
-
Highly active antiretroviral therapy (HAART) is used globally and has significantly reduced the morbidity, mortality, and transmission of human immunodeficiency virus (HIV)-related illness. However, the attendant shortcomings such as toxic side effects, necessity of life-long therapy that is expensive, and inefficiency in eradicating latent viral infections have greatly limited the application of HAART[1, 2]. Alternate strategies that overcome these limitations are urgently needed. RNA-based gene therapies are a candidate, which includes antisense RNA, RNA decoy, ribozyme, RNA interference (RNAi), and Cas9/single guide RNA (sgRNA) technologies. The last three decades have witnessed tremendous progress in the development of these RNA-related approaches for HIV. Their prowess in the inhibition of actively replicating virus or elimination of latent reservoirs of infection has been documented[3, 4]. However, the clinical use of therapeutic RNAs theoretically entails two critical steps: delivery of the RNA to the target tissues and subsequent maintenance of adequate doses. Unfortunately, naked RNAs are inherently susceptible to RNase attack and immune system eradication that severely shortens their half-life in the bloodstream and limits the duration of their in vivo action to minutes. Equally important, the hydrophilicity of RNA molecules reduces their affinity to the membranes of recipient cells and the large molecular weight of naked RNAs impedes their passage across cell membranes, which directly leads to poor cellular uptake or markedly reduced antiviral effect. This poor bioavailability has spurred the need for RNA-based anti-HIV drug delivery systems.
Among the many traditional drug delivery technologies to improve the therapeutic effect of RNAs, viral vectors, nano-polymers, liposomes, and target peptides are commonly used and considered practical. Although many modifications or improvements have been introduced, these traditional delivery platforms have not met the expectations for anti-HIV gene therapy and have remained in the pre-clinical stage. Naturally inherent limitations include the induction of an immune response[5], inability to cross biological barriers[6], lack of in vivo stability[7], and the likelihood of binding to serum proteins[8]. Exosomes are an attractive alternative to these traditional RNA delivery platforms. Exosomes are nanosized intraluminal vesicles secreted by a variety of cell types that are present in almost all biological fluids. They function as natural transporters of bioactive molecules between the exosome-producing and recipient cells, and play a significant and diverse role as an intercellular communicator[9]. Exosomes are specialized in long-distance intercellular communications[10, 11], facilitating the transfer of nucleic acids, such as functional messenger RNA (mRNA) and microRNA (miRNA), for subsequent protein expression in target cells in a highly efficient and economical manner concerning substance exchange[12, 13]. The expectation is that these naturally occurring vesicles can be adopted as delivery vehicles of exogenous RNAs in the presence of in vivo transport obstacles that include extracellular matrix, cell membrane, and the endothelium. In this up-to-date review, we describe how exosomes can be harnessed to overcome the obstacles to the inefficient systemic delivery of anti-HIV RNAs across biological barriers. Most studies on HIV have been performed with HIV type 1. Accordingly, in this article we refer to the virus as 'HIV'.
-
Therapeutic anti-HIV RNAs, including antisense RNA, RNA decoy, ribozyme, small interfering RNA (siRNA), miRNA, and single guide RNA (sgRNA, associated with Cas9) have generated global enthusiasm[3, 14-21]. They all function as small RNAs that can specifically recognize viral genes or proteins using different mechanisms. Antisense RNAs are single-stranded RNA molecules that target complementary HIV RNA in target cells, resulting in RNase H dependent degradation of the hybrid double-stranded mRNA/RNA or inhibition of the target gene expression by blocking ribosome binding[3]. RNA decoys are also single-stranded RNA molecules, normally 15 to 40 bases long, with a high sequence similarity to the cis-acting elements, which results in competitive binding to viral replication-related regulatory proteins to disrupt virus transcription or maturation[14]. Ribozymes are catalytically active small RNA molecules. Hammerhead and the hairpin ribozymes have been most intensively studied. Both contain a target binding domain that is naturally self-cleaving, but which can be engineered to cleave specific sequences by mutation of the substrate recognition sequences. This provides flexibility in targeting diverse HIV genes[15]. RNAi is widely used to silence HIV genes by the RNA-induced silencing complex (RISC), which mediates the degradation of mRNA in a sequence-specific manner. The RISC can be delivered either as a synthetic 21 basepair double-stranded siRNA or as an expressed precursor short hairpin RNA (shRNA)[16]. Some endogenous miRNAs expressed in T lymphocytes, macrophages, and monocytes, as well as chemically synthesized miRNA mimics, can protect against HIV infection by triggering RISC-mediated HIV gene silencing[17]. The Cas9/sgRNA system is comprised of an easily programmable short sgRNA and a multifunctional Cas9 nuclease. This system has been widely heralded in recent years as it can be adapted to selectively target and cut almost any double-strand DNA sequence and disrupt gene expression from virtually any organism[18-20]. Not surprisingly, the application of Cas9/sgRNA has been explored to permanently disable the HIV genome by ablating large segments of integrated proviral DNA in latently infected cells. The current therapies are unable to suppress viral gene transcription from integrated proviral DNA or eliminate the transcriptionally quiescent proviral genomes[21].
During the HIV lifecycle, the uncoated viral RNA, viral transcript, and proviral DNA in the infected host cell present ample interaction sites for therapeutic RNAs to combat HIV. Numerous pre-clinical trials aimed at knockdown or ablation of viral lifecycle-related genes, including gag, pol, env, vif, nef, vpr, vpu, tat, rev, long terminal repeat (LTR), and the virus entry co-receptor CCR5/CXCR4, have provided substantial evidence of the therapeutic potential of RNA-based approaches for the treatment of HIV and acquired immunodeficiency syndrome (AIDS). However, mainly due to the lack of effective vectors[22, 23], most of pre-clinical trials have failed to progress to the clinical stage. Information on current clinical trials is available from the United States National Institutes of Health clinical studies database (www.clinicaltrials.gov) and in Table 1. As shown in Table 1, five RNA species (antisense RNA, RNA decoy, ribozyme, shRNA, and sgRNA) have been adopted in RNA-based therapeutic platforms in clinical anti-HIV gene therapy. These RNA-based clinical interventions depend on autologous or allogeneic transplantation of CD4+ T cells or on CD34+ hematopoietic stem/progenitor cells (HSPCs) transfected ex vivo with genes encoding anti-HIV RNAs. Furthermore, these cell-dependent RNA delivery technologies rely on a selective advantage for the expansion of transduced cells, which is difficult to achieve in subjects with well-controlled HIV levels being treated with traditional regimens because of the low engraftment levels and great difficulty in implementation[24-26]. Scrutiny of the clinical phase data in Table 1 concerning the first posted time and last status highlights the slow and problematic development of these CD4+ or CD34+ cell-mediated RNA deliveries for anti-HIV gene therapy, which has spurred the demand for novel carrier RNA-based drug platforms capable of delivering the therapeutic RNA across the biological barriers to HIV pools or inaccessible HIV-bearing sites, such as macrophages, microglia, and astrocytes in the brain[27, 28].
NCT Number RNA Types Targets Interventions Phases First Posted Last Status NCT00001535 Antisense RNA TAR, rev Syngeneic T cell Phase Ⅰ Nov. 4, 1999 Completed NCT00131560 Antisense RNA env Autologous T cell Phase Ⅱ Aug. 18, 2005 Active NCT00622232 NCT00295477 Antisense RNA env Autologous T cell Phase Ⅰ Feb. 23, 2006 Active Phase Ⅱ NCT00074997 Ribozyme tat, vpr Autologous HSPC Phase Ⅱ Dec. 31, 2003 Active NCT01177059 NCT00002221 Ribozyme tat, rev Autologous PBPC Phase Ⅱ Aug. 31, 2001 Completed NCT01734850 shRNA CCR5 T cell, HSPC Phase Ⅰ Nov. 28, 2012 Active NCT02390297 Phase Ⅱ NCT02378922 shRNA CCR5 Autologous HSPC Phase Ⅰ Mar. 4, 2015 Suspended NCT03164135 sgRNA CCR5 Allogeneic HSPC May. 23, 2017 Recruiting NCT01153646 shRNA tat/rev Autologous T cell Early Phase Ⅰ Jun. 30, 2010 Terminated RNA decoy Tat ribozyme CCR5 NCT00569985 shRNA tat/rev Autologous HSPC Dec. 10, 2007 Active NCT01961063 RNA decoy Tat NCT02337985 ribozyme CCR5 NCT02797470 shRNA CCR5 Autologous HSPC Phase Ⅰ
Phase ⅡJun. 13, 2016 Suspended RNA decoy Tat Note. TAR, trans-activation response element; PBPC, peripheral blood stem cell; HSPC, hematopoietic stem cell; DC, dendritic cells. The data are current as of December 29, 2017 from www.clinicaltrials.gov. Table 1. Clinical Trials of RNA-based Gene Therapies in HIV Infections
-
Exosomes are membrane-bound phospholipid nanovesicles 40-100 nm in diameter, which are actively secreted by most, if not all, mammalian cells. The classical view of exosome biogenesis holds that they form intracellularly by the inward budding of the endosomal membrane to create vesicle-containing multivesicular bodies (MVBs), which eventually fuse with the plasma membrane and release their internal vesicles (the exosomes) into the extracellular medium, thus giving exosomes the same membrane topology as that of the cell of origin[9]. Conversely, the closely related microvesicles (100-500 nm diameter) bud directly from the plasma membrane. Many investigations, especially in the field of drug delivery, have established that extracellular vesicles (EVs) comprise both exosomes and microvesicles, since the complete separation and purification of the two is extremely arduous[29]. According to the International Society for Extracellular Vesicles, characteristics of exosomes are the presence of exosome-associated surface markers including TSG101, Alix, flotillin 1, integrins, cell adhesion molecules CAM, and tetraspanins (CD9, CD63, CD81)[30]. Proteome studies have shed light on the cargo composition of exosomes from different sources and have demonstrated that the similar protein composition of exosomes and parental cells. Aside from proteins, exosomes can intrinsically act as an RNA shuttle service in the body for many coding or noncoding RNA species, including mRNAs, miRNA, Y RNA, transfer RNA fragment, vault RNA, structural RNA, repeat sequences, and RNA transcripts overlapping with protein coding regions[13, 31, 32]. The use of exosome-containing miRNAs in HIV therapy has been extensively studied. In recent years, a growing number of host miRNAs with potent anti-HIV potential has been discovered in host exosomes. These mainly include miR-28, miR-29a, miR-29b, miR-125b, miR-149, miR-150, miR-198, miR-223, miR-324, miR-378, and miR-382[33]. They are not merely the in vivo presence of a natural exosomal model of delivering anti-HIV RNAs. Theoretical predictions have indicated that artificially engineering anti-HIV RNAs into exosomes may be feasible in vitro and the role of these engineered exosomes for in vivo delivery of anti-HIV RNAs may be significant and underappreciated.
In fact, their diverse intrinsic properties could make exosomes an ideal nanocarrier for the in vivo delivery of exogenous RNAs. The first of these intrinsic properties is size. The small size of exosomes allows them to escape phagocytosis by the mononuclear phagocyte system (MPS), which clears particles generally larger than 100 nm in size. The result could be the relatively high level of retention of exosomal carriers in vivo. Secondly, as natural nanocarriers derived from endogenous cells, exosomes are more biocompatible with the immune system than any foreign formulations, and hence cause less cytotoxicity when used in vivo. In addition, allogenic exosomes collected from body fluids or cultured cell supernatants may have a privileged immune status. Supporting this notion, immature dendritic cells (DCs) produce large quantities of exosomes that lack T-cell activators, such as major histocompatibility complex (MHC) Ⅰ, MHC-Ⅱ, and CD86[34], and exosomes derived from CD8+ T cells display potential immunosuppressive activity[35]. Therefore, it can be speculated that the origin of exosomes may be the defining factor for the in vivo exosomal tolerance, both from toxicological and immunological viewpoints. Thirdly, research studies have indicated the avid penetrability of exosomes across biological barriers and the subsequent transfer of their cargoes to target cells. For example, semen-derived exosomes may serve as a potential delivery system because these exosomes can naturally traverse mucosal barriers that hinder other artificially created vesicles or modes of drug delivery[36]. As another example, brain endothelial cell-derived exosomes can be administered safely to the brain and can deliver their payload across the blood-brain barrier (BBB) that essentially restricts therapeutic drugs from entering into the brain[37]. Fourthly, owning the very notable characteristic of an internal microenvironment protected from the extracellular milieu by a phospholipid membrane-bound bilayer, exosomes are naturally endowed with a high affinity for recipient cells that promote the internalization of their payload in target cells via direct membrane fusion, endocytosis, or cell-type specific phagocytosis[38-40]. Lastly, the low affinity of exosomes to the liver or the lack of unwanted accumulation of therapeutic cargoes in the liver explains the favorable toxicity profile as well as the highly efficient delivery to target cells by these vesicles[41, 42]. Over the past two decades, exosomes engineered with therapeutic RNAs in vitro have been demonstrated in many studies to be successful in the transfer of their RNA cargoes in vivo, and the use of exosomes as carriers of therapeutic RNAs for cancers has gathered momentum[43]. Still, the use of exosomal carriers in HIV therapy is in its infancy.
-
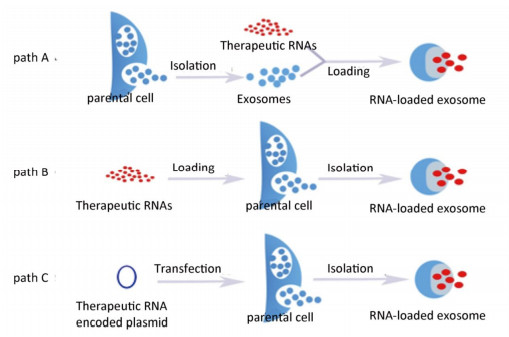
Accumulating evidence indicates that sample preparation, isolation, characterization, and quantification of exosomes are vital in facilitating exosome-mediated drug delivery. A detailed and comprehensive assessment of these procedures is beyond the scope of this review. We refer the reader to other reviews in the literature[44-46]. Once isolated or purified, exosomes can be concentrated, lyophilized, and reconstituted[47]. Three distinct approaches can be utilized to load exosomes with RNA-based anti-HIV therapeutic cargoes (Figure 1). In one approach, which we term path A, loading involves raw exosomes directly isolated from parental cells. The loading can involve incubation at room temperature, freeze/thaw cycles, extrusion, sonication, electroporation, permeabilization with saponin, and treatment with commercial transfection reagent. In the second approach (path B), parental cells are loaded with exogenous RNAs of interest, which are then encased into the exosomes by the inward budding of the endosomal membrane during exosome formation and the subsequent release into a conditioned medium. The final approach (path C) involves the transfection or infection of parental cells with DNA encoding therapeutically active RNAs, which are then recruited into exosomes. Each approach has its advantages and limitations and may be dictated by the type of the therapeutic cargo, cell of exosomal origin, maturity of laboratory techniques, objective of the experiments, etc.
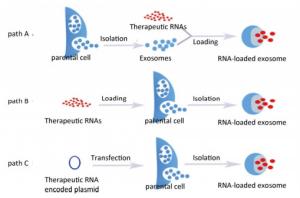
Figure 1. Different strategies for the loading of RNAs into exosomes.
-
Path A is widely used in the packaging of exosomes and is considered the most direct and convenient way to equip exosomes. It eliminates the need for expensive and time-consuming ex vivo transduction compared with path C, and it can yield many cargo-bearing exosomes combine from just several isolations. In this way, the anti-HIV RNAs can be chemically synthesized and the exosomes can be acquired from many cell types similar to the allogeneic immature DCs. The latter cells are an outstanding candidate as exosomes from these cells persist for a long time after being injected into a patient's body and are not immunogenic[34]. In the loading process, the stretching and reshaping of exosomes upon sonication and extrusion enables therapeutic diffusion of RNAs across the relatively tight and highly structured lipid bilayers[47]. Furthermore, treatment with an efficient permeabilization agent like saponin or other commercial transfection reagent may also increase RNA loading into exosomes. However, exosomes carry a negative surface charge, which precludes the formation of complexes of electrostatic RNA molecules. To solve this issue, pre-complexation of siRNA via a cationic liposome-based intermediary followed by the fusion with isolated exosomes has been suggested for their loading with siRNA[48]. Furthermore, an elevated temperature (37 ℃) may be used to improve siRNA loading into exosomes or other types of extracellular vesicles[49]. Recently, O'Loughlin et al. optimized the loading of EVs with cholesterol-conjugated siRNAs (cc-siRNAs) by incubating 15 molecules of cc-siRNA per EV at 37 ℃ for 1 h in a volume of 100 μL, which maximized the retention of siRNA and facilitated the concentration-dependent silencing of human antigen R, which is a therapeutic target in cancer therapy involving EV-treated cells[50]. Notably, some anti-HIV RNAs (antisense RNAs, RNA decoys, and miRNA mimics) are normally single-stranded, and linear RNAs that are extremely unstable exposed to laboratory environments in which pollutant-derived unfavorable physicochemical factors and nucleases may exist. Thus, loading these RNAs into exosomes by path A is inadvisable. However, ribozyme, siRNA, and precursor hairpin RNA (pre-siRNA or pre-miRNA) are relatively stable because of their double-stranded or secondary structure, allowing flexibility in selecting the best way to load exosomes with these RNA molecules. Once anti-HIV RNAs are loaded into exosomes, the RNA can be quantified using specific and customized small RNA quantitative polymerase chain reaction (qPCR) systems, conventional mRNA qPCR analysis, or fluorescently-labeled small RNA systems in intact or disrupted exosomes[41, 51, 52]. According to the exosomal origin, some natural exosomal miRNAs that are stable and present in high concentrations can be used as endogenous controls to normalize other exosome-encased small RNAs and ensure the experimental reliability[52-54].
-
Path B is less commonly used. However, it can be extremely useful for in vivo drug delivery. As an example, Qazi et al. reported DC-derived exosomes loaded with chicken egg ovalbumin using path B efficiently elicited specific transgenic T cell proliferation in vivo, while exosomes loaded with ovalbumin by path A were more efficient in vitro, highlighting the importance of formulation strategies in some cases[55]. To preserve the therapeutic drugs against degradation in parental cells (monocytes and macrophages) and increase loading capacity, drugs can be packaged into a polymer-based nanocontainer before path B[47, 56]. Importantly, the protective nanoparticles are typically restricted in size. The optimal nanoformulation for loading into parental cells features a relatively large size (approximately 200 nm), resulting in improved accumulation in parental cells and drug reshuffling into exosomes. Undoubtedly, this nanotechnology is worth considering for the loading of anti-HIV RNAs, although the size of protective nanoparticles may need to be adjusted. Additionally, equipping exosomes with therapeutic RNAs by path B crucially depends on the conditions used to maintain the vitality of parental cells to obtain large amounts of exosomes.
-
In path C, exosomes containing RNA cargoes can be obtained indirectly with a high level of RNA integrity. This approach is promising for the assembly of exosomal vectors. In this elegant approach, anti-HIV RNAs (e.g., antisense RNA, RNA decoy, ribozyme, miRNA, shRNA, and sgRNA) derived from the genetically altered parental cells can be internalized into exosomes that are then isolated from culture supernatants. Yeo et al. discovered that mesenchymal stem cells (MSCs) can produce a large number of exosomes, and suggested that these cells may be efficient for ex vivo production of exosomes in a clinically applicable scale[57]. Moreover, the uninterrupted production of therapeutically efficacious exosomes is possible using genetically immortalized MSCs[58]. Once the anti-HIV RNA genes are integrated into the immortalized MSCs, the therapeutic RNAs enclosed in the exosomes can be acquired from the culture supernatants continuously, which bypasses the loading procedures in path A. Exosomes from genetically-altered macrophages containing encoded neurotrophic factor as well as its genetic material (DNA and mRNA) can efficiently transfer their contents to neurons in the brain, resulting in protracted (months) long synthesis of neurotrophic factor and prolonged attenuation of illness (over 40 days) in a mouse model of neuroinflammation[59]. Therefore, exosomes secreted from cultured cells transiently transfected with plasmids harboring genes encoding anti-HIV RNAs may simultaneously encase the plasmids and their encoded anti-HIV RNAs and could possibly contribute to long-lasting anti-HIV bioactivity in vivo. In path C, there is no need to obtain autogenous CD4+ T cells or other HIV infected cells from HIV patients as the exosomal origin to avoid the risk of a secondary infection because of reservoirs of latent virus.
-
In some cases, path A, B, and C can be applied in an efficient combination mode when a cocktail of several species of anti-HIV RNAs participates in the loading of exosomes. This is particularly the case when an anti-HIV Cas9/sgRNA system is designed for loading into exosomes, because sgRNA and Cas9 protein can be encased into exosomes independently or can be kept physically separate from each other[60]. The two components differ greatly in physical and chemical properties, such as size and stability, which may require different loading strategies within paths A, B, and C.
Prior to the recognition of exosomes as vectors in the Cas9/sgRNA system, the in vivo application of the Cas9/sgRNA system aimed to either attack the established HIV proviral DNA to cure the infected cells, or to await and then immediately attack the reverse transcribed viral DNA produced in a future infection to prevent the infection of uninfected cells. Both aims focused on the adeno-associated virus (AAV) vector, which was used in two anti-HIV studies for the in vivo delivery by the Cas9/sgRNA system to knock-out HIV LTR and gag genes in transgenic/humanized mice or rats, with great anti-HIV potential due to the lack of immunogenic behavior in vivo[61, 62]. However, the restricted cargo size (approximately 4.5 kb) of AAV is an obstacle for packaging the commonly used Streptococcus pyogenes Cas9 (approximately 4.2 kb)[63-65]. Thus, the insertion of additional components, such as sgRNA, spare oligonucleotide or extra genes, is challenging for single AAV vector-mediated Cas9/sgRNA gene-based delivery[65]. In addition, the Cas9 nuclease should be delivered into cells in protein-based formats other than gene-based delivery that depends on plasmids carrying Cas9 genes[66]. First, unlike gene-based delivery, protein-based delivery has no potential issue of permanently integrating Cas9 gene into the host genome that may disrupt the host gene expression. Next, in gene-based Cas9 delivery, the long-term constitutive expression of Cas9 exacerbates the problem because repeated exposure of Cas9/sgRNA to non-specific genes can lead to large off-target effects, which are one of the major limitations of this genome editing system[67, 68]. The AAV-mediated gene-base delivery of Cas9/sgRNA thus appears limited. The versatile exosomal carriers may be a potential alternative.
Protein-based delivery of Cas9 is preferred when exosomes are involved as delivery vectors, despite the recent report that cancer-derived exosomes loaded with plasmids expressing Cas9/sgRNA system via electroporation can efficiently deliver their cargoes in vivo[69]. However, given the current lack of precedent for the loading of protein-based Cas9 coupled with sgRNAs into exosomes, much attention should be given to the loading procedures. Regarding the distinct molecular size, weight, structure, and charge distribution of Cas9 protein and sgRNAs, these two kinds of cargoes can be processed at different times or spaces with various strategies to maximize loading efficiency. This provides ample flexibility in the application of the Cas9/sgRNA-related exosomal delivery system. For instance, Cas9 protein and synthetic sgRNAs can be loaded directly into exosomes by path A at different times using different physical means, such as incubation at room temperature, freeze/thaw cycles, sonication, or electroporation. To ensure the integrity of Cas9 protein and sgRNAs, priority should be given to moderate measures, such as incubation at room temperature. In addition, loading Cas9 protein by path C via stably transfected Cas9 genes and then loading the isolated Cas9 protein-contained exosomes with a series of well-designed anti-HIV sgRNAs by path A seems workable. Path B may be brought into the combination mode for the testing of a special function. Finally, a smaller sized Cas9 may be beneficial to achieve a relatively high loading efficiency for the limited internal space of exosomes. In this respect, Cas9 protein from Streptococcus aureus is smaller in size than the protein from other sources, such as Streptococcus pyogenes[64, 70].
-
Normally, exosomes spontaneously target recipient cells to some degree through a range of surface adhesion proteins or target ligands. Supporting this point, cancer-derived exosomes function as natural carriers that can efficiently deliver their cargoes to the focus of disease due to the selective accumulation in ovarian cancer tumors of SKOV3 xenograft mice; although the mechanism is unclear, it most likely involves cell tropism[69]. However, experiences from pharmaceutical research have indicated that technological advances in the application of exosomal carriers for RNA delivery should aim at target design. Exosomes are the natural means of communication between adjacent or distant cells with high cellular affinity. Thus, delivery to target sites may lessen the loss of exosomal carriers captured by nontarget cells or adjacent cells at a systemic level. Alvarez-Erviti et al. harnessed the RNA transporting capacity of exosomes in the first proof-of-concept study for the targeted delivery of exogenous siRNA utilizing these vesicles[41]. Immature DCs were transfected with plasmids encoding an exosomal protein, lysosomal-associated membrane protein 2 (Lamp2b), and fused with a 29-mer neuron-specific peptide derived from the rabies virus glycoprotein (RVG) that specifically binds to acetylcholine receptors expressed on neuronal cells and the BBB[71]. RVG-targeted exosomes were subsequently isolated from DC cultures and loaded with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) siRNA by electroporation. Targeted in vivo delivery of GAPDH siRNA inside RVG-targeted exosomes following intravenous injection into tail veins of a mouse model resulted in specific gene knockdown of GAPDH in several regions of mouse brain, such as cortex, striatum, and midbrain[71]. This study highlighted a completely new paradigm for exosome-based targeted delivery of siRNA to the brain and suggested that exosome-based targeted delivery to CD4+ T cells or other latent HIV reservoirs may be an achievable milestone in combating HIV.
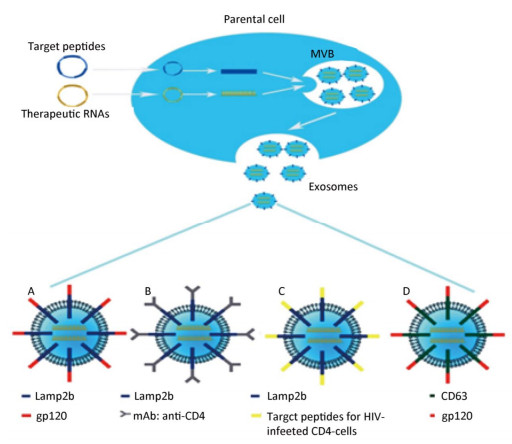
Before the full potential of this biotechnology can be achieved, the feasibility of several technical aspects for RNA-based anti-HIV therapy targeted delivery relying on the exosomal surface-modifying process need to be established (Figure 2). Targeting moieties, such as viral envelope protein gp120 cloned into Lamp2b, should be tested to improve the delivery to CD4+ T cells (Figure 2A). Monoclonal antibody fragments to the ectodomain of CD4 can be introduced to replace gp120 for the binding of exosomes to CD4+ cells (Figure 2B). Furthermore, some peptides should be explored concerning their selective homing to HIV-infected CD4-cells, such as oligodendrocytes and astrocytes, which always serve as latent HIV reservoirs in the brain[72, 73], to extend this technology to brain delivery (Figure 2C). Of course, these targeting moieties including peptides and antibody fragments also can be cloned onto other exosomal surface proteins, such as CD63 other than Lamp2b (Figure 2D). Finally, alternative targeting methods should be tested, in which ligands are captured to the surface of exosomes directly through chemical conjugation, bypassing the time-consuming cloning, followed by recombinant expression procedures when screening a large battery of ligands. Sometimes, to save time and energy, the simultaneous design of targeted and RNA-loaded exosomes is worth trying. For example, the Lamp2b-gp120 fusion protein and anti-HIV shRNA can be encoded in the same plasmid that will be transfected into the parental cells of exosomal origin. Once exosomes are loaded with therapeutic RNAs and target peptides, they can be administered using distinct methods in clinical trials. A study is investigating the use of plant exosomes in the oral delivery of curcumin to normal and colon cancer tissue (NCT01294072). Another study is assessing the effect of intravenously infused microvesicle and exosome therapy on β-cell mass in type 1 diabetes mellitus (NCT02138331).
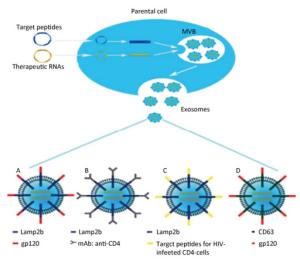
Figure 2. Four target designs of exosomes for anti-HIV RNAs delivery (In this diagram, the RNA loading procedure is assumed to adopt path C).
-
Studies concerning the relationship between exosome and HIV have reported the anti-HIV bioactivity of exosomes from several different origins, even without loading of anti-HIV RNAs. Tumne et al. found that CD8+ T cell-derived exosomes inhibited the transcription of the HIV LTR promoter and elicited a non-cytotoxic suppressive effect on HIV replication in vitro, in a pathway that was seemingly independent of exosomal internalization[74]. Naslund et al. observed that EVs isolated from human breast milk of healthy donors prevented the HIV from infecting monocyte-derived DCs and subsequent transfering to CD4+ T cells, via competitive binding to the DC-SIGN receptor[75]. Another study described that EVs isolated from the semen of healthy men also had the potential to inhibit HIV transmission by disrupting the viral replication in the vaginal epithelium[36]. However, the accumulating data imply that there is little room left for discovery of new origins of exosomes with congenital anti-HIV activity. Thus, research should concentrate on engineered exosomes that carry anti-HIV RNAs as a supplement for the rarity of the naturally anti-HIV exosomes.
Loading exosomes with various types of anti-HIV RNAs can be considered as a supplement for the scarcity of host-derived anti-HIV RNA content of exosomes, although RNA species naturally carried by host exosomes that confer protection against HIV infection are possibly more than simple miRNAs mentioned earlier. Moreover, loading naturally occurring anti-HIV exosomes with artificial anti-HIV RNAs may enhance the therapeutic effect. Equipping the anti-HIV RNA-loaded exosomes with target peptides on their surface is vital to attain higher antiviral efficacy, since it raises the level of therapeutic RNAs enrichment only in target tissues while reducing the off-target effect in nontarget cells, in that it is difficult, perhaps impossible, to create a perfect sequence design of therapeutic RNAs, especially sgRNAs, to completely bypass the conserved transcription factor binding sites to minimize the likelihood of altering host gene expression by bioinformatics screening and off-target prediction[18, 76]. Combined with all the intrinsic merits of exosomes, the target design of RNA-loaded exosomes may achieve the highly effective delivery of therapeutic RNAs to HIV-infected cells or a safe adjuvant therapy. By the Loading brain-penetrable exosomes with the Cas9/sgRNA system and equipping them with target peptides directed to the latent HIV reservoirs in the brain, such as macrophages, microglia, astrocytes, and oligodendrocytes, a BBB-spanning elimination of HIV may soon be possible. Moreover, changing the exosomal surface modification for systemic multi-target delivery of anti-HIV RNAs including sgRNAs and others may enable a systemic double-suppression of viral DNA and RNA. If so, a thorough cure for HIV could be envisioned.
-
One of the most formidable impediments in the loading of exosomes for RNA delivery may be theoretical. During the last two decades, a series of protein members mainly from RAB family[77, 78], tetraspanin family[79, 80], and endosomal sorting complex required for transport (ESCRT) family[81, 82] have been discovered to participate in the exosome formation and cargo selection. The exact mechanism that controls the loading of RNA cargoes into exosomes remains ambiguous and needs to be studied[83]. Thus, it is uncertain whether the RNAs of interest can be efficiently sorted into exosomes by path C, whether the plasmid-encoded target peptides can be positioned on the exosomal surface, and whether all components of a cocktail of various anti-HIV RNA species can be internalized into exosomes by path C at the same time. Notably, some unknown components derived from the parental cells may be encased into exosomes and may be cytotoxic in target cells. All these uncertainties need to be validated in pre-clinical settings.
Another main challenge facing the clinical applications of exosomes are technical aspects. By their very nature, exosomes already encase numerous nucleic acids and proteins from the parental cells, implying that a relatively low loading capacity can be achieved with these carriers[84]. Due to the limited internal space, concentration-dependent downstream applications of exosomes might be limited. To address this issue, further optimization and recombination of the loading procedures of path A need to be explored. Using path B, loading parental cells with a higher concentration of therapeutic agent through cationic peptide-or liposome-mediated fusion may enable the loading of exosomes with relatively high levels of drugs. Concerning path C, strong promoters and enhancers of target genes should be employed in the vector construction process. Another technological obstacle in using exosomal drug formulations in the clinic is whether the production of exosomes is reproducible or scalable[85]. Indeed, it is no exaggeration to say that the exosome yield per cell could impact the final production cost in clinical applications. Thus, the choice of parental cells is crucial. Immortalized mesenchymal stem cells are regarded as an ideal option for the uninterrupted and high yield of exosomes[57, 58]. Optimizing the cultivation conditions could also increase the exosome yield, as might culturing parental cells for an extended time period or the use of low pH[86, 87]. Interestingly, the HIV accessory protein, Nef, which is a potential regulatory factor of the physiological action of endocytosis and exocytosis, has been transduced into parental cells and evidently increases the cellular release of exosomes[88, 89]. Finally, to scale-up exosomal production, specifically designed bioreactor systems have been used, with documented significant increase in the yield of exosomes and EVs compared with the conventional tissue culture flask technique[90, 91].
-
Engineered exosomes may well serve as the next generation of RNA-based anti-HIV drug delivery tool. They have an unparalleled potential in the treatment of HIV/AIDS, which is life-threatening and for which there is not an effective pharmacotherapy. Future breakthroughs will most possibly focus on the production of large amounts of well-characterized exosomal carriers with high loading capacity. Regrettably, the final hurdle in exosome-based therapeutic interventions is the lack of knowledge of all the components that exosomes inherit from the parental cells. Thus, the mechanisms of exosome-mediated in vivo material transfer remain a matter of debate. Ongoing studies on these issues may open the door to uncharted territory where exosomal carriers for RNA-based anti-HIV gene therapies can be freely applied clinically.
Acknowledgments This research was supported by the Youth fund of National Natural Science Foundation of China.
Author Disclosure The authors declare no competing interest here and this article does not contain any studies with human or animal subjects performed by any of the authors.
HTML
RNA-Based Anti-HIV Gene Therapies
Exosomes: Intrinsically FIT To Deliver Anti-HIV RNAS
Loading Exosomes With RNA-based Anti-HIV Therapeutics
Path A
Path B
Path C
Combination of Paths A, B, and C
Target Design For Exosome-mediated Anti-HIV RNA Delivery
Therapeutic Potential of Anti-HIV RNA-loaded Exosomes
Challenges Faced by Exosome-mediated RNA Delivery
Future Directions
Youth fund of National Natural Science Foundation of China 81502974

 Quick Links
Quick Links
 DownLoad:
DownLoad: