
HTML
-
Febrile jaundice is a common indicator of certain infectious diseases, and is a significant public health concern in developing countries, including Sierra Leone. A broad spectrum of pathogens may cause febrile jaundice, including parasites (e.g., malaria, toxoplasmosis, schistosomiasis), bacteria (e.g., leptospirosis, typhoid, typhus, borreliosis), and viruses [e.g., yellow fever virus (YFV), hepatitis virus (HAV, HBV, HCV, HEV), lyssavirus, Marburg virus, Ebola virus, Crimean-Congo hemorrhagic fever virus (CCHFV), hantavirus, Epstein-Barr virus (EBV), rhinovirus, mumps virus, measles virus, rubella virus, and coxsackievirus][1-8]. In low-income countries, the spectrum of causative pathogens is complex owing to poor sanitary conditions.
Outbreaks of Ebola in 2014 drew global attention, but other endemic pathogens have been largely ignored in the post-Ebola era in Sierra Leone. No information is available regarding the causative agents of endemic outbreaks other than those of Ebola, yellow fever, or malaria. The World Health Organization (WHO) has conducted epidemiological surveillance for YFV by antigen detection for over a decade. Infections associated with YFV-negative samples may be caused by as-yet-undefined pathogens.
Using random oligonucleic acids instead of predefined primers, unbiased metagenomic shotgun sequencing (MSS) enables the identification of any virus sequence, known or unknown, local or imported, and even outbreaks of pathogens that have never been reported[9]. In the present study, we collected 96 serum samples from patients with symptoms of febrile jaundice who tested negative for YFV by quantitative reverse transcription polymerase chain reaction (qRT-PCR), and 4 serum samples from healthy individuals without any symptoms. We carried out MSS combined with metagenomic analysis to identify the pathogens that cause febrile jaundice. Of the 100 serum samples investigated, we characterized sequencing reads belonging to 12 viral pathogens and 1 bacterial pathogen, and created a baseline virome spectrum that may aid in the identification of emerging viruses in this region.
-
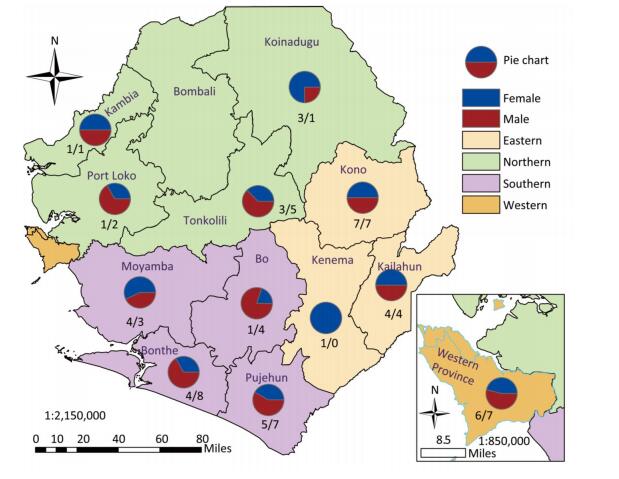
We collected 96 serum samples from patients diagnosed with febrile jaundice (documented axillary temperature of ≥ 37.5 ℃; generalized yellow coloration of the integuments and mucous membranes) and 4 negative controls from sentinel hospitals located in 12 provinces and 3 rural areas (western, eastern, and southern) in Sierra Leone (Figure 1). All samples were collected between July 2016 and June 2017 from patients with a wide age distribution (range 3 months to 75 years) (Table 1). The most frequent signs/symptoms among the 96 patients for which serum samples were analyzed were fever (96) and jaundice (92), followed by, in order of rank, loss of appetite (61), headache (26), diarrhea (23), intense fatigue (21), muscle pain (17), vomiting (13), hemorrhage (8), abdominal pain (7), joint pain (3), and skin rash (2). Five patients (26, 38, 79, 83, 88) died. The durations of illness and seasons of presentation are summarized in Table 1. All 100 samples tested negative for YFV according to qRT-PCR analysis. The qRT-PCR assays were conducted on a Bio-Rad (USA) CFX96 system according to the published protocol[10].
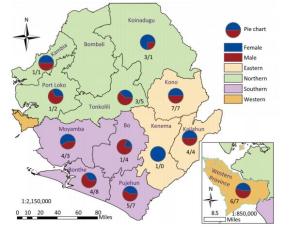
Figure 1. Geographic and gender distribution of 89 serum samples with febrile jaundice.
Characteristics Number of Patients Male (n = 59) Female (n = 41) Total (n = 100) Age (years) < 15 26 17 43 15-25 20 12 32 26-35 7 6 13 > 35 2 5 7 Not given 4 1 5 Duration of illness Less than 7 days 21 15 36 More than 7 days 38 26 64 Seasonal presentation Dry season 37 23 60 Rainy season 22 18 40 Table 1. Demographic and Clinical Characteristics of Patients (n = 100) Presenting with Febrile Jaundice between July 2016 and June 2017 in Sierra Leone
-
For each of the 100 samples, total RNA was extracted from a 200-μL serum sample using an RNeasy Mini Plus Kit (Qiagen, German), and diluted in 30 μL of elution buffer. We synthesized first-strand complementary DNA (cDNA) from 10 μL of eluted total RNA using SuperScript Ⅲ reverse transcriptase (Invitrogen), synthesized second-strand cDNA, and amplified the DNA using a REPLI-g WTA Single Cell Kit (Qiagen) according to the manufacturer's instructions. The amplified templates were then purified with 1.8 × the reaction volume of AMPure beads (Beckman Coulter, Inc.). Library construction was successively performed according to instructions included with the Ion Torrent Hi-Q kit (Thermo Fisher Scientific). Generally, 100 ng of the purified templates was digested for 5 min with the enzymes. Adaptors with sequence barcodes were ligated to the digested templates to distinguish the different libraries. Equal amounts (100 pmole) of each of the 100 libraries were pooled into 4 groups and distributed to 4 Ion PI v2 chips for sequencing. Four negative controls (samples 97, 98, 99, and 100) were put into the 4 pools respectively. Sequencing was performed using Ion PI Chip Kit v2 on an Ion Torrent Proton System (Thermo Fisher Scientific, USA) according to the manufacturer's instructions[11].
-
Raw sequencing reads generated by the Ion Torrent Personal Genome Machine (PGM) system were downloaded in binary alignment map (BAM) format. The raw data were analyzed using our in-house virus identification pipeline (VIP)[12].
First, we preprocessed the raw next-generation sequencing reads by removing the adapter, low-quality, and low-complexity sequences. This was followed by the computational subtraction of host-related reads using Bowtie 2 software. In fast mode, we identified viruses by Bowtie 2 alignment to a nucleotide database: the Virus Pathogen Database and Analysis Resource (ViPR)/Influenza Research Database (IRD). In sense mode, we removed bacteria and related ribosomal RNA (rRNA) reads, and aligned the remaining reads to the virus database. We then aligned unmatched reads to a viral protein database (RefSeq DB) from the National Center for Biotechnology Information (NCBI) using RAP Search. All matched reads were classified under a genus for de novo assembly and phylogenetic analysis.
-
Suspected cases of HCV and HBV were confirmed by fluorescence quantitative PCR[13, 14] using a Bio-Rad CFX96 thermal cycler. The cycle threshold (Ct) values are presented in Supplementary Table S1, available in www.besjournal.com.

Table Supplementary Table S1. Detailed Clinical Manifestation of 96 Febrile Jaundice Cases
Clinical Samples and Pre-preparation
RNA Extraction, cDNA Library Construction, and Ion Torrent Sequencing
Bioinformatics Analysis
Confirmation of HCV and HBV
-
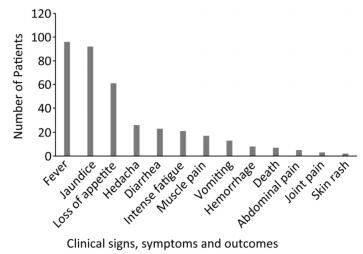
The most prevalent symptoms among the 96 febrile jaundice patients were fever (100%) and jaundice (95.83%), followed by, in order of rank, loss of appetite (63.54%), headache (27.08%), diarrhea (23.96%), intense fatigue (21.88%), muscle pain (17.71%), vomiting (13.54%), hemorrhage (8.33%), abdominal pain (7.29%), death (5.21%), join pain (3.13%), and skin rash (2.08%) (Figure 2).
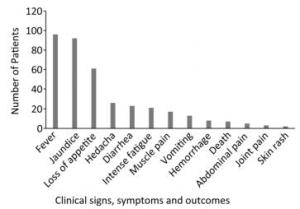
Figure 2. Clinical manifestation distribution among 100 serum samples.
-
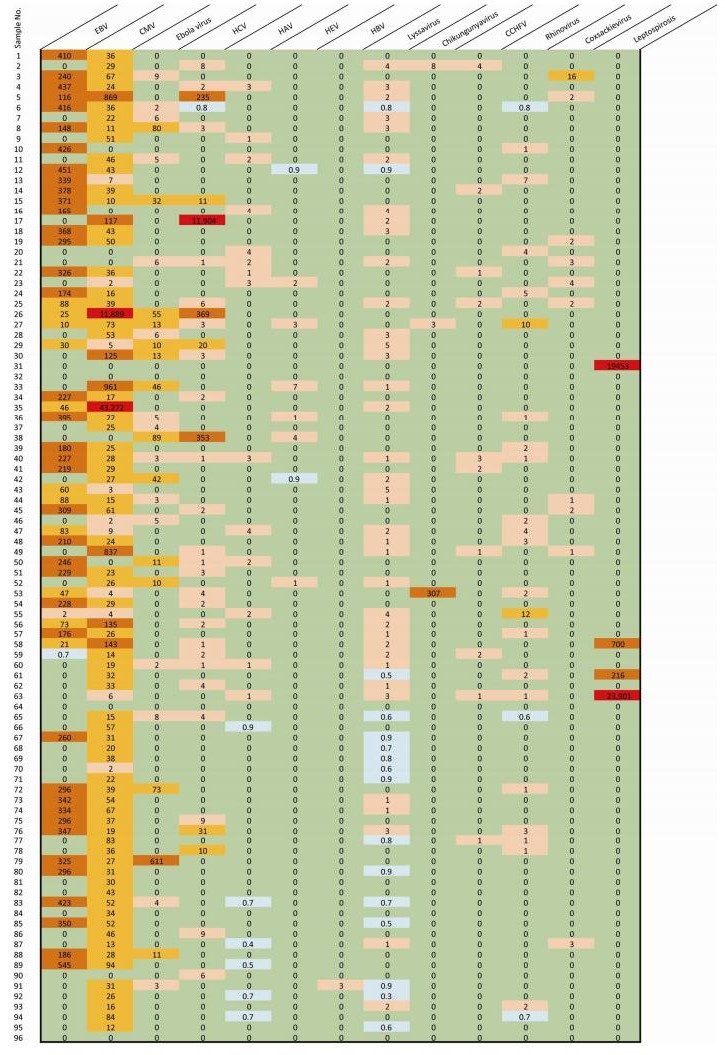
A total of 91 million reads were generated and the average number of reads per sample was 0.9 million. Following bioinformatics analysis, we produced color plots of the percentage of read hits belonging to the different viruses to distinguish the major viral pathogens present among the 96 febrile jaundice patients (Figure 3). The most frequently identified sequencing reads were attributable to CMV, which was detected in 86 samples (89.6%), followed by EBV (55.2%), HCV (34.3%), rhinovirus (28.1%), HAV (20.8%), coxsackievirus (10.4%), Ebola virus (8.3%), HEV (8.3%), lyssavirus (4.2%), chikungunya virus (2.1%), CCHFV (1%), HBV (1%), and leptospirosis (4.2%).

Figure 3. Characterized reads for 12 virus and 1 bacteria in 100 samples.
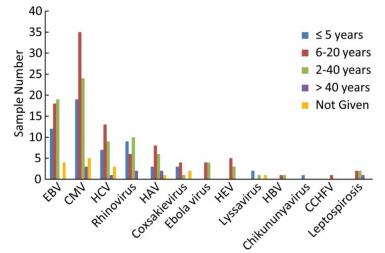
The most abundant sequencing reads were attributable to EBV and were found in 43 samples (44.8%), followed by CMV (33.3%), HCV (5.2%), leptospirosis (4.2%), Ebola virus (3.1%), HAV (3.1%), CCHFV (1%), and rhinovirus (1%). The frequencies of samples for which sequencing reads were attributable to each pathogen according to patient age are shown in Figure 4.
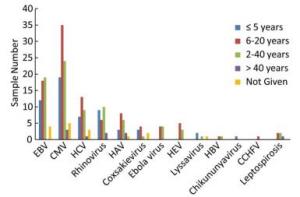
Figure 4. Pathogen distribution in each period of age.
Febrile Jaundice Population Analysis
Serum Viruses
-
Owing to the poor sample storage facilities in Sierra Leone, we excluded samples that were not well preserved and were deemed to be low quality (for example, samples that had not been kept at low temperature, samples for which there was a significant delay between collection and analysis, or samples that had not been maintained in an aseptic state). The 100 collected samples were all preserved at 4 ℃, and were tested within 1 month of collection. Poor sample preservation facilities may lead to the degradation of nucleic acids. All the included patients presented with symptoms of fever and/or jaundice. Only four patients did not have jaundice at the time of presentation. Five patients (26, 38, 79, 83, and 88), who suffered from hemorrhage (Supplementary Table S2, available in www.besjournal.com), died before or after sample collection. According to the sequencing results, EBV, CMV, and HCV were the main pathogens detected in these five deceased patients. Other details of clinical manifestations are shown in Supplementary Table S2.
Sample No. HCV (Ct value) HBV (Ct value) 1 2 neg 3 4 38.1 5 32.5 6 neg 7 8 37.7 9 10 11 12 13 14 15 34.8 16 17 26.2 18 19 20 21 neg 22 23 24 25 38.6 26 30.6 27 36.3 28 29 33.4 30 neg 31 32 33 34 neg 35 36 37 38 31.8 39 40 neg 41 42 43 44 45 37.3 46 47 48 49 neg 50 38.7 51 36.4 52 53 38.8 54 36.9 55 56 neg 57 58 neg 59 neg 60 39.2 61 62 33.7 63 64 65 38.5 66 67 68 69 70 71 72 73 74 75 neg 76 35.2 77 78 34.6 79 80 81 82 83 84 85 86 38.1 87 88 89 90 34.4 91 35.4 92 93 94 95 96 97 98 99 100 Table Supplementary Table S2. Quantitative PCR results of HBV and HCV
Following enrichment of the viral sequences and MSS, over 90 million reads were generated. After bioinformatics analysis, we determined a pathogen spectrum for patients presenting with YFV-negative febrile jaundice. The potentially dominant pathogens were EBV and CMV. Although we found CMV sequencing reads in more samples (86) than EBV sequencing reads (53), read hits for EBV were substantially more numerous than for CMV or for any of the other pathogens in 43 samples. In a very few samples, sequencing reads for CMV (samples 26, 30, 33, 35, 49, 56, and 58), Ebola virus (sample 79), HCV (samples 17 and 38), and chikungunya virus (sample 53) were clearly predominant, indicating the potentially dominant viral pathogen in these samples. Leptospirosis read hits were dominant in four samples (samples 31, 58, 61, and 63), indicating bacterial infection or coinfection with a bacteria and a virus.
These findings suggest a high prevalence of EBV among the population of patients with YFV-negative febrile jaundice investigated in the present study. It has been reported that hepatitis, acute pancreatitis, and acalculous cholecystitis are related to primary acute symptomatic EBV infection; according to the reports, 15%-41% of the EBV-infected samples were from patients with jaundice[15, 16]. EBV has been reported to play an important role in coinfection with HBV, and coinfection is associated with significant jaundice[17]. In the present study, there was little evidence of HBV infection, with the exception of sample 91. This may have been due to the relative inability of the kit to extract DNA. Consequently, it is possible that DNA from viruses such as HBV, which may have been present in low concentrations, was missed. Regardless, the high EBV-positive rate identified in the samples in the present study suggests that consideration of the coexistence of HBV is warranted.
CMV was the most commonly identified virus among the 100 samples (86%). It has been suggested that hepatitis caused by acute CMV infection may be associated with jaundice[16]. Congenital CMV (cCMV) refers to fetal infection in utero[18]. In the present study, 21 patients (21%) with febrile jaundice were under 5 years of age, which suggests cCMV infection.
Viral hemorrhagic fever (VHF) viruses such as Ebola virus, lyssavirus, CCHFV, Rift Valley fever virus (RVFV), dengue virus (DENV), and YFV are associated with a wide spectrum of clinical manifestations including febrile jaundice[19]. In the present study, we found sequencing read hits for Ebola virus in sample 8, and for chikungunya virus in samples 53 and 79. Although the prevalence of Ebola virus has subsided in this region[20], Ebola virus nucleic acid may persist for a long period. Authors who have studied an Ebola
virus survivor have reported that it was possible to detect the Ebola virus nucleic acid in the survivor's semen for at least one year[21-24]. Although the persistence of Ebola virus in the blood has not been reported, it is possible that some individuals contract mild infections that remain clinically undetected[21]. Sequencing reads from other VHF viruses including lyssavirus and CCHFV have also been found in relatively low abundance. Considering the extremely high morbidity associated with lyssavirus infection, the likelihood of identifying lyssavirus among samples in the present study was quite low, although we did detect it in a sample from one patient who died (sample 55).
Hepatitis viruses are thought to be a major cause of febrile jaundice[25-27]. However, in the present study, sequencing reads from HAV, HBV, and HEV were relatively low, with the exception of HCV, for which a large number of sequencing reads were present in samples 17 and 38.
As a prevalent parasitic pathogen in Sierra Leone, malaria is believed to be responsible for febrile jaundice in many cases[28]. However, total RNA extraction and our in-house bioinformatic analysis system focused on viral genomes only, so samples that were weakly positive for viral DNA or RNA may have included an undetected causative pathogen such as malaria.
Owing to restricted laboratory and sample conditions, except for HCV and HBV (Supplementary Table S1) we were unable to retest or confirm the sequencing results using other approaches. The detection of sequencing reads from different viral or bacterial pathogens may contribute to epidemiological research in developing countries in West Africa, because the relative prevalence of viral pathogens has rarely been mentioned in previous reports. Similar studies[4, 28-31] conducted in Tanzania, Ethiopia, eastern Sudan, Kenya, and the Central African Republic have resulted in different perspectives on the pathogenic agents underlying febrile jaundice. These differences may reflect temporal and regional variation in the prevalent viral pathogens-other than YFV or DENV-that cause febrile jaundice.
-
We identified a variety of viral sequencing reads in sera from patients presenting with YFV-negative febrile jaundice in Sierra Leone, and demonstrated the prevalence of potential EBV and other viral or bacterial infections. The findings of this study highlight the current endemic situation and provide a baseline for clinicians and epidemiologic surveillance. The study will also inform appropriate treatment interventions for this patient population.
-
All authors approved the final manuscript and have no conflicts of interest to declare.
-
This work was conducted as part of a Sierra Leone-China Friendship Biological Safety Laboratory technical cooperation project in Sierra Leone, and was supported by the Chinese government. Activities were coordinated by the Sierra Leone Ministry of Health and Sanitation.
-
The authors thank Dr. Foday Dafae (deceased) at the Sierra Leone Ministry of Health and Sanitation for his helpful input, and the staff at the Sierra Leone-China Friendship Biological Safety Laboratory for processing the samples.
the National Key Research and Development Plan of China 2016YFC1200903
the National Key Research and Development Plan of China 2017YFC1200503
the China Mega-Project for Infectious Disease 2017ZX10104001
the China Mega-Project for Infectious Disease 2017ZX100101
the National Key Research and Development Plan of China 2016TFC1202700
the China Mega-Project for Infectious Disease 2017ZX10302301-004

 Quick Links
Quick Links
 DownLoad:
DownLoad: