
HTML
-
E nteroviruses (EVs) are associated with a great variety of manifestations, varying from mild respiratory and gastrointestinal infections, herpangina, and hand-foot-and-mouth disease (HFMD), to more severe diseases leading to mortality[1]. Enterovirus 71 (EV71) and Coxsackievirus A16 (CA16) are the most common EVs causing HFMD[2]. Knowledge of viral genome sequences is very important in epidemiologic investigations, to determine whether specific cases are epidemiologically linked, to identify transmission patterns, and to ascertain the extent of an outbreak[3]. Considering the great variations among different serotypes/genotypes of EVs, whole genome sequencing is the most appropriate way to distinguish different serotypes/genotypes, to detect recent recombination events and to provide a high-resolution view of viral lineages[4].
DNA sequencing technology has made remarkable progress since Sanger sequencing was established in 1977[5]. Since 2005, 454 technology (Roche), Solexa technology (Illumina) and SOLiD technology (Applied Biosystems) have been developed successively as Second-Generation Sequencing (SGS) systems, leading to a new age of high-throughput sequencing[6]. Single molecule sequencing technology, referred to as Third-Generation Sequencing (TGS), represented by SMRT (Pacific Bioscience) technology, offers longer read lengths than the SGS technologies, making it suitable for full-length viral sequencing and many other biological/medical researches[7-8].
The first commercial nanopore sequencer is MinION, developed by Oxford Nanopore Technologies (ONT). It can be defined as a TGS platform considering its single molecule sequencing ability[9], but its technical principles and properties are very different compared with the previous platforms. Thus, this nanopore sequencer is also described as a Fourth-Generation Sequencing (FGS) platform in some publications[10-11]. MinION is a palm-sized device that drives individual DNA/RNA molecules through a nanopore; only a single strand nucleic acid can pass through the pore. Because the electrical properties of the bases A, T, G, and C are different, electrical signals with base specificity can be detected by MinION and sequence information can thus be collected continuously using the MinKNOW software.
MinION is capable of generating reads as long as 882 kb[12], which improves the scaffolding of prokaryotic and eukaryotic genomes and sequencing of bacterial and viral isolates[13-16]. During the Ebola outbreak in west Africa, a set of portable viral MinION-based sequencing systems was transported via standard airline luggage to Guinea[17-18]. In addition, MinION has been used to detect a variety of viruses using amplicons[16-17, 19-20], hybridization capture[21-22] or unbiased metagenomics approaches[23-24]. Though the read accuracy of MinION is lower than that of short-read sequencers such as MiSeq or HiSeq, the deeper genome coverage depths can be used to generate an accurate consensus sequence, achieving > 99% accuracy post-data analysis[16].
MinION may provide new opportunities in EV diagnostics by rapid whole genome sequencing and determination of EV serotypes/genotypes, for example during an outbreak of HFMD. In this study, we established a whole genome sequencing method for EV71 and CA16 using MinION. We explored the accuracy, minimum sequencing time, discrimination and high-throughput sequencing ability of MinION, and compared the data with Sanger sequencing results.
-
One EV71 and 10 CA16 isolates used in this study were from swab or stool samples collected from HFMD patients < 5-year-old. The samples were inoculated onto rhabdomyosarcoma cells provided by the WHO Global Poliovirus Specialized Laboratory, USA, and originally purchased from the American Type Culture Collection (Manassas, VA, USA). Cell cultures were harvested after complete cytopathic effect was observed. The titers of CA16 and EV71 were 105.5CCID50/mL and 106.5 CCID50/mL, respectively. All the CA16 strains belonged to the B1 genotype with nucleotide similarity ranges 90.7%-98.5%, and the EV71 strain belonged to the C4a evolutionary branch in the C4 genotype. This work was approved by the Ethics Review Committee of the National Institute for Viral Disease Control and Prevention and was supported by a long-term surveillance program for HFMD.
Viral RNA was extracted from the viral isolates using a QIAamp Viral RNA Mini Kit (Qiagen) and stored at -80 ℃ until further use. The EV71 and CA16 amplicons were amplified using the RT-PCR protocol established by the National Polio Laboratory of China. Briefly, the viral RNA was converted to cDNA by a random-priming strategy. The cDNA was added to 4-10 separate PCR tubes with a SuperScript One-Step RT-PCR System for Long Templates Kit (Invitrogen) using gene specific primers. The PCR program used was: 2 min at 94 ℃, then 40 cycles of 15 s at 94 ℃, 30 s at 56 ℃, and 1 min at 68 ℃, with a final extension at 68 ℃ for 7 min.
-
The Ligation Sequencing Kit 1D R9 Version (ONT, SQK-LSK108) was applied to prepare the EV71 and CA16 amplicon libraries. The Native Barcoding Kit 1D (ONT, EXP-NBD103) was applied to tag the amplicons derived from the 10 different CA16 viral strains. Briefly, library preparation from CA16 strains was as follows: (1) All of the amplicons were purified using 1.8-times the volume of Agencourt AMPure XP (Beckman Coulter) and quantified using the Qubit dsDNA HS Assay Kit (Invitrogen) according to the manufacturers' instructions. (2) To calculate the quantity per barcode, the total input quantity of DNA (1.5 μg) was divided by the number of barcodes being used. The PCR products were kept separately at this stage. (3) End-repair was performed using NEBNext Ultra Ⅱ End-repair/dA-tailing Module (New England BioLabs) by adding 7 μL Ultra Ⅱ End-Prep buffer and 3 μL Ultra Ⅱ End-Prep enzyme mix, and incubated at 20 ℃ for 5 min and then 65 ℃ for 5 min. (4) The end-repaired DNA was cleaned up with Agencourt AMPure XP. (5) The DNA was quantified with a Qubit fluorimeter so as to recover about 700 ng of material. (6) The barcoding reaction was then performed by adding 22.5 µL end repaired DNA, 2.5 µL of one of the barcodes and 25 µL Blunt/TA Ligase Master Mix (New England BioLabs), followed by incubation at room temperature for 10 min. (7) The barcoded DNA was cleaned up with Agencourt AMPure XP and quantified with the Qubit fluorimeter. (8) Amplicons were homogenized and a total of 700 ng equimolar barcoded DNA was pooled into 51 µL. (9) Ligation was performed using the NEBNext Quick Ligation Module (New England BioLabs) by adding 20 µL BAM, 20 µL NEBNext Quick Ligation Reaction Buffer, and 10 µL Quick T4 DNA Ligase with incubation at room temperature for 10 min. (10) Adapted DNA was cleaned up with Agencourt AMPure XP, and ABB (ONT) was used for washing instead of ethanol. (11) The adapted library was eluted and quantified with the Qubit fluorimeter to recover about 200 ng of material.
The library was then ready for sequencing on the MinION. The library preparation of EV71 was generally the same as the process above, but the barcoding step was omitted (Figure 1).
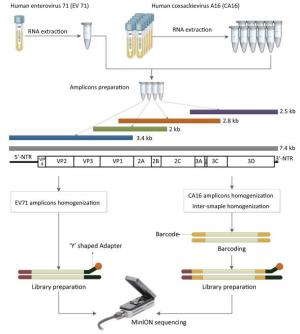
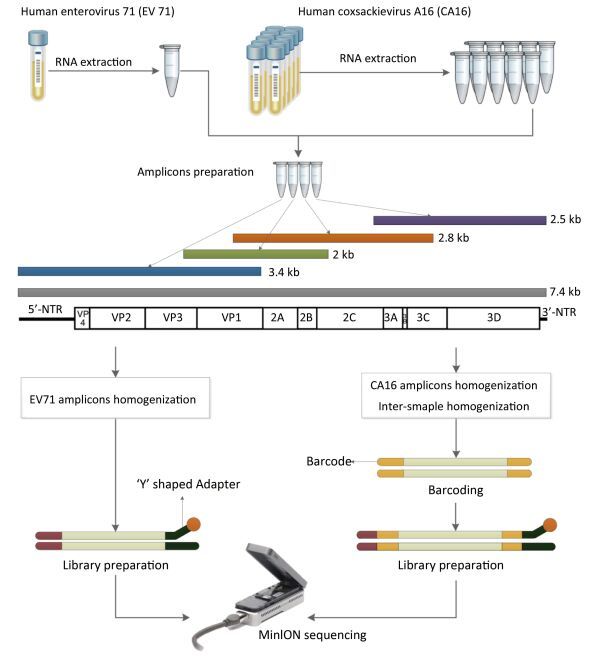
Figure 1. The workflow of Nanopore sequencing of the single sample and multiple samples. The EV71 and CA16 genomes were amplified using the RT-PCR protocol established by the National Polio Laboratory of China. A total of 4-10 individual PCRs using overlapping primers were carried out for amplicon preparation. Amplicons were quantified and homogenized before pooling together. Multiple samples were barcoded. The detailed process of the library preparation and MinION sequencing is described in the main text.
-
During the library incubation, the MinION was assembled with a Flow Cell Mk 1 Spot-ON (ONT, FLO-MIN 107 R9) and was connected to a computer (Windows 10; two Xeon E5-2640 v4 processors, 128 GB DDR4, 2, 133 MHz, 1 TB solid-state drive) via a USB 3.0 hub. The sequencing library was loaded into the flow cell using Library Loading Bead Kit R9 Version (ONT, EXP-LLB001) according to the manufacturer's instructions. A 48 h sequencing plus basecaller protocol was selected in the MinKNOW software and the sequencing process was stopped according to the experimental needs. For the single sample sequencing of EV71, the MinION running time was set for 19 h. For the multiple-sample sequencing of CA16, MinION was set for 6 h.
-
Platform QC (quality control) was performed before the sequencing step to assess the number of nanopores that were available in the flow cell. The MinKNOW software was set up to run platform QC using the script 'NC_Platform_QC.py'. The total number of pores available was reported in the notification panel. After sequencing was finished, 150 µL of Washing Solution A was added through the priming port of the flow cell and we waited for 10 min. Then, 500 μL of Storage Buffer was added through the priming port of the flow cell. The platform QC was performed again after the flow cell washing.
-
Raw read data were obtained by MinKNOW software and base-called via the 1D basecalling plus barcoding workflow of Metrichor. Mapping with the Sanger sequencing results was performed using BWA[25]. SAMtools was used to convert the data into bam format. Mapped data were applied in IGV software after filtering with filter_fastq.py (ONT support). Consensus sequences were assembled using SAMtools and BCFtools[26]. IGV software was used for visualization[27]. For the single sample sequencing of EV71, the length distribution was analyzed using the reads generated at eight different time points. Scatter charts and curve graphs were plotted using R. Sequence coverage was computed with BEDTools using the total data generated from the beginning of sequencing to three different time points[28]. The sequencing accuracy of the single and multiple samples was validated by comparing the consensus sequence with the Sanger reference sequence. The relationship between the number of reads and the sequencing time was analyzed and plotted according to the generation of FASTQ files.
Viral Strains and Amplicon Preparation
Library Preparation
MinION Sequencing
Platform QC and Flow Cell Washing
Data Analysis
-
The core component of MinION is a flow cell with 2, 048 wells-four groups each consisting of 512 channels[29]. In QC of the brand-new flow cell used for EV71 sequencing, a total of 1, 040 active pores were detected. These pores were split into four groups, containing 482, 353, 161, and 44 pores respectively. After 19 h of sequencing, a total of 361 active pores were detected on the flow cell. The numbers of active nanopores in the flow cell used for CA16 were similar to those in the cell used for EV71-about 1, 000 before sequencing and 350 after sequencing.
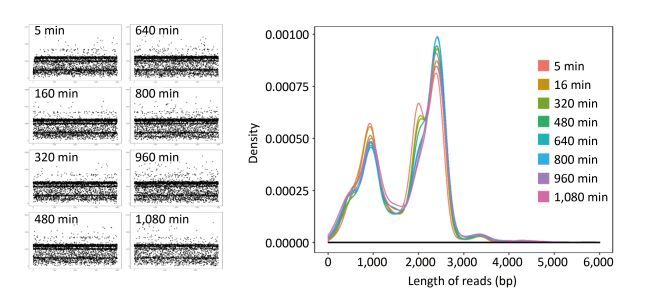
For single-sample sequencing of EV71, the sequencing run time was about 19 h. A total of 233 FASTQ files and about 932, 000 reads were generated. For multiple-sample sequencing of CA16, the running time was about 6 h. A total of 265 FASTQ files and about 1, 060, 000 reads were generated. In Figure 2, the curves in different colors respectively represent the length distribution of 4, 000 reads generated in the 5th, 160th, 320th, 480th, 640th, 800th, 960th, and 1, 080th minute during EV71 sequencing. The shape of the eight curves was almost identical and the scatter plots (Figure 2) were similar to each other. The lengths of the reads were mostly distributed around 1 kb, 2 kb, 2.8 kb, and 3.4 kb, and the abundance of 3.4 kb reads was lower than the others.
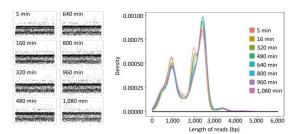
Figure 2. The read distribution at eight different time points during EV71 sequencing. The total sequencing run time for EV71 was about 19 h. The curves in different colors represent the length distribution of 4, 000 reads generated in the 5th, 160th, 320th, 480th, 640th, 800th, 960th, and 1, 080th min of sequencing, respectively. Every spot in the scatter plot represented one read (4, 000 for each plot) and Y axis represented the length of reads.
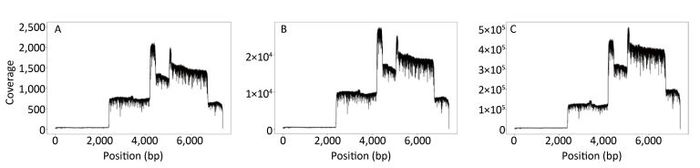
The sequence coverage profiles for EV71 were plotted using the data generated from the first min, from the first 14 min, and from the total data after 19 h, respectively (Figure 3A-C). Comparing the three plots, the coverage ratio at each position of the genome was similar, although the sequencing depths of three data sets were different. Nevertheless, each of them reached 100% genome coverage. The coverage depth for the non-structural protein regions was higher than that for the structural protein regions, due to the higher number of reads generated from the corresponding amplicons.

Figure 3. EV71 genome coverage. Sequence coverage profiles for EV71 plotted using the total data generated from the beginning of the sequencing to three different time points. (A) The data generated in the first min after MinION started running. (B) The data generated in the first 14 min, when the identity of the consensus sequence reached 99% compared to the Sanger reference sequence. (C) The data generated in the total run time of 19 h.
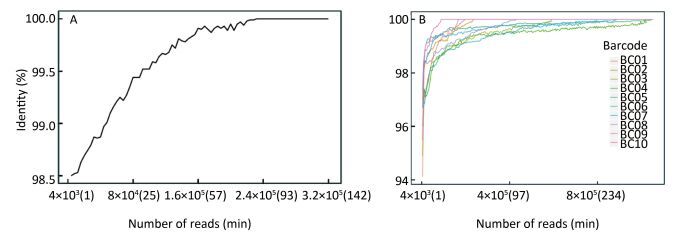
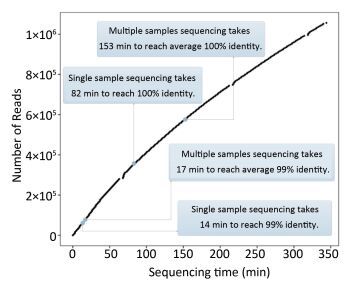
As Figure 4A shows, the consensus sequence assembled from the first generated FASTQ file (containing the first 4, 000 reads of EV71) showed 98.5% identity with the Sanger reference sequence. It took 14 min to reach 99% identity, and 82 min to reach 100% identity. For the multiple-sample sequencing of CA16 (Figure 4B), the consensus sequences from the first generated FASTQ files showed 94.12%-97.33% identity with the Sanger reference sequences. It took an average 17 min (range 4-36 min) to reach 99% identity and 153 min (19-330 min) to reach 100% identity for the 10 samples. Using a brand-new flow cell, about 1, 060, 000 reads were generated during 6 h of sequencing (Figure 5), which means the MinION could generate about 3, 000 reads per min.

Figure 4. The accuracy of Nanopore sequencing of the single EV71 sample and multiple CA16 samples. (A) For the single-sample sequencing of EV71, the consensus sequence from the first generated FASTQ file showed 98.5% identity with the Sanger reference sequence. It took 14 min to reach 99% identity, and 82 min to reach 100% identity. (B) For the multiple-sample sequencing of CA16, the consensus sequences showed an average 96.11% (94.12%-97.33%) identity with the Sanger reference sequence. It took an average of 17 min (range 4-36 min) to reach 99% identity, and 153 min (19-330 min) to reach 100% identity for the 10 samples.
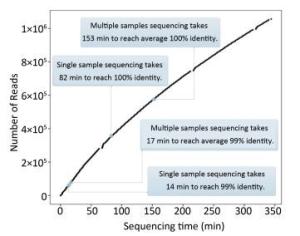
Figure 5. The relationship between the number of reads and the sequencing time. Using a brand-new flow cell, about 1, 060, 000 reads were generated during 6 h of sequencing, which means the MinION could generate about 3, 000 reads per min. But the change of curve slope indicated that the sequencing rate declined with time, which might be due to the loss of hundreds of active pores on running for several hours. This figure was plotted according to the generation of FASTQ files, which means the basecalling rate was also considered. The data curves for FASTQ and FAST5 files are almost identical to each other (data not shown).
-
The MinION system has been widely used to sequence various pathogens in many clinical situations because of its long reads, portability, real-time accessibility of sequenced data, and very low initial costs[20, 24, 30-31]. Compared to metagenomic sequencing, amplicon sequencing is a cost effective and sensitive approach for determining genomes of known pathogens and has been used for diverse viruses[32]. However, information is lacking on the MinION sequencing of EVs.
For viral amplicon preparation, because of the high variability of viral genomes, much effort has been made to find perfect conservative primer binding sites. In our study, we introduced many degenerate bases in the primers to increase the probability of PCR success and lowered the annealing temperature to improve the efficiency of long fragment amplification. The PCR products required for MinION sequencing are usually longer than those for SGS[6, 31]. As shown in Figure 1, the EV71 library consisted of four overlapping amplicons of 2, 2.5, 2.8, and 3.4 kb respectively. In addition to the target products, unexpected fragments of 1 kb were also amplified as seen in electrophoresis results (supplemental Figure 1 availble in www.besjournal.com). This non-specific amplification was attributable to the use of degenerate primers and the adjustment of the annealing temperature. The electrophoresis result was consistent with the read distribution in sequencing, where a 1 kb peak was observed (Figure 2), indicating that MinION sequencing accurately reflected the characteristics of the library.

Before the sequencing, the number of active nanopores on the flow cell was 1, 040. As shown in Figure 3, the reads generated in the first minute covered the entire genome of EV71. The distribution of read coverage after 19 h was not significantly different from that at the beginning of the sequencing. However, at the end of the 19 h process, the number of active nanopores remaining on the flow cell was only 361. The number of active nanopores will continue to decrease during even longer-term sequencing, which may eventually reduce the sequencing efficiency of the MinION. Interestingly, about 650 active pores were exhausted on brand-new flow cells used for sequencing both EV71 and CA16 despite the different running times (19 and 6 h respectively). This suggests that the service life of the flow cell may be more related to the data volume than the running time. The more frequently DNA strands pass through the nanopore, the more possible it is the nanopore will exhaust.
Previous researchers have observed higher mean read lengths during the first 8 h of MinION operation, suggesting the initial stages of a run are better for the generation of longer reads than the later stages[33]. However, our results indicated that the sequencing and basecalling performance was stable for 19 h (Figure 2). Although 679 active pores were lost during 19 running hours, the flow cell showed no obvious bias in terms of fragment length. This may be due to the different samples and library types used in our study (viral amplicons), or because the quality and method of the flow cell have continued to improve in recent years.
The even mix of different amplicons may reduce the sequencing bias. The four amplicons of EV71 were quantified and homogenized prior to pooling together. However, the Qubit-based quantitative method cannot eliminate interference from non-target PCR products, which explains the obvious difference in the coverage depth of the reads in different genomic positions (Figure 3). The lowest coverage depth area determines the minimum sequencing time and computational resources necessary to accurately obtain whole genome information. In viral genomes, the structural protein coding region(s) are generally the most variable due to selective pressure from the environment or host. These hypervariable regions are considered crucial sites that relate to the pathogenicity of the virus, and might determine the genotype/serotype of the virus. However, the hypervariable region(s) are much difficult to amplify or enrich than conserved regions. Taking our study as an example, the capsid sequence of EVs is more correlated with genotype than other genomic regions, but the codon degeneracy and sequence diversity make it difficult to amplify[1, 3]. Even with the use of multiple degenerate primers and lower annealing temperature, the abundance of the expected PCR product remains low, which makes the coverage depth of the corresponding genomic region relatively low (Figure 3). But, with a reasonable sequencing time, MinION can ensure sufficient sequencing depth for each genomic region and generate a consensus sequence with 100% identity to the Sanger result (Figure 4).
To determine the relationship between the sequencing accuracy and sequencing time, we analyzed total data generated every minute from when MinION started running. As shown in Figure 4, the accuracy of the EV71 consensus sequence increased over time and reached 100% identity to the reference sequence in 82 min, which means the following 17 running hours were not necessary. Theoretically, the minimum running time would be shorter if all four amplicons were homogenized with an equal amount. According to the read coverage derived in the first 14 min (Figure 3B), each amplicon should be sequenced at least 514 times to achieve 99% accuracy. Ideally, for a perfectly homogenized library with a known number of amplicons and samples, in a single run, the minimum sequencing time can be inferred roughly from to the relationship shown in Figure 5. Taking the CA16 experiment as an example, at least 20, 560 reads (514 reads × 4 amplicons per genome × 10 samples per run) would be needed to achieve 99% accuracy, and the minimum sequencing time needed to achieve this was 7 min (20, 560 reads / 3, 000 reads per min).
The relationship between the sequencing accuracy and sequencing time is also affected by the size of the amplicon and the quality of library. It might also relate to the base composition of the library sequences, which need to be further explored in our future research. In the presence of the 3.4 kb amplicons that were amplified with low PCR efficiency and non-specific fragments of 1 kb (Figure 2), 17 min was actually needed to reach 99% identity (Figure 5) rather than the theoretical minimum sequencing time of 7 min. Gel electrophoresis for amplicon size selection was attempted in our study to improve the library quality; however, extended hands-on operation time and lower recovery of long amplicons deteriorated the library quality. Even in the case of the removal of unexpected products, target products were lost as well (data not shown). Therefore, for viral amplicon sequencing, we do not recommend the use of electrophoresis, unless the amplification efficiency of each primer is high and the target product is abundant, which is not common for long fragment PCR of viral genomes.
Compared to SGS platforms, nanopore sequencing is a rapid and simple way for accurate viral whole genome identification. In this study, the accuracy of MinION was 98.5% for the single EV71 sample and 94.12%-97.33% for CA16 samples in the first minute of sequencing, which is already sufficient for preliminary identification of pathogens. The turnaround time from library preparation to the assembly of consensus sequences was < 1 h, making the total turnaround time of the whole process (sample in, result out) < 3.5 h. By contrast, it may take days to achieve the same result on SGS platforms[6].
Our research demonstrated that the MinION nanopore sequencer is suitable for whole genome sequencing of EVs with sufficient accuracy and has the great potential, as a fast, reliable and convenient method for routine use in the laboratory. Future study will be testing this method with clinical samples. Thus, MinION might offer a new option for pathogen detection and genome sequencing in places with limited facilities[17, 34]. In many local research facilities and primary medical institutions in developing countries, it is difficult to establish and maintain a high-throughput sequencing platform, which usually requires significant amounts of laboratory space, specialized equipment and large-scale computing resources[16]. By using an automated device like VolTRAX, the risk of contamination and error caused by manual operation of MinION will be further reduced. When library preparation becomes simpler and more consistent, MinION will be more powerful in the sequencing of whole viral genomes and many other samples.
-
No conflict of interest to declare.
-
MA Xue Jun, XU Wen Bo, WANG Ji, and DONG Xiao Ping designed the study; WANG Ji prepared the library and performed the sequencing experiment; KE Yue Hua analyzed the sequencing data; ZHANG Yong designed the primers and cultured the viral strains; HUANG Ke Qiang prepared the amplicons; WANG Lei and SHEN Xin Xin prepared the reagents and the labware; MA Xue Jun and XU Wen Bo supervised the research; WANG Ji wrote the manuscript. The first three authors in the author list contributed equally to this research.
the National key research and development plan 2016YFC1200900
the National key research and development plan 2016TFC1202700
Beijing Municipal Science & Technology Commission project grant numbers D151100002115003
Guangzhou Municipal Science & Technology Commission project grant numbers 2015B2150820

 Quick Links
Quick Links
 DownLoad:
DownLoad: