
-
We studied effects of nutrient quercetin on cytochromes' P450 1A (CYP1A) activities (measured spectrofluorimetrically using 7-ethoxy-resorufin for CYP1A1 and 7-methoxy-resorufin for CYP1A2 as substrates), on mRNA levels (measured by RT-PCR), and on DNA-binding activities (evaluated by an electrophoretic mobility shift assay) of proteins regulating CYP1A expression in untreated and benzo (α) pyrene (BaP)-treated rats. Wistar rats received quercetin, BaP, or both once daily for 1-3 days. Quercetin did not influence CYP1A1 in untreated rats but inhibited BaP-mediated CYP1A induction on the transcriptional level decreasing positive input (AhR functional activity) and increasing negative input (AhRR/ARNT expression and Oct-1 and C/EBP functional activities).
Cytochromes Р450, subfamily 1А (CYP1A1 and CYP1A2) metabolize widespread anthropogenic xenobiotics and environmental pollutants, such as aromatic amines and polycyclic aromatic hydrocarbons (PAHs), in particular, benzo (α) pyrene (BaP). Subfamily CYP1A catalyzes both reactions of detoxification of PAHs resulting in formation of water-soluble metabolites that are excreted by kidneys and the reactions of toxification of PAHs leading to formation of carcinogenic, mutagenic, and toxic derivatives, which can damage cellular components[1].
CYP1A enzymes are rather conserved evolutionarily and are at least 80% homologous in rodents and humans. In healthy untreated mammals CYP1A1 is predominantly expressed in the intestine, lungs, placenta, and kidneys, while CYP1A2 is mainly expressed in the liver[2]. Both CYP1A enzymes are induced by planar aromatic compounds such as the majority of PAHs (some PAHs can also inhibit CYP1A1 activity[3]) and arylamines via the signaling pathway of aryl hydrocarbon receptor (AhR), which is well known and described in detail elsewhere[1].
The inducibility of CYP1A together with weak constitutive expression in the liver implies the existence of negative regulatory mechanisms. CYP1A expression can be downregulated by the AhR repressor protein (AhRR), which competes with AhR for binding to Ah receptor nuclear translocator (ARNT) and forms heterodimer AhRR-ARNT capable of binding to an xenobiotic respensive element (XRE) but unable to activate gene expression[1]. In addition, transcription factors Oct-1 and С/EBPβ can inhibit CYP1A1 expression by binding to the 5' region of the CYP1A1 gene independently of AhR. Nonetheless, in general, negative regulation of CYP1A is not very well studied.
In the past decades, structurally diverse non-PAH-like CYP1A inducers including vitamins, indoles, flavonoids, and various metabolites[1] were discovered. They weakly enhance CYP1A expression either via the AhR-dependent signaling pathway by serving as AhR ligands or by activating AhR indirectly via other signaling molecules. Nevertheless, in many cases, molecular mechanisms underlying CYP1A induction by non-PAH inducers have yet to be elucidated. Many weak CYP1A1 inducers, such as some vitamins and flavonoids, attenuate activation of AhR-dependent genes by PAH and arylamines[4]. The molecular mechanisms behind this phenomenon are often unknown.
Among weak potential inducers of CYP1A1 and AhR ligands, the natural flavonoid quercetin[1] is of particular interest. In recent years, quercetin has been extensively studied as a potential pharmacological agent[5]. Published data on the influence of quercetin on CYP1A are incomplete and inconsistent and have mostly been obtained in in vitro studies. Nevertheless, it is known that quercetin can modulate activity of CYP1A2 in people[6]. Because CYP1A enzymes are involved in chemically induced carcinogenesis, modulation of activities of these enzymes by quercetin may influence the fate and toxicity of PAH-like compounds in humans and should be studied.
Here we conducted an in vivo experiment to study the effects of quercetin on the activity and mRNA level of CYP1A, mRNA level of some components of the AhR-dependent signaling pathway, and on the DNA-binding activity of negative regulators of AhR and CYP1A in the rat liver of untreated and BaP-treated rats.
Experiments were performed on three-month-old male Wistar rats. Breeding, housing, maintenance, and experimental procedures were performed in compliance with the EU Directive of 22 September 2010 (2010/63/EU) and approved by the Ethics Committee of the Institute of Molecular Biology and Biophysics. Rats received quercetin (80 mg/kg) per os or BaP (5 mg/kg) intraperitoneally or both BaP and quercetin. The control group received vegetable oil. The rats received these agents once a day for 1-3 d. Samples were collected 3, 6, 12, and 24 h (single administration) or 72 h (3-day administration) after the first treatment (for more details see Supplemental File S1 available in www.besjournal.com). CYP1A activities in hepatic microsomes prepared by differential ultracentrifugation were measured by spectrofluorimetry using 7-ethoxy-and 7-methoxy resorufins as substrates for CYP1A1 and CYP1A2, respectively. mRNA levels of CYP1A and CYP1A regulatory proteins (AhR, Arnt, AhRR, and C/EBPβ) were assessed by semiquantitative multiplex RT-PCR. Functional activities of AhR, C/EBP, and Oct-1 were evaluated by an electrophoretic mobility shift assay (EMSA) in nuclear extracts. Protein concentrations in samples were measured by the Lowry (microsomes) or Bradford (nuclear extracts) methods. All calculations were performed in the Statistica software package (StatSoft, Inc., USA). Differences between groups were assessed by analysis of variance (ANOVA) with the Newman-Keuls post hoc test. Differences were regarded as significant for P values below 0.05. Methods used in the study are described in detail in Supplemental File S1.
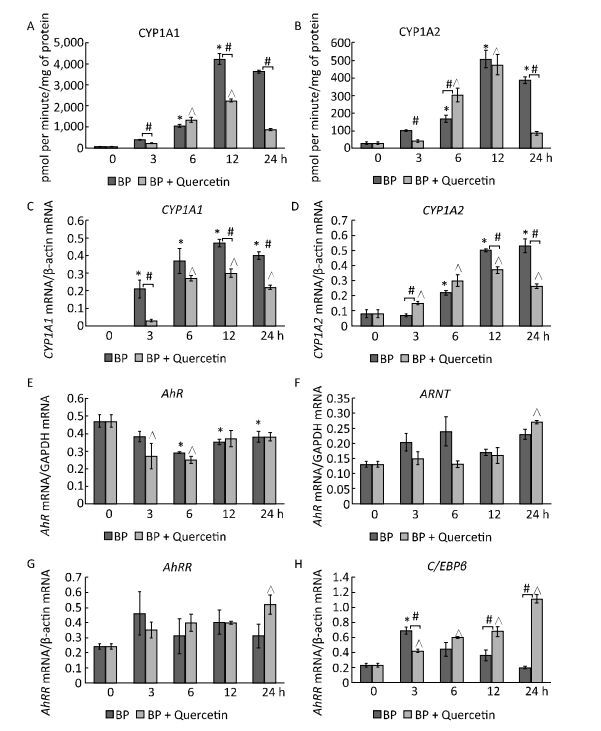
In untreated animals, quercetin failed to influence the CYP1A1 activity and mRNA levels of CYP1A1 and CYP1A2 (Figure 1A, C, D) but enhanced CYP1A2 activity (Figure 1B). The latter effect seems to be mediated by post-transcriptional mechanisms because the mRNA level of CYP1A2 remained unchanged (Figure 1D). For example, some CYPs (CYP2B4, CYP2B1/2, and CYP2E) are activated via phosphorylation by protein kinase A[7]. It is also possible that the degradation of protein CYP1A2 proceeds more slowly than the degradation of CYP1A2 mRNA. In this case, it is possible that CYP1A2 was induced by quercetin transcriptionally, but on the 3rd day, the mRNA level returned to normal, while the protein level remained elevated.
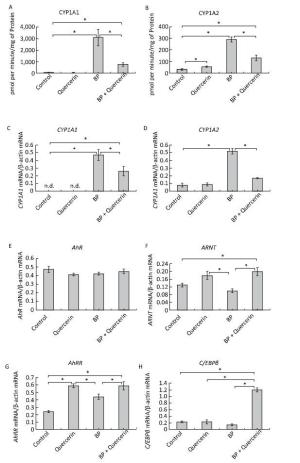
Figure 1. CYP1A activity (A, B), CYP1A (C, D), AhR (E), ARNT (F), AhRR (G), and C/EBPβ (H) mRNA levels. Daily for 3 d, rats received quercetin in vegetable oil (80 mg/kg) or BaP in vegetable oil (5 mg/kg) or both. The control group received vegetable oil. Differences were assessed by ANOVA with the Newman-Keuls post hoc test. The data are presented as mean ± SEM. Samples from four animals were analyzed in duplicate; *P < 0.05.
Activity of CYP1A and its mRNA levels were lower in rats receiving both BaP and quercetin in comparison with the rats that received only BaP; that is, quercetin downregulated the BaP-induced expression of CYP1A (Figure 1A-D). Literature data describing the effects of quercetin on CYP1A1 are inconsistent and were obtained in vitro. In a study by Zhang and coauthors[8], quercetin did not influence the activity of a luciferase reporter controlled by the CYP1A1 promotor in MCF-7 and HepG2 cells. In contrast, another research group observed an increase in enzymatic activity and/or expression of CYP1A1 in MCF-7 and Caco-2 cells[9] and human keratinocytes treated with quercetin[10]. Niestroy and coauthors showed that in BaP-treated Caco-2 cells, quercetin increased mRNA levels of CYP1A1 but decreased the mRNA level of CYP1A2[9], whereas other investigators reported a decrease in both expression and activity of 2, 3, 7, 8-tetrachlorodi-benzo-p-dioxin (TCDD)-induced CYP1A1 in MCF-7 and Caco-2 cells in response to quercetin[8]. The discrepancies between published data and our results may be caused by differences in experimental procedures and models, for instance, in duration of quercetin treatment: in our study during the first several hours, quercetin effects were variable (Supplemental Figure S2, available in www.besjournal.com).
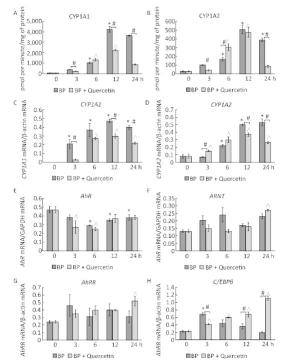
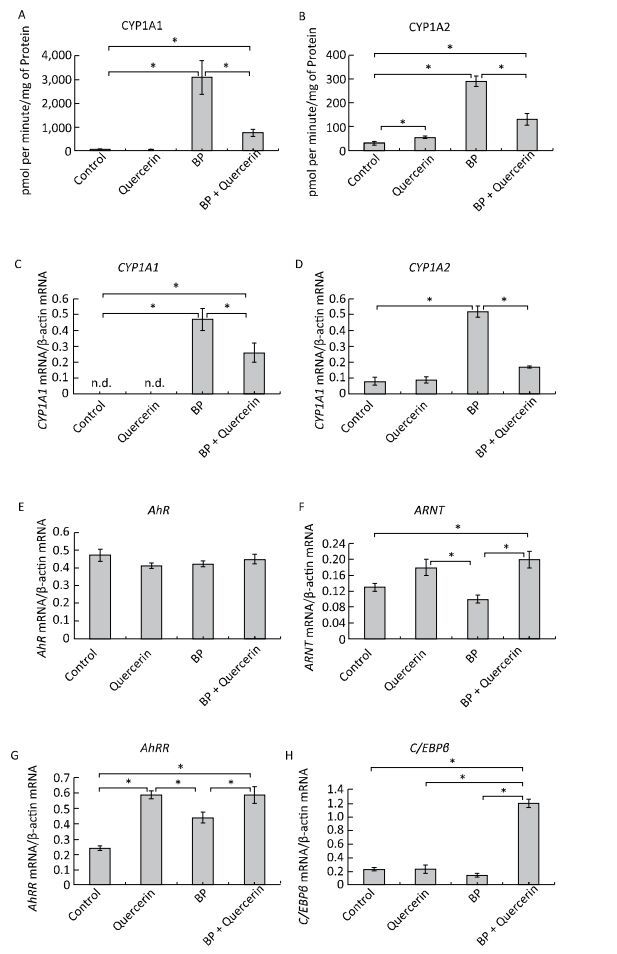
Figure Figure S2. Time-dependent changes in the activity and mRNA levels of CYP1A and in mRNA levels of CYP1A-regulating proteins during treatment with BaP or BaP in combination with quercetin. CYP1A1 and CYP1A2 activity (A, B), CYP1A1 and CYP1A2 mRNA levels (C, D), AhR (E), ARNT (F), and AhRR (G) mRNA levels (components of the AhR-dependent signal transduction pathway), and the C/EBPβ (H) mRNA level (the factor involved in negative regulation of CYP1A). Rats (groups of four animals) received BaP in vegetable oil (5 mg/kg) or both BaP (5 mg/kg) and quercetin (80 mg/kg in vegetable oil). The control group received vegetable oil. The rats received the chemicals once and were euthanized after 3, 6, 12, or 24 h. Differences among the groups were assessed by analysis of variance (ANOVA) with the Newman-Keuls post hoc test. The data are presented as mean ± SEM. Samples from four animals were analyzed in duplicate; *P < 0.05: significant differences between the control group and the BaP-treated group; ^P < 0.05: significant differences between the control group and the group treated with BaP plus quercetin; #P < 0.05: significant differences between BaP-treated group and the group treated with BaP plus quercetin.
We further analyzed the mRNA levels and functional activity of CYP1A1 regulators to gain insights into possible mechanisms underlying the observed effects. In untreated animals, neither quercetin nor BaP influenced mRNA levels of AhR (in agreement with literature data[9]), ARNTor C/EBPβ (Figure 1E, F, H) although quercetin upregulated AhRR mRNA (Figure 1G). Taken together, these data can explain the inability of quercetin to activate CYP1A expression despite being an AhR ligand[1]: AhRR forms the AhRR-ARNT complex, which binds to an XRE and blocks expression of AhR-dependent genes via competition with the AhR-ARNT complex[1]. Unexpectedly, we failed to detect upregulation of AhRR in response to BaP, in contradiction to multiple other reports; this result indicates that the dose of BaP here was insufficient to activate AhRR.
In BaP-treated animals, quercetin increased mRNA levels of AhRR, ARNT, and C/EBPβ (Figure 1F-H) but not AhR (Figure 1E). AhRR and ARNT upregulation can contribute to the inhibitory effect of quercetin on BaP-activated CYP1A expression via formation of the AhRR-ARNT complex competing with the AhR-ARNT complex (see above). A fivefold upregulation of C/EBPβ mRNA (Figure 1H), in BP+quercetin-treated animals may also contribute to the inhibitory effect of quercetin on BaP-activated CYP1A expression because C/EBPβ is known to be involved in downregulation of CYP1A. These data are in partial agreement with published ones: Niestroy and coauthors observed upregulation of AhRR mRNA in BaP+quercetin-treated Caco-2 cells, but they also reported an increase of the AhRR mRNA level in BaP-treated samples and downregulation of ARNT mRNA in BaP-treated and BaP+quercetin-treated groups in comparison with vehicle-treated cells[9].
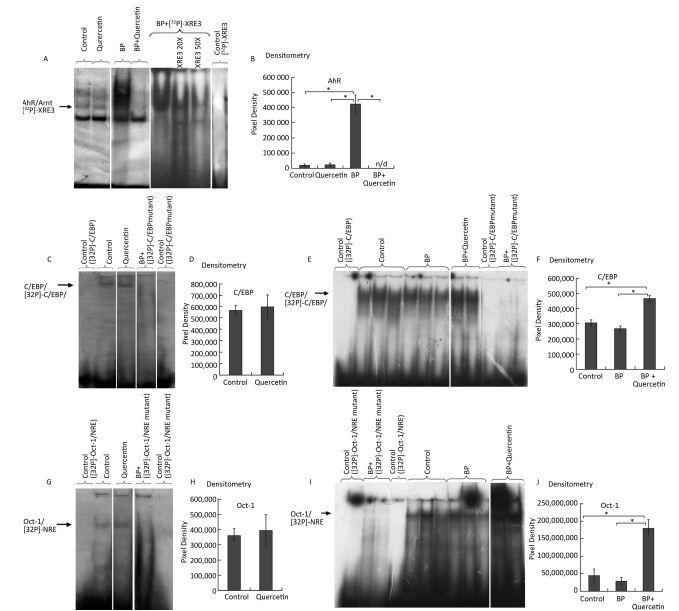
We carried out an EMSA of DNA-binding activity of AhR-ARNT and of CYP1A1's negative regulators Oct-1 and C/EBP in animals treated for 3 days with vehicle, quercetin, BaP, or BaP + quercetin. Quercetin administration to untreated rats did not enhance binding of the AhR-ARNT complex to XRE (Figure 2A, B), C/EBP to the C/EBP response element (Figure 2C, D), or Oct-1 to NRE (Figure 2G, H). At the same time, quercetin decreased BaP-induced DNA-binding activity of AhR (Figure 2A, B) and increased BaP-induced DNA-binding activity of transcription factors C/EBP and Oct-1 (Figure 2E, F, I, J).

Figure 2. EMSA of the binding of AhR-ARNT to XRE3 (A, B), of C/EBP to the C/EBP response element (C-F), and Oct-1 to NRE (G-J) in nuclear extracts from the liver of untreated rats (control) and of the rats treated with quercetin (80 mg/kg), benzo (a) pyrene (BaP; 25 mg/kg), or both for 3 d. Specificity of the bands was confirmed by means of mutated oligonucleotides or an excess of an unlabeled oligonucleotide. Digital density of the bands was measured in pixels and expressed in arbitrary units. The data are presented as mean ± SEM. Samples from 3 animals were analyzed in duplicate; *P < 0.05.
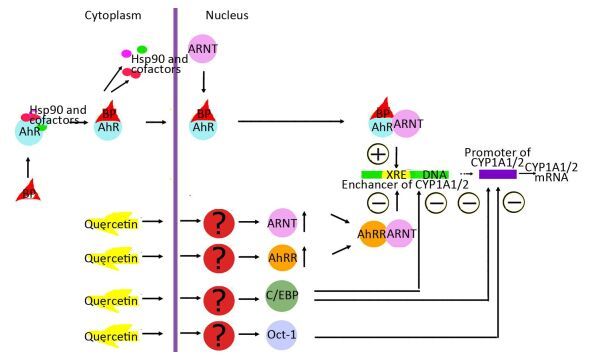
This finding suggests that quercetin at least partly attenuates BaP-induced expression of CYP1A by inhibiting the functional activity of AhR and increasing functional activity of transcription factors negatively regulating AhR-responsive genes: С/EBPβ and Oct-1. These results are also consistent with upregulation of С/EBPβ mRNA in the liver of rats receiving quercetin+BaP (Figure 1H). We summarized possible mechanisms of quercetin-mediated attenuation of BaP-induced CYP1A activation in Figure 3. Taken together, our data indicate that in the rat liver, quercetin suppresses BaP-induced activation of CYP1A on the transcriptional level by reducing positive input from AhR and by inducing negative input from AhR-AhRR, Oct-1, and CEBP signaling pathways.
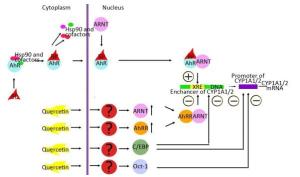
Figure 3. Possible mechanisms of quercetin-driven inhibition of the benzo (α) pyrene (BaP)-induced AhR-dependent signaling pathway.








HTML
-
Acrylamide, tris base (2-Amino-2-(hydroxymethyl) propane-1, 3-diol), NADPH, ammonium persulfate, bovine serum albumin, ethyleneglycoltetraacetic acid (EGTA), poly[dI-dC], spermidine, dithiothreitol (DTT), phenylmethanesulfonylfluoride (PMSF), quercetin, and spermine were purchased from ICN (USA); ammonium sulfate from Helicon (Russia); N, N'-methylenebisacrylamide, sodium dodecyl sulfate (SDS), dimethyl sulfoxide (DMSO), HEPES, and TEMED from Serva GmbH (Germany); and 2-mercaptoethanol and MgCl2 from Janssen Chimica (Belgium). Glycine and ethylenediaminetetraacetic acid (EDTA) were acquired from Merck (Germany); RNASecure Reagent from Ambion, Inc. (USA); 7-ethoxyresorufin, 7-methoxyresorufin, M-MuLV reverse transcriptase, and RNasin®m from Promega Corporation (USA); and agarose from Life Technologies (Scotland). Glycerol was purchased from Panreac Quimica S.A.U. (Spain); [α-32P]ATP and [γ-32P]ATP from Amersham Pharmacia Biotech (USA); DNA molecular weight markers from Sibenzyme (Russia); and Taq polymerase, T4 Polynucleotide Kinase, dNTP, and sucrose from Medigen (Russia). The VektoRNK-ekstraktsiya RNA isolation kit was acquired from Vector-Best (Russia), and oligonucleotides for analysis of CYP1A1, CYP1A2, AhR, ARNT, AhRR, C/EBPβ, β-actin, XRE3, NRE, and C/EBP and random hexanucleotide primers from BIOSSET (Russia) and Medigen (Russia). All other reagents were of analytical grade.
-
Experiments were performed on three-month-old male Wistar rats (weighing 150-200 g) from the stock maintained at the Animal Facility of the Institute of Cytology and Genetics, SB RAS, (Novosibirsk, Russia). The animals were housed in plastic cages 58 × 37 × 25 cm in groups of four under standard conditions (12:12 h light/dark regimen; food and water available ad libitum). Breeding, housing, maintenance, and experimental procedures were performed in compliance with the EU Directive of 22 September 2010 (2010/63/EU). The use of animals in experiments was approved by the Ethics Committee of the Institute of Molecular Biology and Biophysics of the Siberian Branch of the Russian Academy of Science. The size of experimental groups was four rats and was chosen on the basis of previous studies in order to achieve statistical power of 0.8.
Rats received quercetin in vegetable oil (80 mg/kg in a volume of 130-160 μL depending on body weight) per os or BaP in vegetable oil (5 mg/kg in a volume of 130-160 μL depending on body weight) intraperitoneally or both BaP (5 mg/kg) and quercetin (80 mg/kg). The latter dose was chosen on the basis of published data showing that this dose of quercetin increases CYP1A activity in Wistar rats[1]. The control group received vegetable oil. Rats received these agents once a day for 3 d (72 h) in cases where decapitation was carried out in 3 d, or once in cases where decapitation was carried out in 3, 6, 12, or 24 h after the treatment (Figure S1). After administration of the substances under study was completed, the rats were anesthetized with CO2and decapitated.
-
These microsomes were prepared by differential ultracentrifugation[2]. Rat livers were transcardially perfused with a cold buffer consisting of 1.15% KCl and 20 mM Tris-HCl (pH 7.4), then dissected and mechanically homogenized in the same buffer. The liver homogenates were centrifuged at 4 °С for 20 min at 10, 000 × g, and the resulting supernatants were centrifuged at 4 °С for 60 min at 105, 000 × g. The final pellets were resuspended in 0.1 mol/L KH2PO4 buffer (pH 7.4) containing 20% glycerol. Protein concentrations were measured by the Lowry method[3], with bovine serum albumin as a standard.
-
Selective activities of the CYP1A isoforms 7-ethoxyresorufin-O-deethylase (CYP1A1) and 7-methoxyresorufin-O-demethylase (CYP1A2) were measured by the spectrofluorometric method[2] on the basis of the rate of formation of resorufin—a product of О-dealkylation of highly specific substrates 7-ethoxyresorufin and 7-methoxyresorufin—for CYP1A1 and CYP1A2, respectively. The reaction mixture contained 0.4 ml of a buffer (50 mmol/L HEPES, 15 mmol/L MgCl2, 1 mmol/L EDTA, рН 7.6), 20 μg of microsomal protein, 1 μmol/L substrate and 1 mmol/L NADPH. The rate of formation of resorufin was determined at wavelengths 530 nm (excitation) and 585 nm (emission) on the spectrofluorimeter Hitachi MPF-4 (Japan). As a standard for construction of the calibration curve, we used resorufin. Each experiment was repeated twice with samples from four rats.
-
Total RNA was isolated from the rat liver using the VektoRNK-ekstraktsiya RNA isolation kit (Vector-Best, Russia) based on the phenol-chloroform extraction method. RNA pellets were dissolved in 1 mmol/L sodium citrate buffer (pH 6.5) containing 1 × RNASecure Reagent. The RNA concentration was determined by UV spectrophotometry, and RNA integrity was verified by agarose gel electrophoresis with ethidium bromide staining. The reaction mixture for reverse transcription consisted of 400 ng of total RNA, reaction buffer (50 mmol/L Tris-HCl pH 8.3, 75 mmol/L KCl, 3 mmol/L MgCl2, and 10 mmol/L DTT), 1 mmol/L dNTPs, 200 U of M-MuLV reverse transcriptase, 4 µg of random hexamer primers, and 25 U of RNasin®in a 25-µL final volume. cDNA synthesis was carried out at 37 ℃ for 120 min.
-
We used the following PCR primers: for CYP1A1, forward 5'-CTGGTTCTGGATACCCAGCTG-3' and reverse 5'-CCTAGGGTTGGTTACCAGG-3', amplicon size 331 bp[4]; for CYP1A2, forward 5'-GCAGGTCAACCATGATGAGAA-3' and reverse 5'-CGGCCGATGTCTCGGCCATCT-3', amplicon size 334 bp[5]; for AhR, forward 5'-TCCATGTACCAGTGCCAGG-3' and reverse 5'-ATATCAGGAAGAGGCTGGGC-3', amplicon size 212 bp[6]; for ARNT, forward 5'-GTCTCCCTCCCAGATGATGA-3' and reverse 5'-AAGAGCTCCTGTGGCTGGTA-3', amplicon size 218 bp[6]; for AhRR, forward 5'-aaagtcagcatccctccttg-3' and reverse 5'-cccatcagatcctttggatg-3', amplicon size 161 bp[7]; for C/EBPβ, forward 5'-CGCCAAGCCGAGCAAGAAGC-3' and reverse 5'-CACCTTGTGCTGCGTCTCCA-3', amplicon size 150 bp[8]; for GAPDH, forward 5'-TTCAACGGCACAGTCAAGG-3' and reverse 5'-CATGGACTGTGGTCATGAG-3', amplicon size 340 bp[9]; for β-actin, forward 5'-CGTTGACATCCGTAAAGACCTCTA-3' and reverse 5'-TAAAACGCAGCTCAGTAACAGTCCG-3', amplicon size 290 bp[10]. β-Actin or GAPDH (housekeeping genes) served as an internal control to confirm equal loading and good quality of cDNA for each sample. Multiplex PCR was carried out in a 20-μL reaction mixture containing 1 × PCR buffer (150 mmol/L Tris-HCl pH 8.3, 50 mmol/L KCl), 0.25 mmol/L dNTPs, 0.25 μmol/L target gene primers and 0.25 μmol/L β-actin primers, 2 U of Taq polymerase, ~500 ng of cDNA, and 3.5 mmol/L MgCl2 for all primers except AhR primers, for which 2.5 mmol/L MgCl2was used. The PCR program started with initial denaturation at 95 ℃ for 3 min followed by cycles described in Table 1 and ending with final extension at 72 ℃ for 4 min.
Genes CYP1A1 + β-actin CYP1A2 + β-actin ARNT + β-actin AhR + GAPDH AhRR + β-actin C/EBPβ + β-actin Cycles (26 + 26) (30 + 26) (34 + 24) (30 + 30) (36 + 26) (30 + 28) Denaturation Annealing Elongation 95℃, 15" 60℃, 15" 72℃, 15" 95℃, 15" 60℃, 15" 72℃, 15" 95℃, 15" 60℃, 15" 72℃, 15" 95℃, 15" 60℃, 15" 72℃, 15" 95℃, 15" 60℃, 15" 72℃, 15" 95℃, 15" 60℃, 15" 72℃, 15" Table 1. PCR cycling conditions
The optimal number of amplification cycles (indicated in Table 1) for each primer pair was determined in a preliminary experiment by the "primer-dropping" method to stay within the exponential phase of the amplification curve and is indicated in Table 1. In all cases, except for CYP1A1 and AhR, target genes were amplified for several cycles (4 for CYP1A2; 10 for ARNT and AhRR, and 2 for C/EBPβ) before the addition of primers for the house-keeping gene.
Each sample was amplified twice with samples from four rats. PCR products were separated by electrophoresis in a 2% agarose gel in 1 × TBE buffer and stained with ethidium bromide. PCR bands were visualized using UV light, photographed by means of a DNA Analyzer Video System (Lytech, Russia), and analyzed in the TotalLab software (TotalLab, UK). The level of expression of a target gene was determined as a ratio of optical density of the band corresponding to the amplicon from the target gene and optical density of the band corresponding to the amplicon from β-actin.
-
Nuclear extracts from the liver of the experimental animals (the same rats from which the liver was collected for RNA analysis) were prepared according to a previously published method[11] with modifications described elsewhere[12]. Briefly, the livers were transcardially perfused with a buffer consisting of 1.15% KCl and 20 mmol/L Tris-HCl pH 7.4, cut into fragments, and mechanically homogenized in sucrose buffer (10 mmol/L HEPES, pH 7.6, 25 mmol/L KCl, 0.15 mmol/L spermine, 0.5 mmol/L spermidine, 1 mmol/L EDTA, 2.05 mol/L sucrose, and 10% glycerol). The homogenates were layered on top of 5 mL of sucrose buffer and centrifuged at 4°С in a SW28 rotor (Optima L-90K Ultracentrifuge, Beckman Coulter, USA) at 24, 000 rpm for 40 min. The pelleted nuclei were resuspended in 4 mL of lysis buffer (10 mmol/L HEPES, pH 7.6, 100 mmol/L KCl, 3 mmol/L MgCl2, 0.3 mol/L EDTA, 1 mmol/L DTT, 10% glycerol, and 0.1 mmol/L PMSF) and lysed by the addition of 0.4 mL of a supersaturated (NH4)2SO4 solution. Chromatin was precipitated by centrifugation in the SW65 rotor at 38, 600 rpm for 90 min. Nuclear proteins were precipitated by (NH4)2SO4 (0.252 g per milliliter of the protein solution) and collected by centrifugation in the SW65 rotor at 36, 200 rpm for 20 min. The resulting pellets were dissolved in 0.2-0.5 mL of dialysis buffer (25 mmol/L HEPES, pH 7.6, 80 mmol/L KCl, 0.1 mmol/L EDTA, 0.2 mmol/L EGTA, 1 mmol/L DTT, 10% glycerol, and 0.1 mmol/L PMSF). Nuclear extracts were dialyzed against 100 volumes of the buffer (three times for 30-45 min each), split into aliquots, and stored at-70℃. Protein concentration was determined by the Bradford method[13]. Bovine serum albumin served as a standard.
-
To assess the binding of nuclear proteins to specific DNA sequences, we used the following oligonucleotides: for XRE3 sequences, forward 5′-TGCACGGAGTTGCGTGAGAAGAGCCATGCA-3′ and reverse 3′-ACGTgcctcaacgcactcttctcggtACGT-5′[14]; for C/EBP consensus sequences, forward 5′-TGCAGATTGCGCAATCTGCA-3′ and reverse 3′-ACGTCTAACGCGTTAGACGT-5′[15]; for C/EBP mutant sequences, forward 5′-TGCAGAGACTAGTCTCTGCA-3′ and reverse 3′-ACGTCTCTGATCAGAGACGT-5′[15]; for Oct-1 consensus sequences, forward 5′-TGCATGTCGAATGCAAATCACTAGAATGCA-3′ and reverse 3′-ACGTACAGCTTACGTTTAGTGATCTTACGT-5′[16]; and for Oct-1 mutant sequences, forward 5′-TGCATGTCGAATGCAAGCCACTAGAATGCA-3′ and reverse 3′-ACGTACAGCTTACGTTCGGTGATCTTACGT-5′[16]. The oligonucleotides were radioactively labeled using a standard protocol[17]. Briefly, 20 pmol of oligonucleotides for XRE3 sequences were incubated with 20 µCi of α32P-dATP and 2 U of Klenow polymerase in a buffer consisting of 50 mmol/L Tris-HCl pH 7.6, 10 mmol/L MgCl2, and 5 mmol/L DTT. The oligonucleotides were rid of unbound α32P-dATP on G-50 spin columns or DEAE filters. Nucleoprotein complexes were formed under the following conditions: 1× DNA binding buffer (25 mmol/L HEPES, pH 7.6, 80 mmol/L KCl, 0.1 mmol/L EDTA, 1 mmol/L DTT, 10% glycerol, 4-5 µg of protein of nuclear extracts [depending on the oligonucleotide being tested], 0.83 µg of salmon sperm DNA, and a 32P-labeled DNA probe [400-1000 cpm/reaction]). All components of the reaction mixture except for the labeled oligonucleotide were mixed in a total volume of 11 µL and preincubated on ice for 10 min to block nonspecific binding. After that, the 32P-labeled DNA probe was added, and the samples were incubated at room temperature for 10 min. To assess specificity of the DNA-protein complexes, either mutant oligonucleotides were used or a 20-, 50-, or 75-fold excess of an unlabeled oligonucleotide was added to the reaction mixture. The resulting DNA-protein complexes were analyzed by electrophoresis in a 4% nondenaturing polyacrylamide gel in 0.5× TBE buffer (44.5 mmol/L Tris base, 44.5 mmol/L boric acid, and 1 mmol/L EDTA). After the electrophoresis, the gel was dried, and the DNA-protein complexes were visualized by autoradiography. Each experiment was repeated twice with samples from three rats.
-
All calculations were performed in the Statistica software package (StatSoft, Inc., USA). Differences between groups were assessed by analysis of variance (ANOVA) with the Newman-Keuls post hoc test. Differences were regarded as significant for p values below 0.05.
1. Rahden-Staron I, H Czeczot, and M. Szumilo. Induction of rat liver cytochrome P450 isoenzymes CYP 1A and CYP 2B by different fungicides, nitrofurans, and quercetin. Mutat Res. 2001; 498, 57-66.
2. Burke MD, et al. Ethoxy-, pentoxy-and benzyloxyphenoxazones and homologues: a series of substrates to distinguish between different induced cytochromes P-450. Biochem Pharmacol. 1985; 34, 3337-45.
3. Lowry OH, et al. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951; 193, 265-75.
4. Morris DL and Davila JC. Analysis of rat cytochrome P450 isoenzyme expression using semi-quantitative reverse transcriptase-polymerase chain reaction (RT-PCR). Biochem Pharmacol. 1996; 52, 781-92.
5. Walker NJ, et al. Characterization of the dose-response of CYP1B1, CYP1A1, and CYP1A2 in the liver of female Sprague-Dawley rats following chronic exposure to 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin. Toxicol Appl Pharmacol. 1999; 154, 279-86.
6. Lindros KO, et al. Selective centrilobular expression of the aryl hydrocarbon receptor in rat liver. J Pharmacol Exp Ther. 1997; 280, 506-11.
7. Korkalainen M, Tuomisto J and Pohjanvirta R. Primary structure and inducibility by 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin (TCDD) of aryl hydrocarbon receptor repressor in a TCDD-sensitive and a TCDD-resistant rat strain. Biochem Biophys Res Commun. 2004; 315, 123-31.
8. Tsavachidou D, et al. Gene array analysis reveals changes in peripheral nervous system gene expression following stimuli that result in reactivation of latent herpes simplex virus type 1: induction of transcription factor Bcl-3. J Virol. 2001; 75, 9909-17.
9. Kami D, et al. Gremlin enhances the determined path to cardiomyogenesis. PLoS One. 2008; 3, e2407.
10. Adams NH, PE Levi, and E Hodgson. Regulation of cytochrome P-450 isozymes by methylenedioxyphenyl compounds. Chem Biol Interact. 1993; 86, 255-74.
11. Gorski K, M Carneiro, and U Schibler. Tissue-specific in vitro transcription from the mouse albumin promoter. Cell. 1986; 47, 767-76.
12. Shapiro DJ, et al. A high-efficiency HeLa cell nuclear transcription extract. DNA. 1988; 7, 47-55.
13. Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976; 72, 248-54.
14. Denison MS and RM Deal. The binding of transformed aromatic hydrocarbon (Ah) receptor to its DNA recognition site is not affected by metal depletion. Mol Cell Endocrinol. 1990; 69, 51-7.
15. Wang H, et al. Platelet-activating factor and endotoxin activate CCAAT/enhancer binding protein in rat small intestine. Br J Pharmacol; 2001, 133, 713-21.
16. Malone CS, et al. B29 gene silencing in pituitary cells is regulated by its 3' enhancer. J Mol Biol. 2006; 362, 173-83.
17. Maniatis T, EF Fritsch, and J Sambrook. Molecular cloning: a laboratory manual. Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory. 1982.
MATERIALS AND METHODS
Reagents
Animals
Preparation of Rat Liver Microsomes
Enzymatic Assays
RNA Isolation and Reverse Transcription
Polymerase Chain Reaction (PCR)
Preparation of Nuclear Protein Extracts
Electrophoretic Mobility Shift Assay (EMSA)
Statistical Analysis
This work was supported by the Russian Foundation for Basic Research project No. 05-04-48327-а

 Quick Links
Quick Links
 DownLoad:
DownLoad: