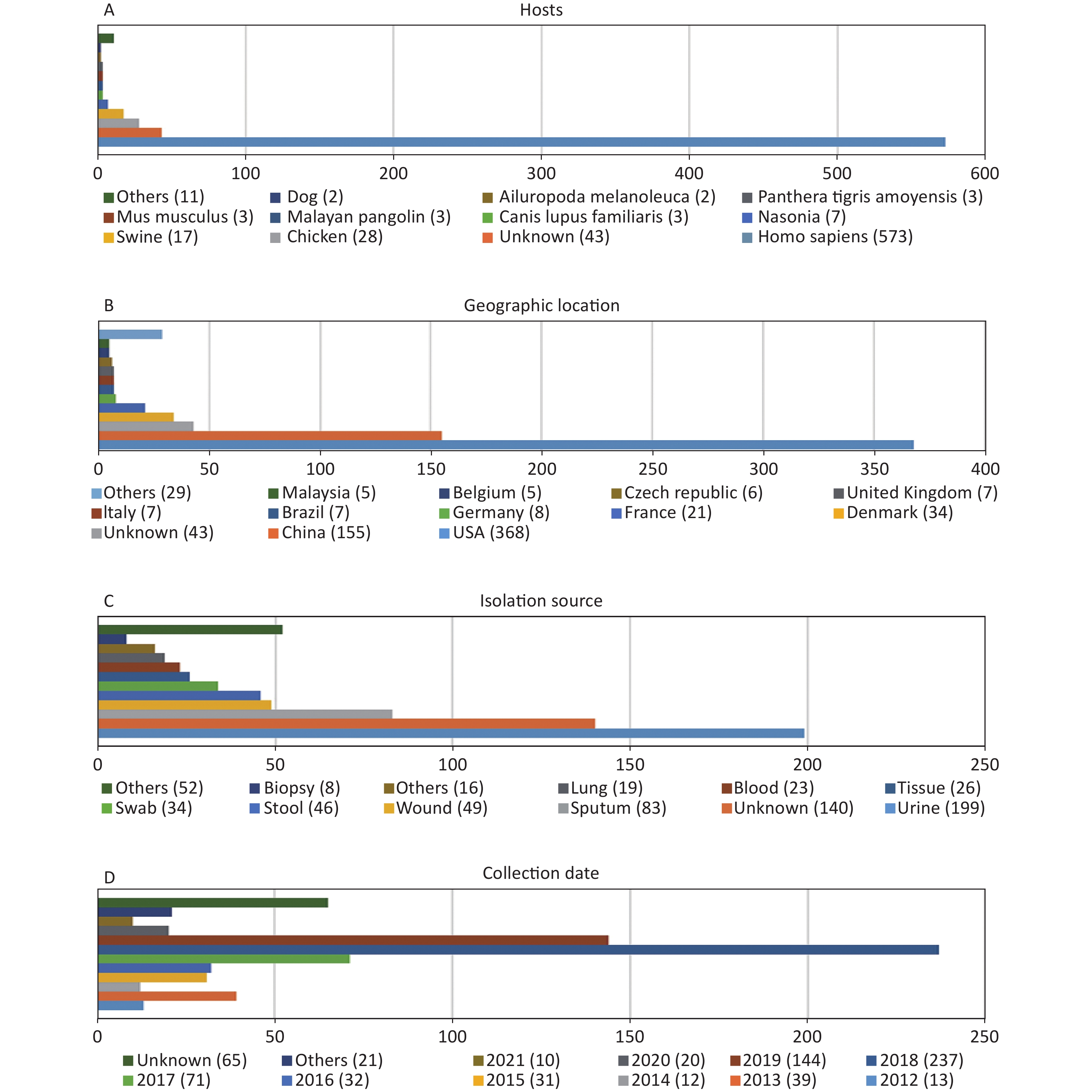
http://www.ncbi.nlm.nih.gov/genome/ ). Of these genomes, 45 were not included due to the low assembly quality (Supplementary Table S1 available in www.besjournal.com). The genome sequences of 12 P. mirabilis strains were excluded during the cgMLST scheme validation process (Supplementary Table S1 ). According to our cgMLST scheme, reference genome HI4320 was not included. Finally, 695 P. mirabilis strains were included in the analysis according to our cgMLST scheme. Information regarding all strains is provided inSupplementary Table S2 (available in www.besjournal.com). As shown in Figure 1, the majority of P. mirabilis isolates were collected from humans (82.4%), most samples were collected in the USA or China (75.3%), urine and sputum samples were the most common (40.6%), and most samples were collected in 2018 or 2019 (54.8%).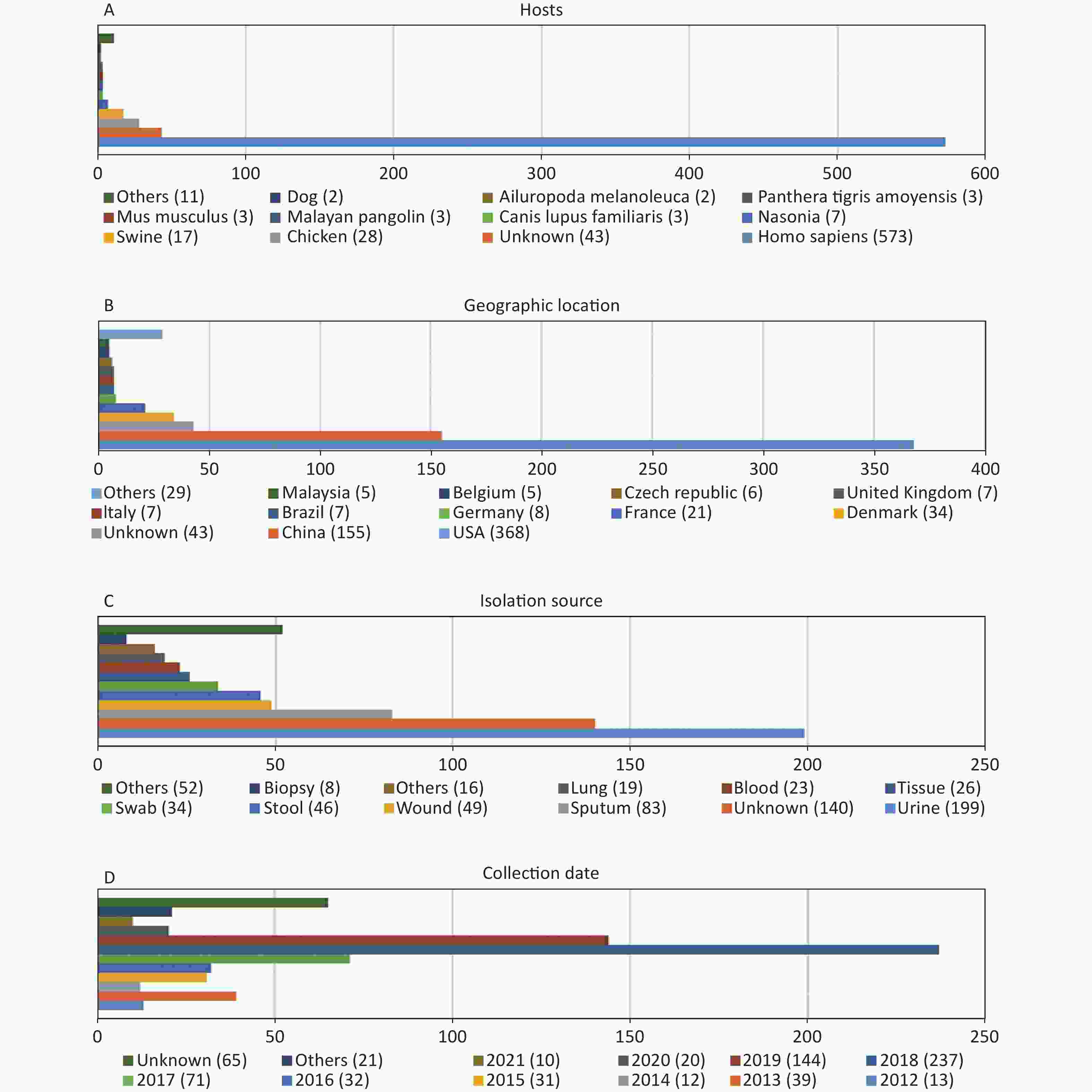
Figure 1. Characterization of the P. mirabilis isolates included in this study. (A) Hosts of isolated P. mirabilis. “Others” represents host types with only one strain. (B) Geographic distribution of available genomes. “Others” represents countries with less than five strains. (C) Isolation source distribution of isolates. “Others” represents sources with less than eight strains. (D) Temporal distribution of P. mirabilis isolates. “Others” represents years when less than two strains were collected.
-
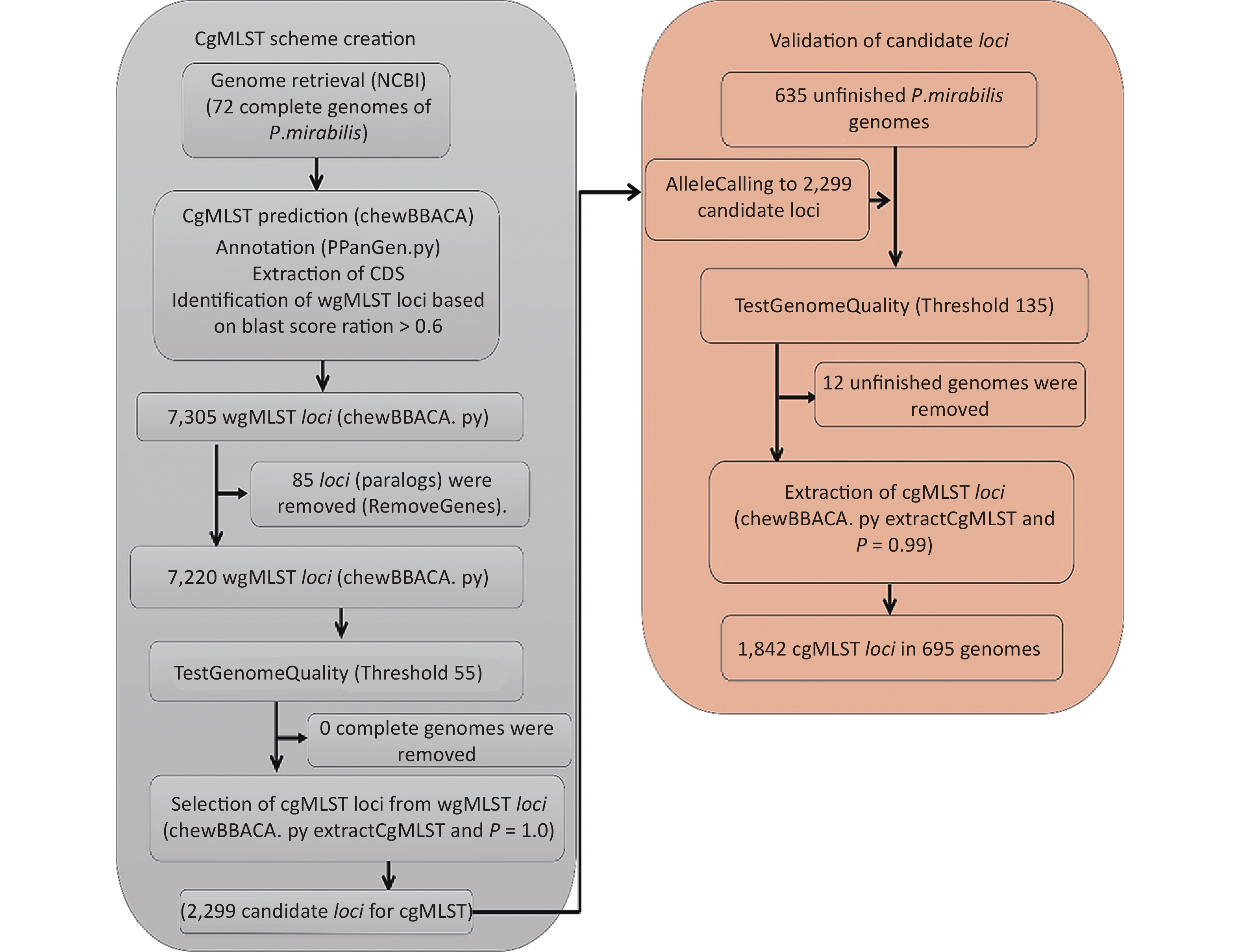
A flowchart of the cgMLST scheme of P. mirabilis (
https://github.com/B-UMMI/chewBBACA ) is provided in Figure 2. In total, 72 complete genome sequences were retrieved. A total of 7,305 target loci were generated and annotated using the wgMLST dataset. Using the RemoveGenes operation to remove possible paralogous loci from the wgMLST scheme, we discarded 85 loci. We filtered out an additional 4,921 loci using the genome quality test (threshold 55). A total of 2,299 candidate loci for cgMLST were retained, which were present in 72 complete genome sequences (Supplementary Figure S1 available in www.besjournal.com). During the creation of the scheme, no genome sequence was removed.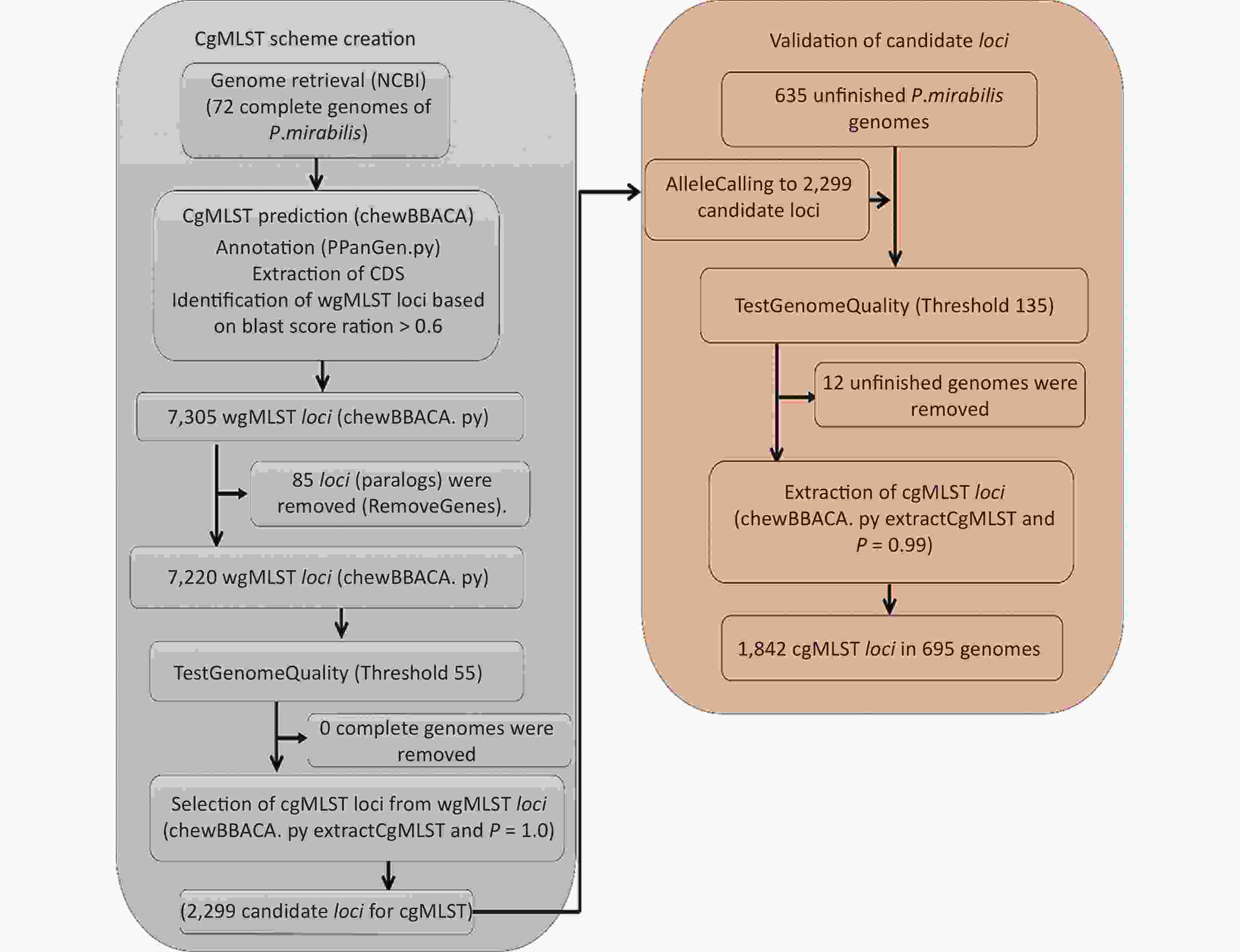
Figure 2. Flowchart describing the development of the cgMLST scheme for P. mirabilis using chewBBACA (
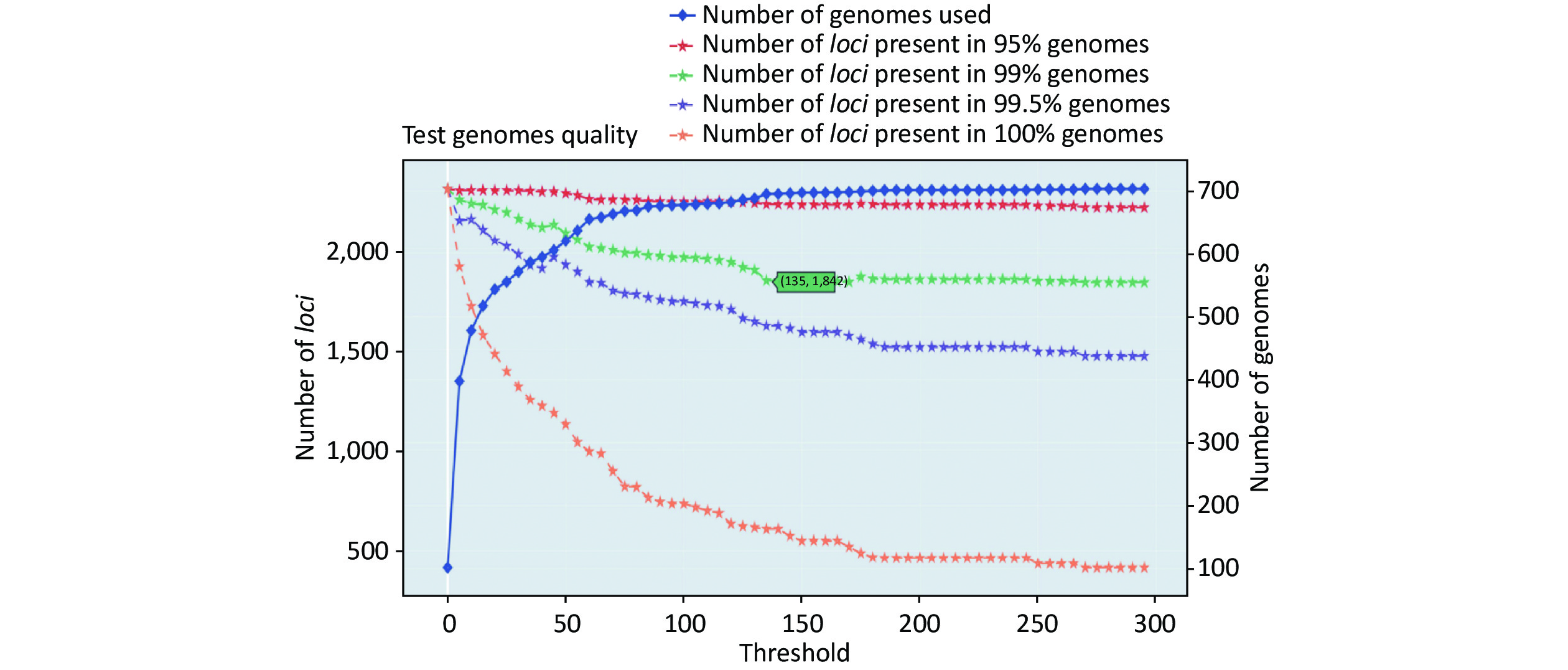
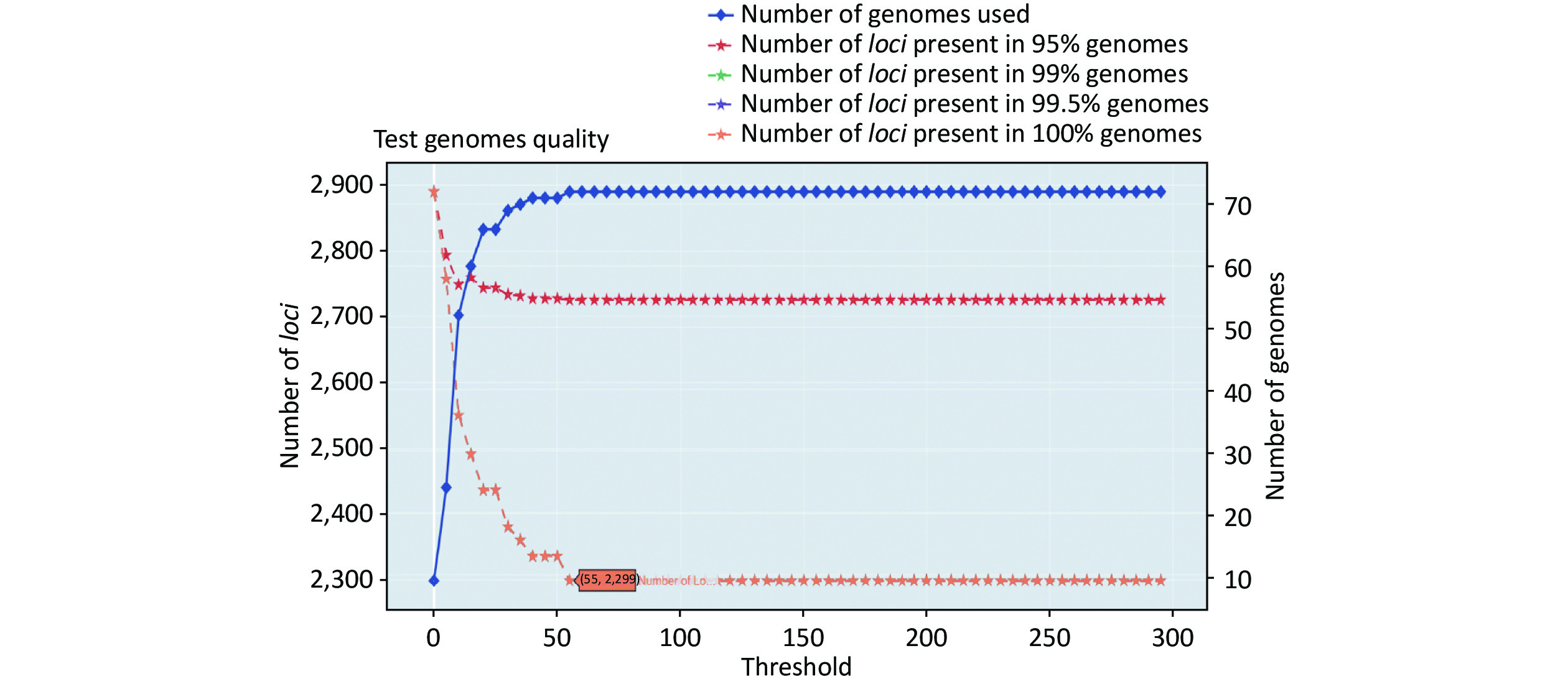
https://github.com/B-UMMI/chewBBACA ).We used the 635 unfinished P. mirabilis genomes for the validation of the cgMLST scheme. The TestGenomeQuality operation was used to select loci present in a predetermined percentage (99%) of the analyzed strains from the cgMLST scheme (Supplementary Figure S2 available in www.besjournal.com). During the validation step, 12 unfinished genomes were removed. In the end, 1,842 cgMLST loci were selected. Following this step, a cgMLST scheme consisting of 1,842 gene targets was defined, covering 49.91% of the 3,690 open reading frames predicted for the reference strain HI4320.
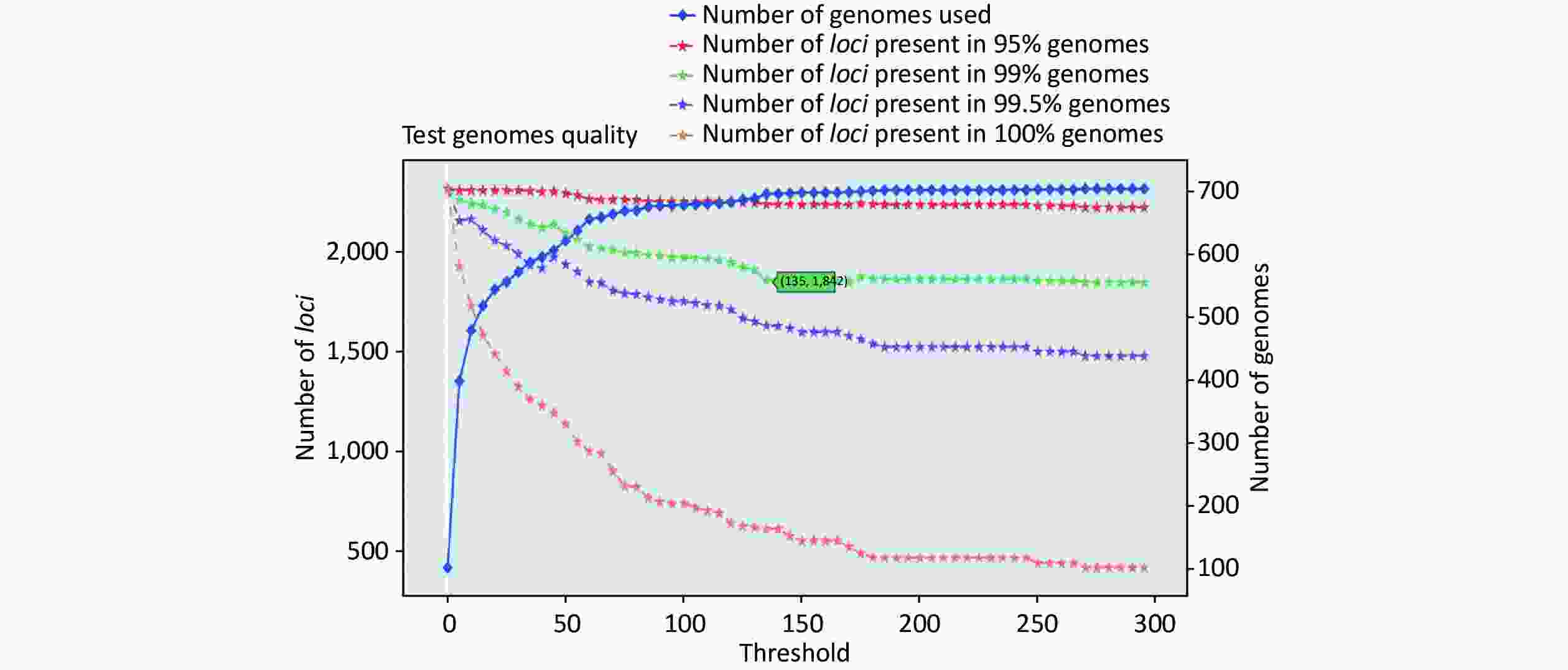
Figure S2. Number of loci and number of genomes at every exclusion threshold level for the 635 validation strains’ genomes and 72 completed genomes that were used to construct the cgMLST scheme
-
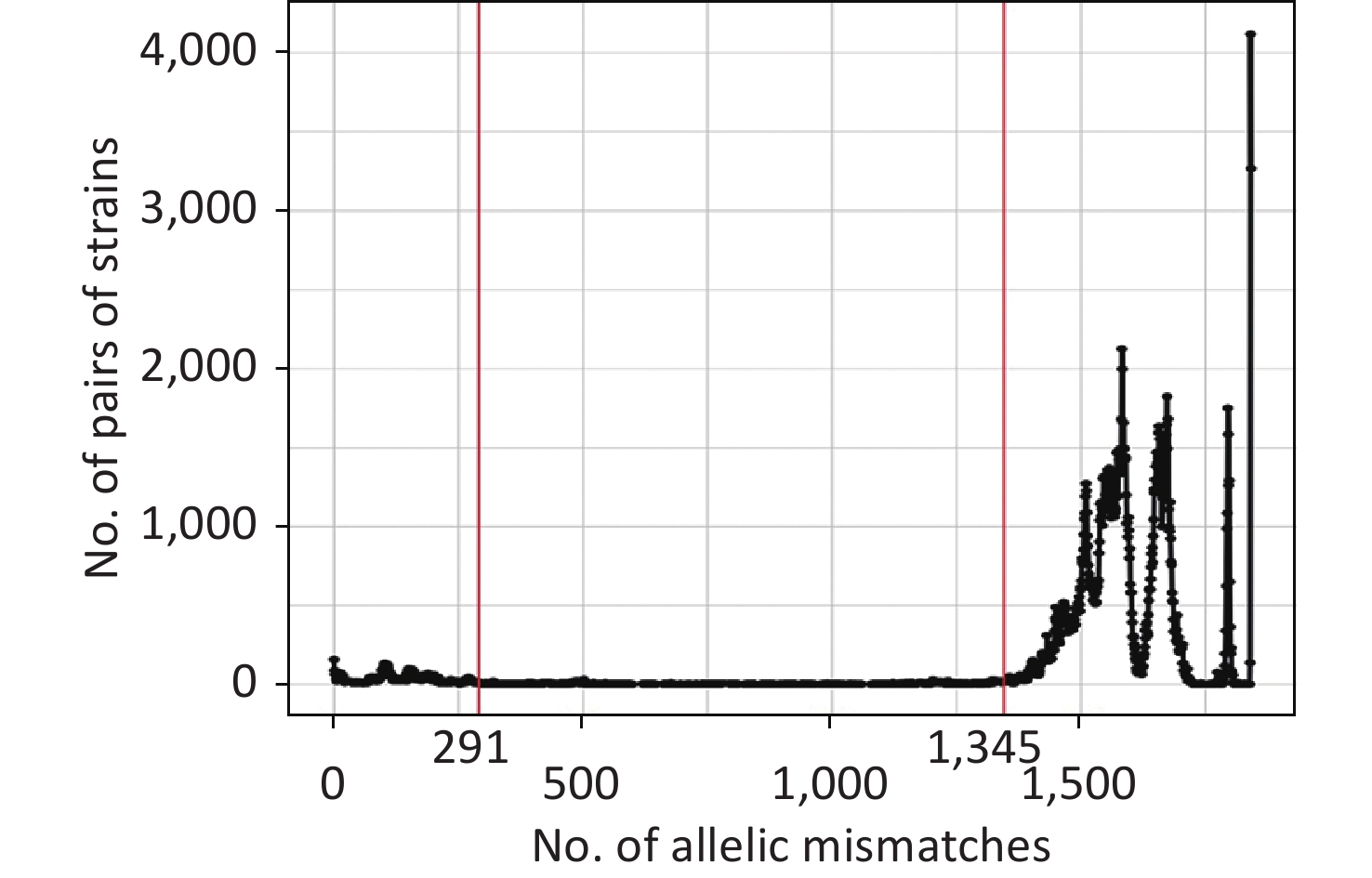
The allelic difference threshold analysis showed a high number of genome pairs with < 291 mismatches or > 1,345 mismatches, and almost no genome pairs had 291–1,345 mismatches (Figure 3). Finally, we defined the P. mirabilis cgMLST CGs as a set of cgMLST profiles having ≤ 291 allelic mismatches with at least one other member of the group. The final 205 CGs were identified based on single-linkage clustering of the cgMLST allele profile.
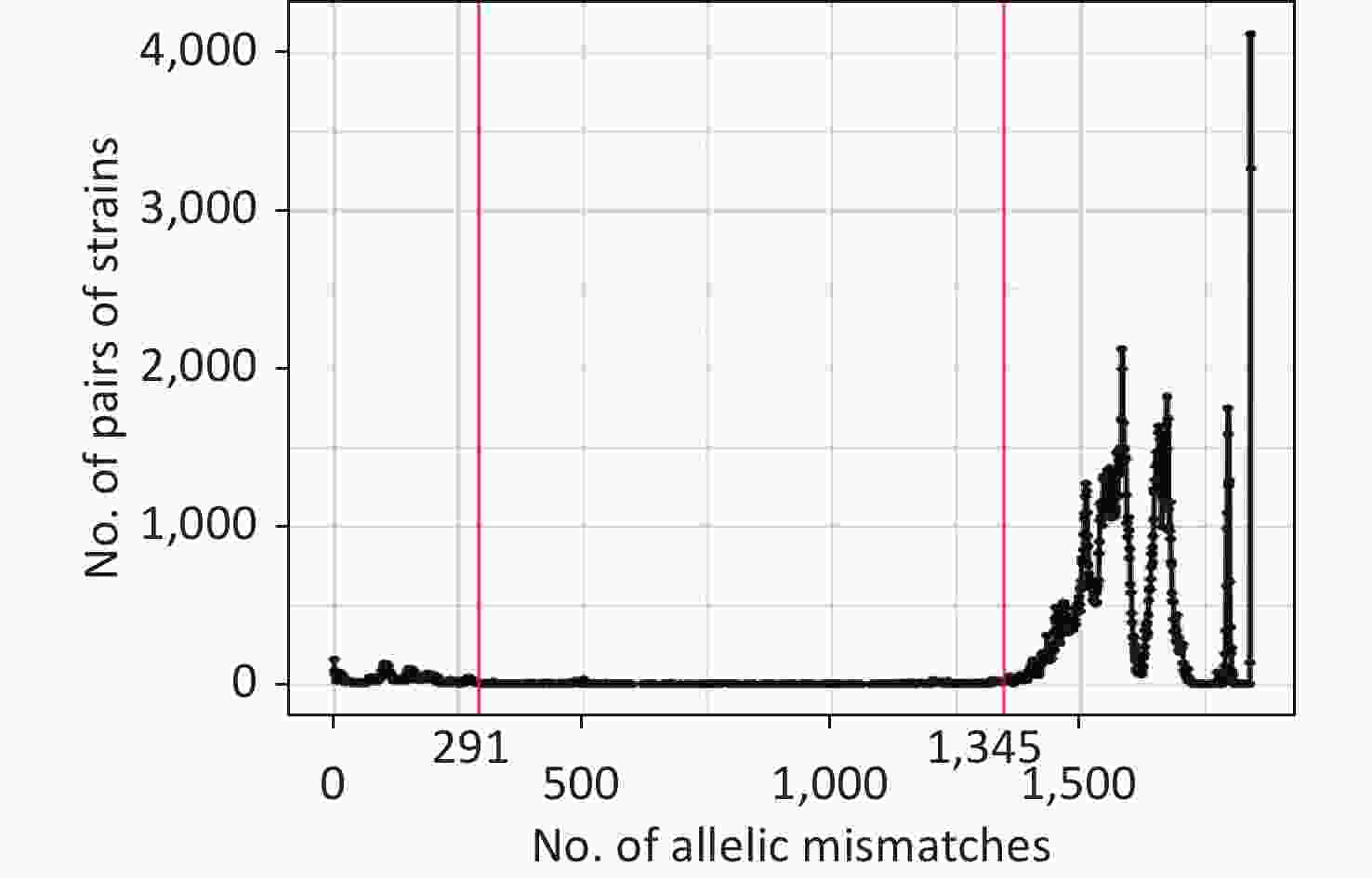
Figure 3. Distribution of the number of paired allele mismatches in P. mirabilis (i.e., the number of different loci for a given pair of strains). The allelic mismatch cutoff value of 291 proposed for the CG definition is shown in red.
-
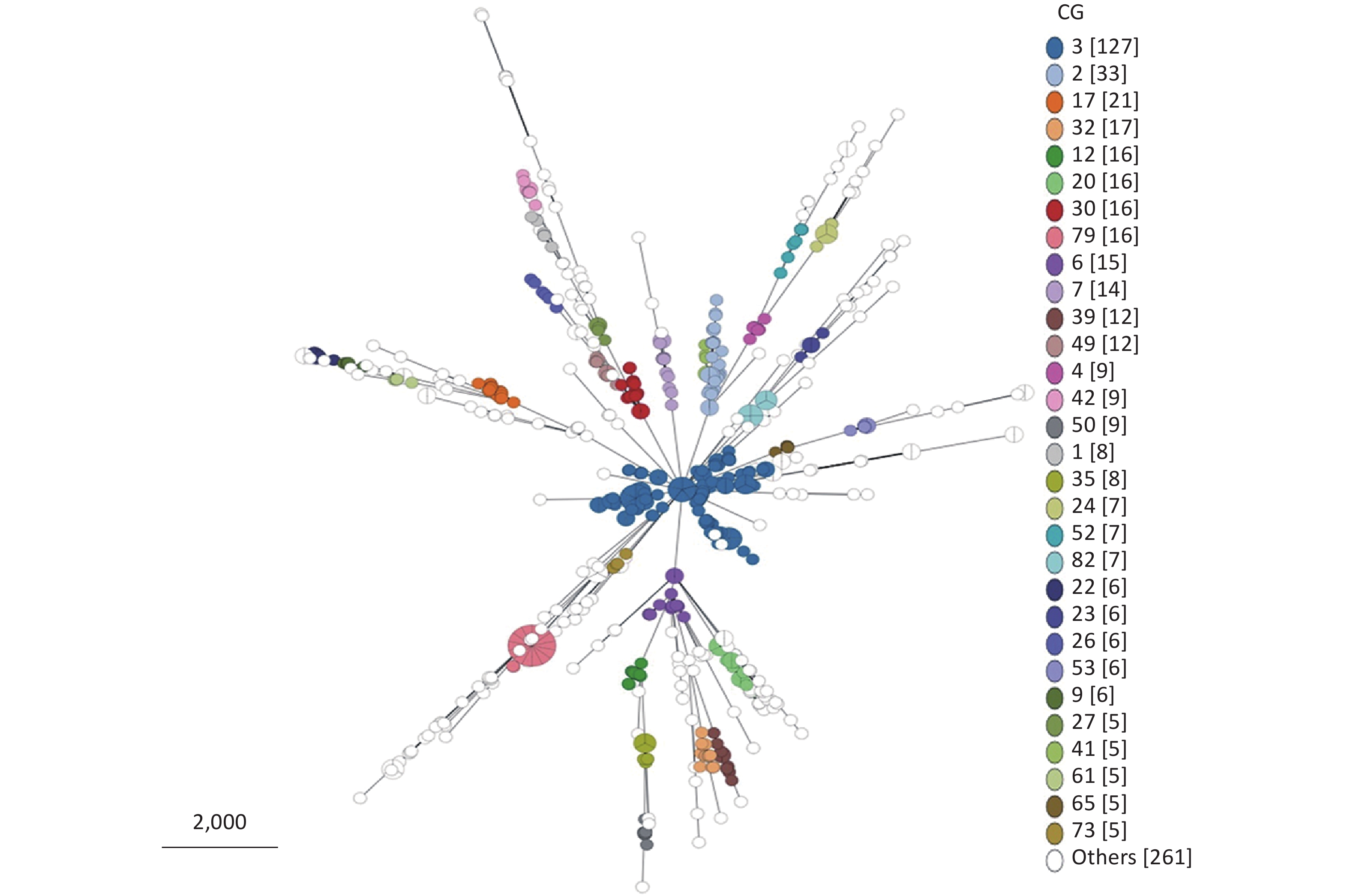
From 695 P. mirabilis strains that conform to the cgMLST scheme, 205 CG types were identified (
Supplementary Table S2 ). Owing to the limitation of the MST visualization, Figure 4 only exhibits the CGs (ranked by the number of strains included in the top 30) distributed in clusters. As shown in Figure 4, CGs exhibited good clustering. In other words, most P. mirabilis strains have been well typed according to our cgMLST scheme.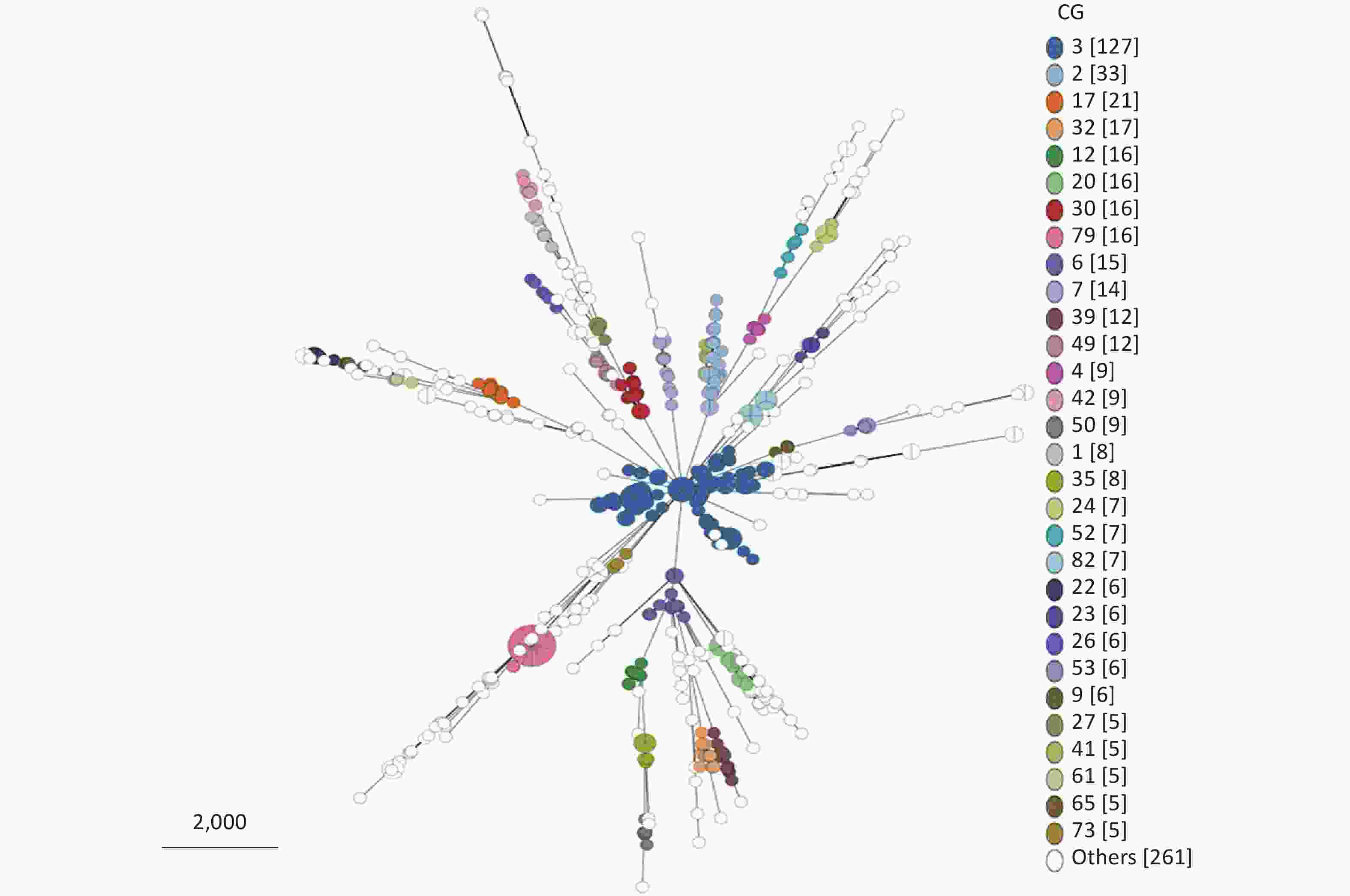
Figure 4. Minimum spanning tree (MST) of CGs generated using the 695 P. mirabilis strains based on the cgMLST scheme. Due to the limitation of the visualization of the MST, only the CG types containing the top 30 strains are displayed. “Others” represents the CG types containing less than five strains.
-
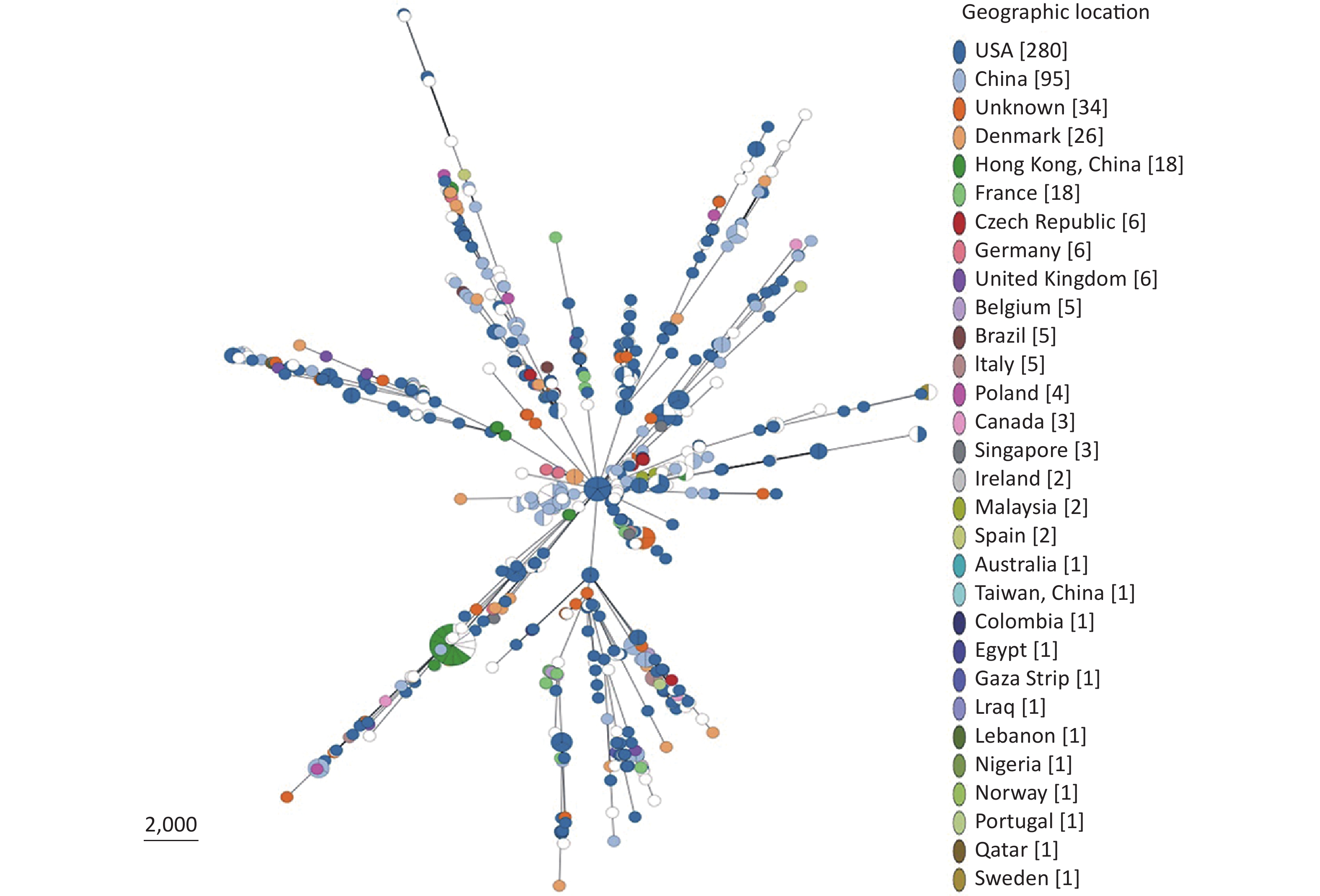
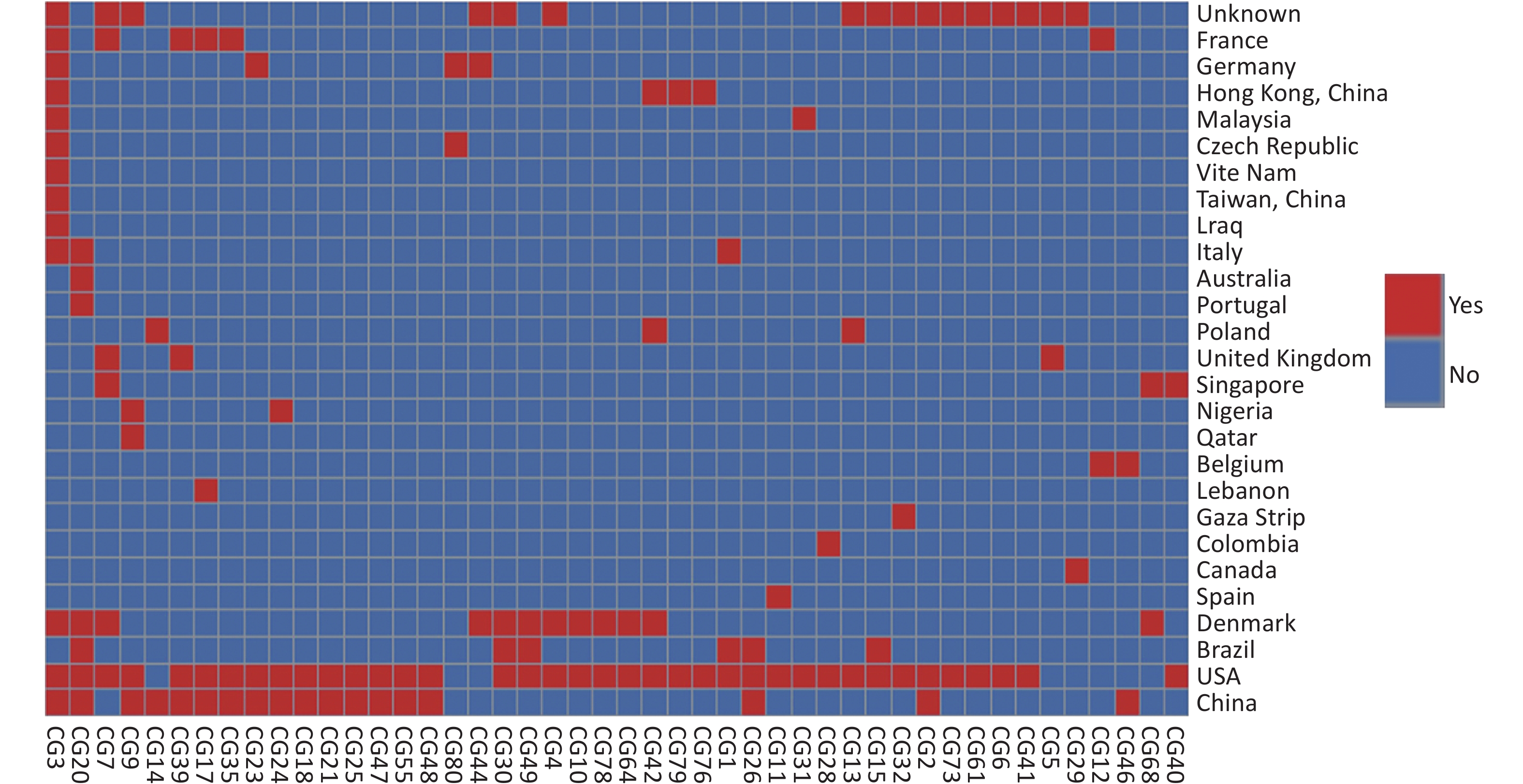
Based on our cgMLST scheme, a minimal generative tree of 695 P. mirabilis strains from different countries or regions showed no close relationships at the geographic level (Figure 5). Overall, it could be seen that P. mirabilis presented a clear global spread, and the root of the MST originated from the USA. However, there are differences between shared CGs and unique CGs in different countries or regions. In total, 46 shared CGs were distributed in 26 countries or regions. CG3 was present in at least 13 countries or regions, CG20 was present in seven countries, and CG7 was present in five countries (Supplementary Figure S3, available in www.besjournal.com). Moreover, 159 unique CGs were distributed in 16 countries (
Supplementary Table S3 , available in www.besjournal.com). The USA had 105 unique CGs, China had 10 unique CGs, and Denmark had 13 unique CGs. Overall, there were regional differences in the distribution of CGs.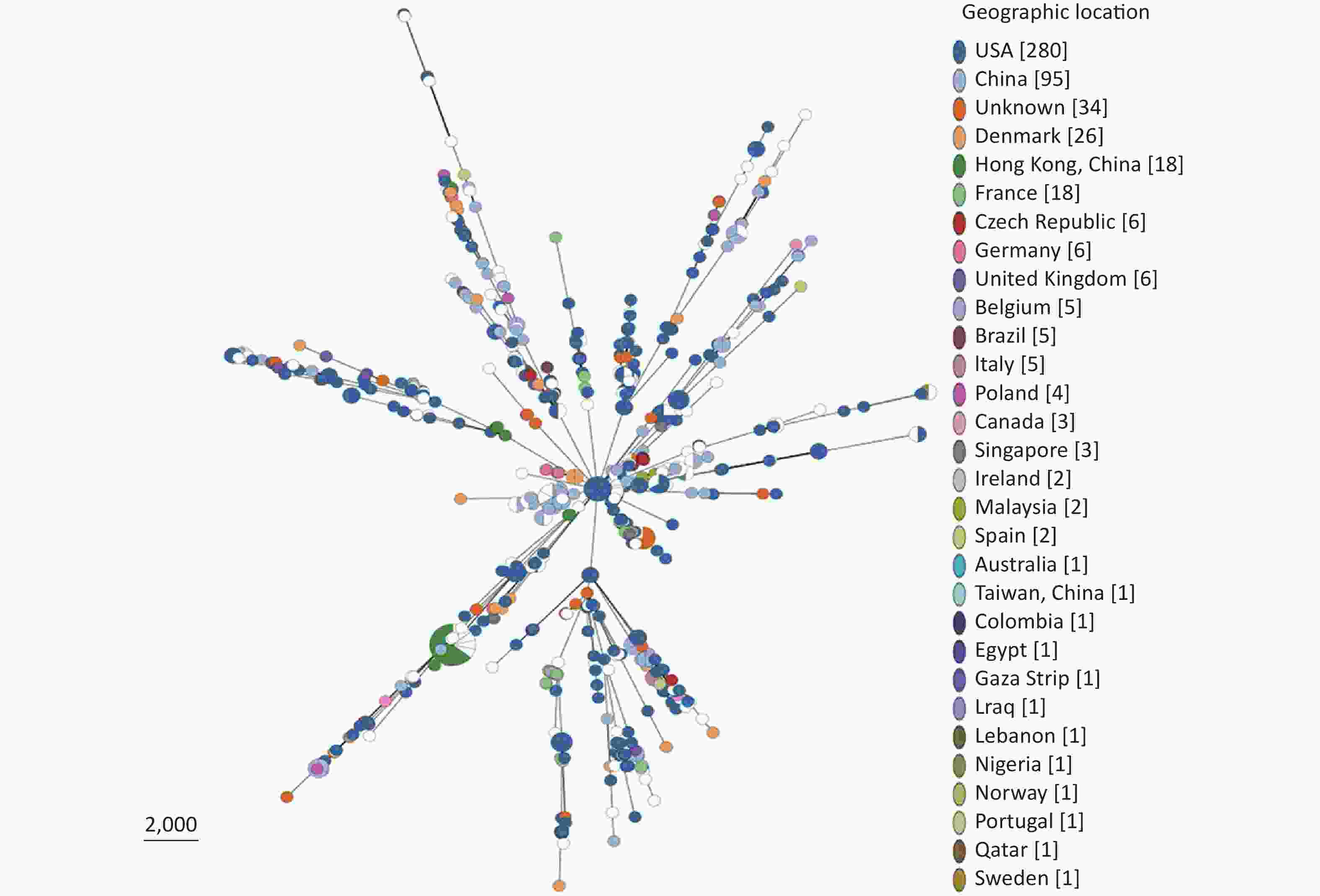
Figure 5. Minimal spanning tree analysis of cgMLST profiles of 695 P. mirabilis strains isolated from different countries or regions. The genome sequences were downloaded from the NCBI database (
https://www.ncbi.nlm.nih.gov/ genome/ ). Colors indicate the geographic locations of the isolates. -
In total, 64 virulence gene types were found in 695 P. mirabilis strains.
Supplementary Table S4 (available in www.besjournal.com) presents the distribution of the 64 virulence gene types in the 205 CGs. The carrier rate of virulence genes cheB, cheY, flgG, flgH, fliI, fliN, flip, gmhA/lpcA, kdsA, lpxC, lpxD, and luxS was 100%. The carrier rate of virulence genes cheW, flgC, flhC, flhD, fliA, fliG, fliM, fliZ, galU, htpB, ompA, and rfaD was > 95%. It is worth noting that CG20 carries nine unique virulence types, namely, papC, papD, papE, papF, papG, papH, papI, papJ, and papK. These nine unique types of virulence genes were present only in CG20 and were P fimbrial operons that cause severe urinary tract infections [22-24]. -
The subtypes of 156 ARGs were found in 695 P. mirabilis strains.
Supplementary Table S5 (available in www.besjournal.com) presents the distribution of the subtypes of 156 ARGs among the 205 CGs. The ARG CRP was present in all CGs. The ARGs H-NS, acrB, cpxA, and tet(J) were present in more than 95% of CGs of P. mirabilis. The total number of ARG subtypes present in different CGs ranged from six to 95. CG3, CG20, CG29, CG32, CG6, CG49, CG41, CG80, CG12, CG7, CG4, CG1, CG61, CG68, CG48, CG28, CG107, and CG11 not only have a large number of shared ARG subtypes, but also unique ARG subtypes. CG3 has 77 shared ARG subtypes and 18 unique ARG subtypes. CG20 has 37 shared ARG subtypes and five unique ARG subtypes. -
The spread of multidrug-resistant P. mirabilis [4-8] is an urgent public health problem. Proper typing of P. mirabilis is important. MLST [9, 10] and PFGE [11-13] are almost incapable of high-resolution typing of P. mirabilis. CgMLST is based on comparison of bacterial whole genome sequences, and has proven to be a promising means for disease outbreak investigation, source tracing, surveillance of bacterial pathogens, and contamination control [25-27]. cgMLST is based on comparison of a complete or draft de novo genome assembly with a scheme consisting of a series of loci and a collection of related allele sequences. This is a suitable and unbiased means to identify possible clusters from a sample of the entire species, and does not require any outbreak-specific reference [28]. To the best of our knowledge, no previous studies have proposed a cgMLST scheme for P. mirabilis typing.
GbG approaches are becoming increasingly popular in bacterial typing [29, 30], bacterial genome epidemiology [31], and outbreak investigation [31, 32]. However, the lack of open source software for scheme definition and allele calling has limited the widespread use of the cgMLST approach. Interestingly, the chewBBACA suite can address this limitation. The chewBBACA suite was designed to help users create and evaluate novel whole genome or core genome GbG schemes and make allelic calls to bacterial strains of interest [17]. We used chewBBACA to call alleles of P. mirabilis and created and validated the first cgMLST scheme for P. mirabilis. In the end, the proposed cgMLST scheme for P. mirabilis typed 100% (707/707) of the genomes available for this research considering at least 95% of the cgMLST target genes present. We proposed the public cgMLST scheme for P. mirabilis based on 72 complete genome sequences and validated the scheme using 1,842 target loci in 635 unfinished genome sequences. The proposed 1842-locus cgMLST scheme delineated the 695 P. mirabilis strains into 205 distinct CGs. CG3 (18.3%) was the most dominant, followed by CG2 (4.7%), CG17 (3.0%), and CG32 (2.4%). Several CGs showed interesting regional distribution characteristics.
The cgMLST approach, which extends the MLST concept to the core genome, has proven to be a useful high-resolution typing scheme in other bacterial species [29, 30, 33, 34]. In contrast to the genus-wide Leptospira core genome MLST for strain taxonomy [30], we established a cgMLST scheme at the species level. Currently, the genus Proteus consists of five species (P. mirabilis, P. vulgaris, P. penneri, P. myxofaciens, and P. hauseri), with P. mirabilis being the main pathogenic species [35]. P. mirabilis is generally more susceptible to antimicrobials than P. penneri, P. hauseri, and P. vulgaris [35, 36]. Therefore, a cgMLST scheme at the species level is necessary. Unlike the cgMLST scheme of de Sales RO et al. for Pseudomonas aeruginosa [29], our cgMLST scheme allows for analysis of ARGs and virulence genes. The identification of CGs of high-risk P. mirabilis is important for public health.
Virulence genes papC, papD, papE, papF, papG, papH, papI, papJ, and papK were only in CG20. These nine virulence genes encode adhesive pili of the chaperone-usher family and they form the important P. mirabilis pmf fimbrial operon. These virulence genes in P. mirabilis may be the cause of severe urinary tract infections [22-24, 37-39]. In addition, CG20 has 42 shared ARG subtypes and five unique ARG subtypes. ARGs AAC(3)-Id [40, 41], aadA7 [42,43], dfrA15 [44,45], and vanRA [46] have been found in multidrug-resistant P. mirabilis. A recent study from Italy revealed that the coexistence of ARGs and virulence factors could lead to the emergence of a new population of resistant clones, which poses a major challenge for addressing antimicrobial resistance [47]. We believe that P. mirabilis deserves more attention. In addition, CG3 has 77 shared ARG subtypes and 18 unique ARG subtypes. CG3 was widely distributed on all continents, and its major resistance genes were classified as β-lactams (e.g., cephalosporin and carbapenem), which is consistent with the prevalence of β-lactamase-carrying P. mirabilis strains reported in many countries and regions [48-50]. Therefore, attention should be paid to these CGs of P. mirabilis.
There are some limitations in this study that must be pointed out. First, our analysis of the ARGs and virulence genes of the CGs of P. mirabilis was only preliminary. We simply used the easy and fast bioinformatics pipeline (ABRicate) and did not perform relevant experimental studies such as gene validation using polymerase chain reaction and quantitative PCR. The main reason for this is that the sequences of all 695 P. mirabilis strains were downloaded from NCBI, which became the main reason why we were unable to perform many experiments. Second, the main aim of the cgMLST scheme is not only typing, but also outbreak tracing. We did not apply the cgMLST scheme to epidemiological practice in this study. Therefore, further research will be conducted in the future to confirm that our cgMLST scheme is well suited not only for molecular typing of P. mirabilis, but also for epidemiological practice of P. mirabilis. Despite these current limitations, this is still the first study that performed high-resolution molecular typing of P. mirabilis using WGS technology combined with bioinformatics pipelines (chewBBACA), our results support the prevention and control of P. mirabilis infections.
-
In summary, we carried out high-resolution molecular typing of P. mirabilis from all over the world and found that the CGs of P. mirabilis showed significant regional distribution differences. We identified a total of 205 CGs in 695 P. mirabilis strains. The proposed cgMLST scheme for P. mirabilis typed 100% (707/707) of the available isolates, and at least 95% of these selected loci were present in the genome. We also found that some P. mirabilis CGs carried a large number of shared ARGs (or virulence genotypes) and unique ARGs (or virulence genotypes). To the best of our knowledge, this is the first report of high-resolution molecular typing of P. mirabilis using whole genome sequencing technology combined with a bioinformatics pipeline (chewBBACA). We have published our scheme, associated codes, and files on GitHub (
https://github.com/Natasha22222222/cgMLST-Proteus-mirabilis ) to facilitate further discussion and contribute to the establishment of cgMLST for P. mirabilis. -
The datasets for this study can be found in the NCBI (National Center for Biotechnology Information) genome sequence repository (
http://www.ncbi.nlm.nih.gov/genome/ ). The RefSeq assembly accession of all included P. mirabilis sequences are provided inSupplementary Table S2 . -
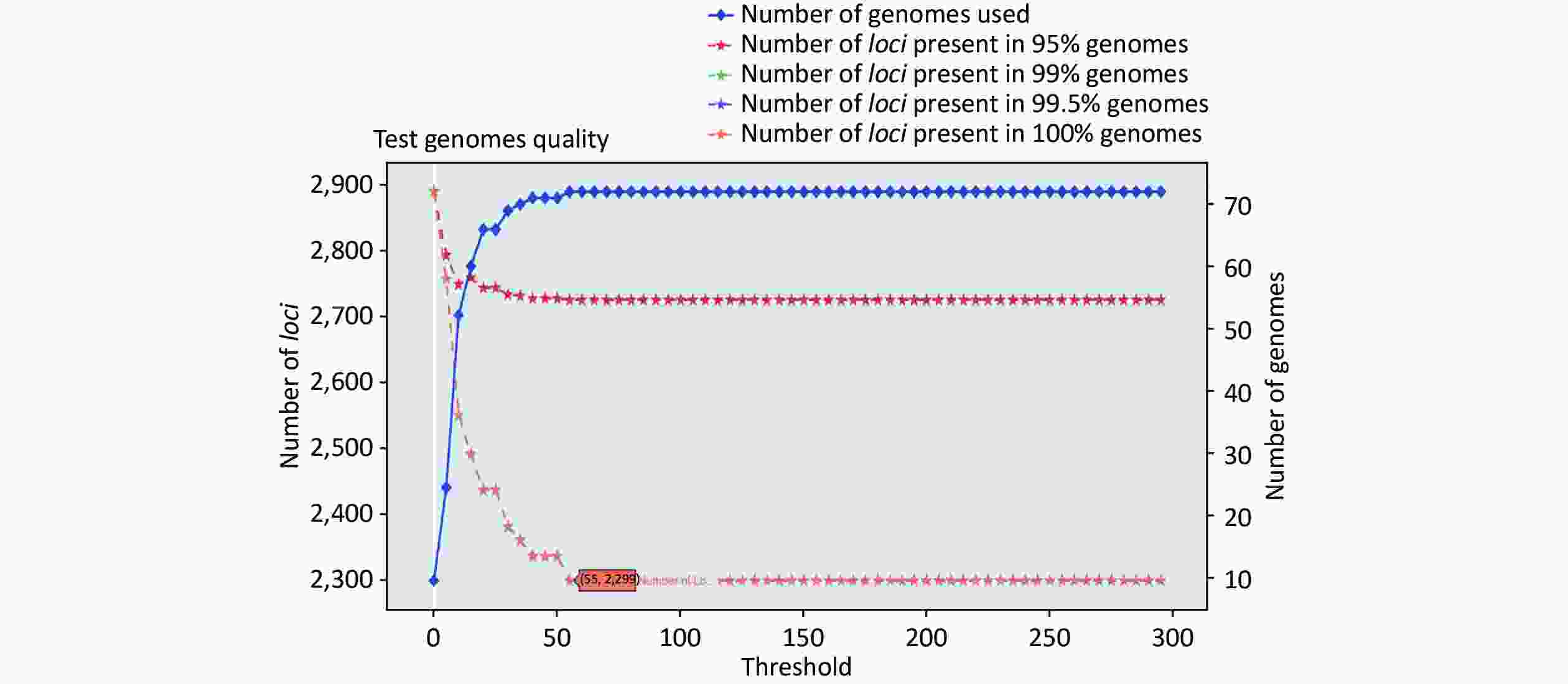
Figure S1. Number of loci and number of strains at every exclusion threshold level for the 72 complete genomes that were used to create the cgMLST scheme
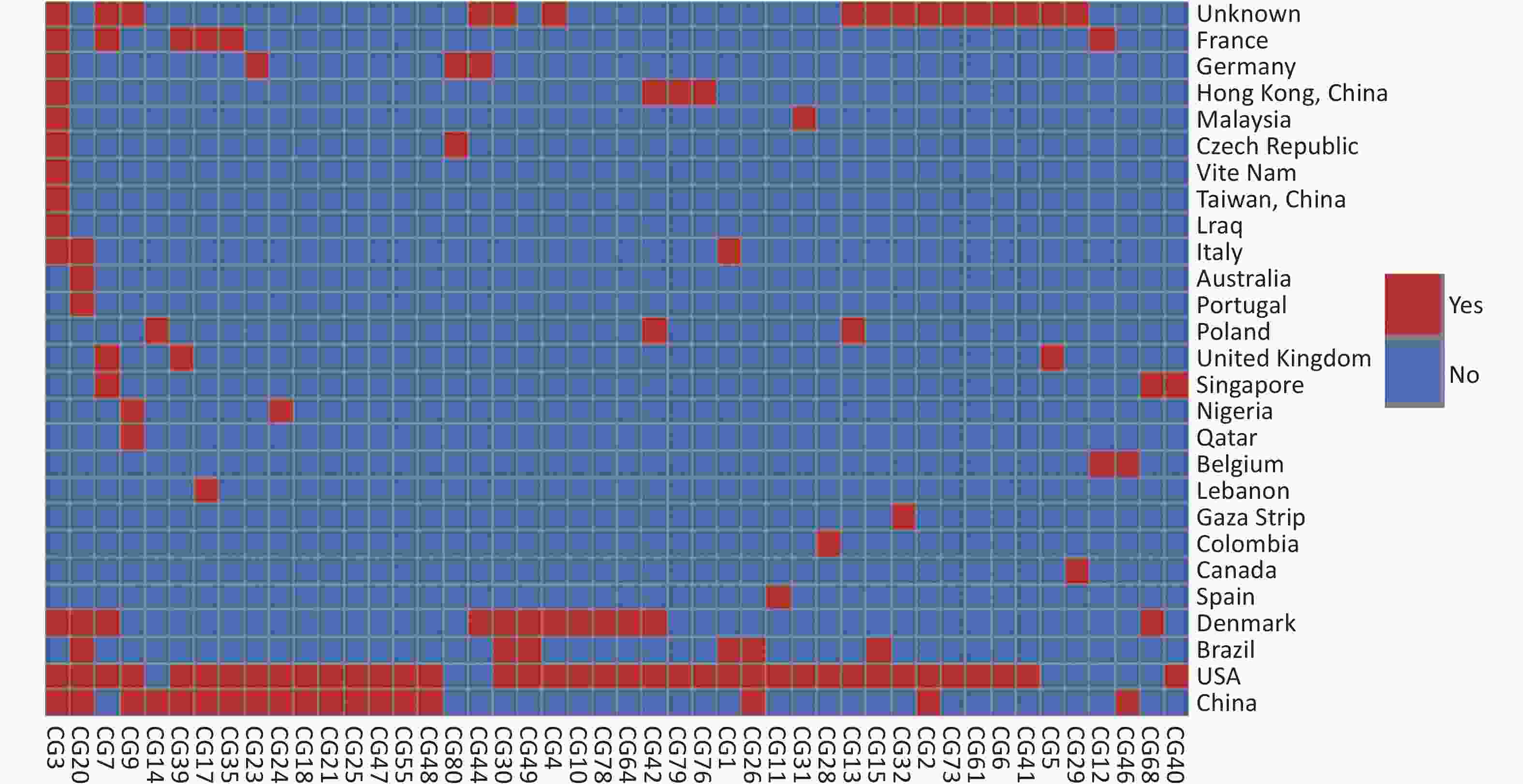
Figure S3. Heatmap of 46 shared CGs in 26 countries or regions
doi: 10.3967/bes2023.040
-
Abstract:
Objective A core genome multilocus sequence typing (cgMLST) scheme to genotype and identify potential risk clonal groups (CGs) in Proteus mirabilis. Methods In this work, we propose a publicly available cgMLST scheme for P. mirabilis using chewBBACA. In total 72 complete P. mirabilis genomes, representing the diversity of this species, were used to set up a cgMLST scheme targeting 1,842 genes, 635 unfinished (contig, chromosome, and scaffold) genomes were used for its validation. Results We identified a total of 205 CGs from 695 P. mirabilis strains with regional distribution characteristics. Of these, 159 unique CGs were distributed in 16 countries. CG20 and CG3 carried large numbers of shared and unique antibiotic resistance genes. Nine virulence genes (papC, papD, papE, papF, papG, papH, papI, papJ, and papK) related to the P fimbrial operon that cause severe urinary tract infections were only found in CG20. These CGs require attention due to potential risks. Conclusion This research innovatively performs high-resolution molecular typing of P. mirabilis using whole-genome sequencing technology combined with a bioinformatics pipeline (chewBBACA). We found that the CGs of P. mirabilis showed regional distribution differences. We expect that our research will contribute to the establishment of cgMLST for P. mirabilis. -
Key words:
- Proteus mirabilis /
- CgMLST /
- Genotyping /
- Clonal evolution /
- ChewBBACA
注释:1) CONFLICT OF INTEREST: -
Figure 1. Characterization of the P. mirabilis isolates included in this study. (A) Hosts of isolated P. mirabilis. “Others” represents host types with only one strain. (B) Geographic distribution of available genomes. “Others” represents countries with less than five strains. (C) Isolation source distribution of isolates. “Others” represents sources with less than eight strains. (D) Temporal distribution of P. mirabilis isolates. “Others” represents years when less than two strains were collected.
Figure 2. Flowchart describing the development of the cgMLST scheme for P. mirabilis using chewBBACA (
https://github.com/B-UMMI/chewBBACA ).
S2. Number of loci and number of genomes at every exclusion threshold level for the 635 validation strains’ genomes and 72 completed genomes that were used to construct the cgMLST scheme
Figure 3. Distribution of the number of paired allele mismatches in P. mirabilis (i.e., the number of different loci for a given pair of strains). The allelic mismatch cutoff value of 291 proposed for the CG definition is shown in red.
Figure 4. Minimum spanning tree (MST) of CGs generated using the 695 P. mirabilis strains based on the cgMLST scheme. Due to the limitation of the visualization of the MST, only the CG types containing the top 30 strains are displayed. “Others” represents the CG types containing less than five strains.
Figure 5. Minimal spanning tree analysis of cgMLST profiles of 695 P. mirabilis strains isolated from different countries or regions. The genome sequences were downloaded from the NCBI database (
https://www.ncbi.nlm.nih.gov/ genome/ ). Colors indicate the geographic locations of the isolates.
S1. Number of loci and number of strains at every exclusion threshold level for the 72 complete genomes that were used to create the cgMLST scheme
-
[1] Jamil RT, Foris LA, Snowden J. Proteus mirabilis infections. StatPearls Publishing. 2022. [2] Berger SA. Proteus bacteraemia in a general hospital 1972-1982. J Hosp Infect, 1985; 6, 293−8. doi: 10.1016/S0195-6701(85)80133-X [3] Luzzaro F, Perilli M, Amicosante G, et al. Properties of multidrug-resistant, ESBL-producing Proteus mirabilis isolates and possible role of β-lactam/β-lactamase inhibitor combinations. Int J Antimicrob Agents, 2001; 17, 131−5. doi: 10.1016/S0924-8579(00)00325-3 [4] D'Andrea MM, Literacka E, Zioga A, et al. Evolution and spread of a multidrug-resistant Proteus mirabilis clone with chromosomal AmpC-type cephalosporinases in Europe. Antimicrob Agents Chemother, 2011; 55, 2735−42. doi: 10.1128/AAC.01736-10 [5] Harada K, Niina A, Shimizu T, et al. Phenotypic and molecular characterization of antimicrobial resistance in Proteus mirabilis isolates from dogs. J Med Microbiol, 2014; 63, 1561−7. doi: 10.1099/jmm.0.081539-0 [6] Mazzariol A, Kocsis B, Koncan R, et al. Description and plasmid characterization of qnrD determinants in Proteus mirabilis and Morganella morganii. Clin Microbiol Infect, 2012; 18, E46−8. doi: 10.1111/j.1469-0691.2011.03728.x [7] Mokracka J, Gruszczyńska B, Kaznowski A. Integrons, β-lactamase and qnr genes in multidrug resistant clinical isolates of Proteus mirabilis and P. vulgaris. APMIS, 2012; 120, 950−8. doi: 10.1111/j.1600-0463.2012.02923.x [8] Wei QH, Hu QF, Li SS, et al. A novel functional class 2 integron in clinical Proteus mirabilis isolates. J Antimicrob Chemother, 2014; 69, 973−6. doi: 10.1093/jac/dkt456 [9] Dziri O, Alonso CA, Dziri R, et al. Metallo-β-lactamases and class D carbapenemases in south-east Tunisia: Implication of mobile genetic elements in their dissemination. Int J Antimicrob Agents, 2018; 52, 871−7. doi: 10.1016/j.ijantimicag.2018.06.002 [10] Marques C, Belas A, Franco A, et al. Increase in antimicrobial resistance and emergence of major international high-risk clonal lineages in dogs and cats with urinary tract infection: 16 year retrospective study. J Antimicrob Chemother, 2018; 73, 377−84. doi: 10.1093/jac/dkx401 [11] Kanayama A, Kobayashi I, Shibuya K. Distribution and antimicrobial susceptibility profile of extended-spectrum β-lactamase-producing Proteus mirabilis strains recently isolated in Japan. Int J Antimicrob Agents, 2015; 45, 113−8. doi: 10.1016/j.ijantimicag.2014.06.005 [12] Lin MF, Liou ML, Kuo CH, et al. Antimicrobial susceptibility and molecular epidemiology of Proteus mirabilis isolates from three hospitals in Northern Taiwan. Microb Drug Resist, 2019; 25, 1338−46. doi: 10.1089/mdr.2019.0066 [13] Mathur S, Sabbuba NA, Suller MTE, et al. Genotyping of urinary and fecal Proteus mirabilis isolates from individuals with long-term urinary catheters. Eur J Clin Microbiol Infect Dis, 2005; 24, 643−4. doi: 10.1007/s10096-005-0003-0 [14] de Been M, Pinholt M, Top J, et al. Core genome multilocus sequence typing scheme for high-resolution typing of Enterococcus faecium. J Clin Microbio, 2015; 53, 3788−97. doi: 10.1128/JCM.01946-15 [15] Pearce ME, Alikhan NF, Dallman TJ, et al. Comparative analysis of core genome MLST and SNP typing within a European Salmonella serovar Enteritidis outbreak. Int J Food Microbiol, 2018; 274, 1−11. doi: 10.1016/j.ijfoodmicro.2018.02.023 [16] Martak D, Valot B, Sauget M, et al. Fourier-transform infrared spectroscopy can quickly type gram-negative bacilli responsible for hospital outbreaks. Front Microbiol, 2019; 10, 1440. doi: 10.3389/fmicb.2019.01440 [17] Maiden MCJ, Bygraves JA, Feil E, et al. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci USA, 1998; 95, 3140−5. doi: 10.1073/pnas.95.6.3140 [18] Silva M, Machado MP, Silva DN, et al. chewBBACA: a complete suite for gene-by-gene schema creation and strain identification. Microb Genom, 2018; 4, 1−7. [19] Hyatt D, Chen GL, Locascio PF, et al. Prodigal, prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics, 2010; 11, 119. doi: 10.1186/1471-2105-11-119 [20] Rasko DA, Myers GSA, Ravel J. Visualization of comparative genomic analyses by BLAST score ratio. BMC Bioinformatics, 2005; 6, 2. doi: 10.1186/1471-2105-6-2 [21] Feil EJ. Small change: keeping pace with microevolution. Nat Rev Microbiol, 2004; 2, 483−95. doi: 10.1038/nrmicro904 [22] Jacobsen SM, Stickler DJ, Mobley HLT, et al. Complicated catheter-associated urinary tract infections due to Escherichia coli and Proteus mirabilis. Clin Microbiol Rev, 2008; 21, 26−59. doi: 10.1128/CMR.00019-07 [23] Li X, Mobley HLT. MrpB functions as the terminator for assembly of Proteus mirabilis mannose-resistant Proteus-like fimbriae. Infect Immun, 1998; 66, 1759−63. doi: 10.1128/IAI.66.4.1759-1763.1998 [24] Tsai YL, Chien HF, Huang KT, et al. cAMP receptor protein regulates mouse colonization, motility, fimbria-mediated adhesion, and stress tolerance in uropathogenic Proteus mirabilis. Sci Rep, 2017; 7, 7282. doi: 10.1038/s41598-017-07304-7 [25] Franz E, Gras LM, Dallman T. Significance of whole genome sequencing for surveillance, source attribution and microbial risk assessment of foodborne pathogens. Curr Opin Food Sci, 2016; 8, 74−9. doi: 10.1016/j.cofs.2016.04.004 [26] Jagadeesan B, Gerner-Smidt P, Allard MW, et al. The use of next generation sequencing for improving food safety: translation into practice. Food Microbiol, 2019; 79, 96−115. doi: 10.1016/j.fm.2018.11.005 [27] Ronholm J, Nasheri N, Petronella N, et al. Navigating microbiological food safety in the era of whole-genome sequencing. Clin Microbiol Rev, 2016; 29, 837−57. doi: 10.1128/CMR.00056-16 [28] Deneke C, Uelze L, Brendebach H, et al. Decentralized investigation of bacterial outbreaks based on hashed cgMLST. Front Microbiol, 2021; 12, 649517. doi: 10.3389/fmicb.2021.649517 [29] de Sales RO, Migliorini LB, Puga R, et al. A core genome multilocus sequence typing scheme for Pseudomonas aeruginosa. Front Microbiol, 2020; 11, 1049. doi: 10.3389/fmicb.2020.01049 [30] Guglielmini J, Bourhy P, Schiettekatte O, et al. Genus-wide Leptospira core genome multilocus sequence typing for strain taxonomy and global surveillance. PLoS Negl Trop Dis, 2019; 13, e0007374. doi: 10.1371/journal.pntd.0007374 [31] Mäesaar M, Mamede R, Elias T, et al. Retrospective use of whole-genome sequencing expands the multicountry outbreak cluster of Listeria monocytogenes ST1247. Int J Genomics, 2021; 2021; 6636138. [32] Silva M, Machado MP, Silva DN, et al. chewBBACA: a complete suite for gene-by-gene schema creation and strain identification. Microb Genom, 2018; 4, e000166. [33] Bialek-Davenet S, Criscuolo A, Ailloud F, et al. Genomic definition of hypervirulent and multidrug-resistant Klebsiella pneumoniae clonal groups. Emerg Infect Dis, 2014; 20, 1812−20. doi: 10.3201/eid2011.140206 [34] Moura A, Criscuolo A, Pouseele H, et al. Whole genome-based population biology and epidemiological surveillance of Listeria monocytogenes. Nat Microbiol, 2017; 2, 16185. doi: 10.1038/nmicrobiol.2016.185 [35] O'Hara CM, Brenner FW, Miller JM. Classification, identification, and clinical significance of Proteus, Providencia, and Morganella. Clin Microbiol Rev, 2000; 13, 534−46. doi: 10.1128/CMR.13.4.534 [36] Thornsberry C, Yee YC. Comparative activity of eight antimicrobial agents against clinical bacterial isolates from the United States, measured by two methods. Am J Med, 1996; 100, 26S−38S. doi: 10.1016/S0002-9343(96)00105-2 [37] Burall LS, Harro JM, Li X, et al. Proteus mirabilis genes that contribute to pathogenesis of urinary tract infection: identification of 25 signature-tagged mutants attenuated at least 100-fold. Infect Immun, 2004; 72, 2922−38. doi: 10.1128/IAI.72.5.2922-2938.2004 [38] Massad G, Mobley HLT. Genetic organization and complete sequence of the Proteus mirabilis pmf fimbrial operon. Gene, 1994; 150, 101−4. doi: 10.1016/0378-1119(94)90866-4 [39] Opletal L, Ločárek M, Fraňková A, et al. Antimicrobial activity of extracts and isoquinoline alkaloids of selected papaveraceae plants. Nat Prod Commun, 2014; 9, 1709−12. [40] Lei CW, Chen YP, Kang ZZ, et al. Characterization of a novel SXT/R391 integrative and conjugative element carrying cfr, blaCTX-M-65, fosA3, and aac(6')-Ib-cr in Proteus mirabilis. Antimicrob Agents Chemother, 2018; 62, e00849−18. [41] Leulmi Z, Kandouli C, Mihoubi I, et al. First report of blaOXA-24 carbapenemase gene, armA methyltransferase and aac(6')-Ib-cr among multidrug-resistant clinical isolates of Proteus mirabilis in Algeria. J Glob Antimicrob Resist, 2019; 16, 125−9. doi: 10.1016/j.jgar.2018.08.019 [42] Siebor E, de Curraize C, Varin V, et al. Mobilisation of plasmid-mediated blaVEB-1 gene cassette into distinct genomic islands of Proteus mirabilis after ceftazidime exposure. J Glob Antimicrob Resist, 2021; 27, 26−30. doi: 10.1016/j.jgar.2021.07.011 [43] Sung JY, Kim S, Kwon G, et al. Molecular characterization of Salmonella genomic island 1 in Proteus mirabilis isolates from Chungcheong province, Korea. J Microbiol Biotechnol, 2017; 27, 2052−9. doi: 10.4014/jmb.1708.08040 [44] Ahmed AM, Hussein AIA, Shimamoto T. Proteus mirabilis clinical isolate harbouring a new variant of Salmonella genomic island 1 containing the multiple antibiotic resistance region. J Antimicrob Chemother, 2007; 59, 184−90. [45] Schultz E, Barraud O, Madec JY, et al. Multidrug resistance Salmonella genomic island 1 in a Morganella morganii subsp. morganii human clinical isolate from france. mSphere, 2017; 2, e00118−17. [46] Markovska R, Schneider I, Keuleyan E, et al. Dissemination of a multidrug-resistant VIM-1- and CMY-99-producing Proteus mirabilis clone in bulgaria. Microb Drug Resist, 2017; 23, 345−50. doi: 10.1089/mdr.2016.0026 [47] Fasciana T, Gentile B, Aquilina M, et al. Co-existence of virulence factors and antibiotic resistance in new Klebsiella pneumoniae clones emerging in south of Italy. BMC Infect Dis, 2019; 19, 928. doi: 10.1186/s12879-019-4565-3 [48] Palzkill T, Thomson KS, Sanders CC, et al. New variant of TEM-10 beta-lactamase gene produced by a clinical isolate of proteus mirabilis. Antimicrob Agents Chemother, 1995; 39, 1199−200. doi: 10.1128/AAC.39.5.1199 [49] Pitout JDD, Thomson KS, Hanson ND, et al. β-Lactamases responsible for resistance to expanded-spectrum cephalosporins in Klebsiella pneumoniae, Escherichia coli, and Proteus mirabilis isolates recovered in South Africa. Antimicrob Agents Chemother, 1998; 42, 1350−4. doi: 10.1128/AAC.42.6.1350 [50] Verdet C, Arlet G, Redjeb SB, et al. Characterisation of CMY-4, an AmpC-type plasmid-mediated β-lactamase in a Tunisian clinical isolate of Proteus mirabilis. FEMS Microbiol Lett, 1998; 169, 235−40. -
 22263Supplementary Materials.pdf
22263Supplementary Materials.pdf

-

 点击查看大图
点击查看大图
计量
- 文章访问数: 1131
- HTML全文浏览量: 505
- PDF下载量: 66
- 被引次数: 0

 Quick Links
Quick Links