
-
Silicosis is characterized by the development of progressive pulmonary fibrosis as a result of inhaling free SiO2. Treatment is limited as to date only a limited number of effective therapies are known. This fibrotic process usually takes between 15 and 20 years to develop and is dependent on the amount of SiO2 inhaled. Its main symptoms are coughing, breathlessness, and general weakness, and diagnosis is primarily facilitated by chest X-ray. Sufferers tend to be diagnosed only in the late stages, when the lungs present considerable fibrotic nodules, and consequently, an important loss of respiratory function [1]. Based on increasing evidence from epidemiological and experimental animal studies, the International Agency for Research on Cancer (IARC) has classified crystalline silica as a Group 1 carcinogen (1997), which could lead to lung cancer (LC)[2]. Early detection and intervention of silica-induced pulmonary damage will help to reduce the incidence and progression of silicosis-related diseases, with the ultimate goal of avoiding lung tumorigenesis[3]. Unfortunately, to date, no effective biomarker has been identified for early diagnosis.
Advances in high-throughput expression profiling, especially interactive microarray analysis, has enhanced the availability and accuracy of big data results. Identification of genes that are differentially expressed in response to a toxic agent and subsequent careful bioinformatics analysis may provide valuable information regarding the molecular targets and pathological mechanisms of these agents. However, as a result of the difficulties in early diagnosis and obtaining tissue samples, the integrative analysis of multiple factors that may contribute to the development of silicosis has proven to be a major challenge. Therefore, the pathogenic mechanisms of silicosis remain poorly understood.
Because of the limited in silico studies conducted on silicosis, we aimed to integrate the available gene expression datasets in this study. We used a comprehensive bioinformatics approach to identify mutual (differentially expressed genes) DEGs, subjected them to functional enrichment and (protein-protein interaction) PPI network delineation[4-5]. We then selected the most relevant gene, Slc26a4, was selected for further validation ex vivo. Our results demonstrate the potential of SLC26A4 as a biomarker of SiO2-induced toxicity in rats after chronic exposure.
The expression profiles GSE32147 and GSE30178 were downloaded from the GEO database, hosted by the National Center of Biotechnology Information (NCBI). This data was collected using the GPL6101 Illumina ratRef-12 v1.0 expression bead chip platform. CDF strain male Fisher 344 rats were exposed to crystalline silica (15 mg/m3, 6 h/day) for 5 consecutive days. To mimic the early stages of silicosis, rats were euthanized at 16 h after completion of the exposure program. These animals were sacrificed and the upper lobe of right lung was taken for RNA isolation. The final dataset was comprised of 32 samples: 16 silica exposure (SE) samples and 16 normal controls (exposed to filtered air) (NC).
The probe names were changed back to their representative gene symbols based on the Affy probe annotation files. DEGs from each data set were first analyzed using the GEO2R method from the GEO database. Then we set the threshold values for further evaluation at |log2 fold change (logFC)| ≥ 1, an adjusted P value < 0.01 and FDR < 0.01. The common DEGs for each data set that met our basic criteria were then subjected to GO enrichment and KEGG pathway analysis using the Database for Annotation, Visualization and Integrated Discovery (DAVID, https://david.ncifcrf.gov/). The cutoff value for functional enrichment was set at P < 0.05. Subsequently, the proteins associated with each of the DEGs were annotated using the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING, http://string-db.org/cgi/input.pl) (medium > 0.7), and the PPI networks were constructed using Cytoscape 3.5. In this study, the degree of centrality was calculated using Cytoscape plugin CytoHubba. This value is a fundamental parameter in network theory and was used to evaluate the nodes in our network. Degree of centrality is defined as the number of adjacent links; that is, the number of interactions that connect one protein to its neighbors.
Human lung adenocarcinoma A549 cells were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA) and grown in Dulbecco’s modified Eagle’s medium (DMEM; GIBCO, Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (ScienCell, San Diego, CA, USA), 100 U/mL of penicillin, 100 mg/mL of streptomycin in a humidified incubator at 37 °C in an atmosphere of 5% CO2. The A549 cell line is a human type II alveolar epithelial (AT II) cell line and is routinely used to study of pulmonary fibrosis. For the SLC26A4 small interfering (si) RNA experiment, A549 cells were divided into three groups: control; SiO2 200 μg/mL; SLC26A4 siRNA+SiO2 200 μg/mL. Prior to SiO2 stimulation, cells were transfected with SLC26A4 siRNA for 48 h according to the manufacturer’s instructions (Kangcheng, Shanghai, China). After incubation, cells were exposed to SiO2 for 24 h.
Protein expression was evaluated by western blot. Transferred blots were incubated sequentially with 5% non-fat milk, primary antibodies against SLC26A4, Collagen IV (COL IV), Alpha Smooth Muscle Actin (α-SMA), Fibronectin (FN) (CST, USA), β-actin (Abcam, UK) and HRP conjugated secondary antibodies (CST, USA). Protein bands were visualized using an enhanced chemiluminescence detection kit (Beyotime, China) and imaged on a gel imaging system (Bio-rad, USA). Relative protein level was determined using the images’ integrated density (IntDen) and analyzed on Image J software 1.52a (http://imagej.nih.gov/ij/).
Finally, the online tool, KM-plotter, was used to evaluate the correlation between SLC26A4 expression and LC patient survival. Overall survival (OS) was estimated using the Kaplan-Meier method and survival differences were further validated between subtypes including lung adenocarcinoma (LA) as well as lung squamous cell carcinoma (LSCC). The log rank P-value and hazard ratio (HR) were calculated with a 95% confidence interval.
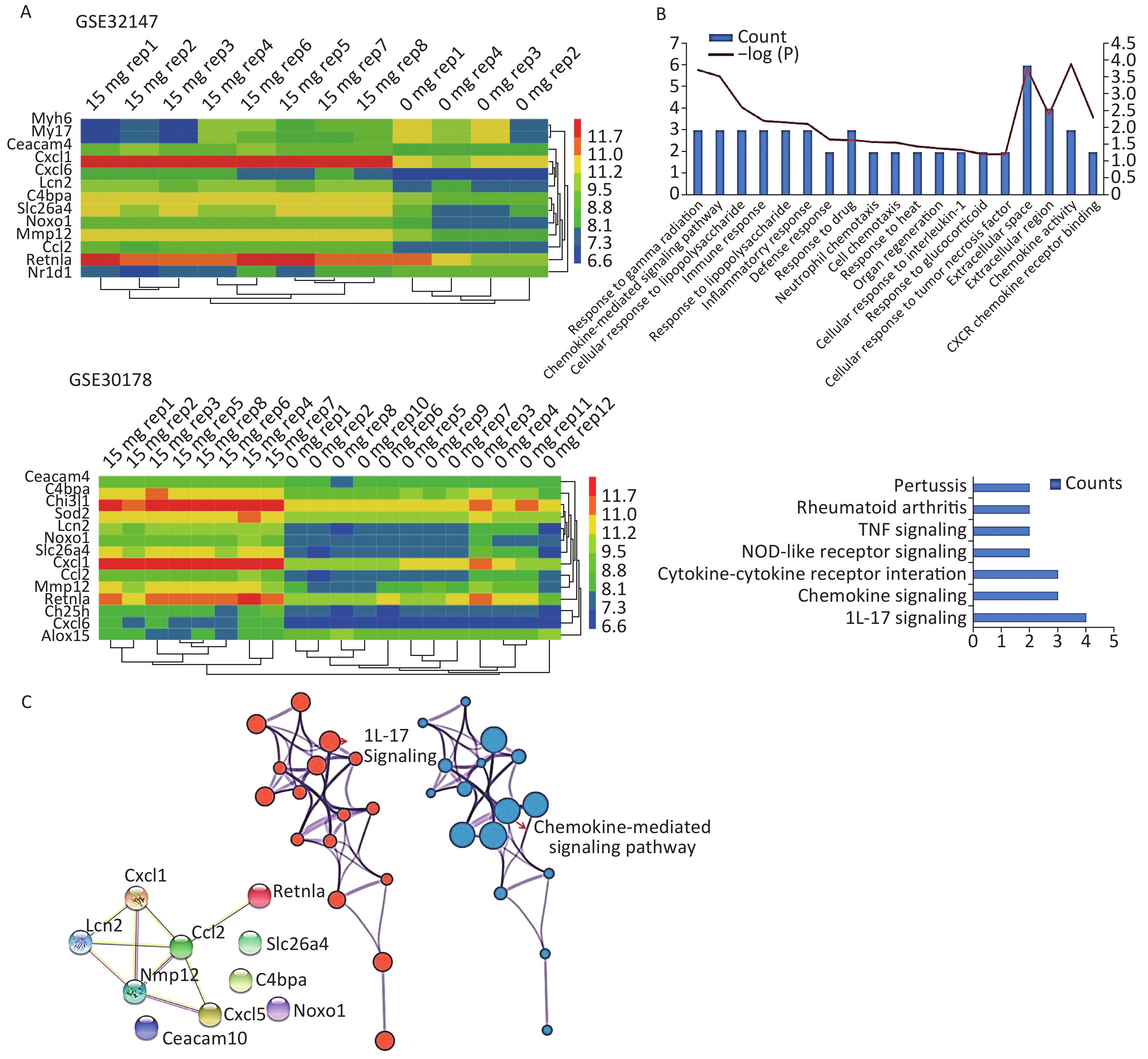
We applied GEO2R to identify DEGs from both GSE32147 and GSE30178. The value distribution plot indicated that the data were median-centered across samples, and thus could be cross compared (Supplementary Figure S1 available in www.besjournal.com). Ultimately, we identified 13 (10 up-regulated and 3 down-regulated) genes from GSE32147 and 14 (13 up-regulated and 1 down-regulated) genes from GSE30178 when comparing SE and NC groups. Organizing the expression profiles into heatmaps, we were able to visualize the expression patterns of the DEGs (Figure 1A).
Figure S1. The distribution of the values for the samples in each dataset. (A) GSE32147, (B) GSE30178.
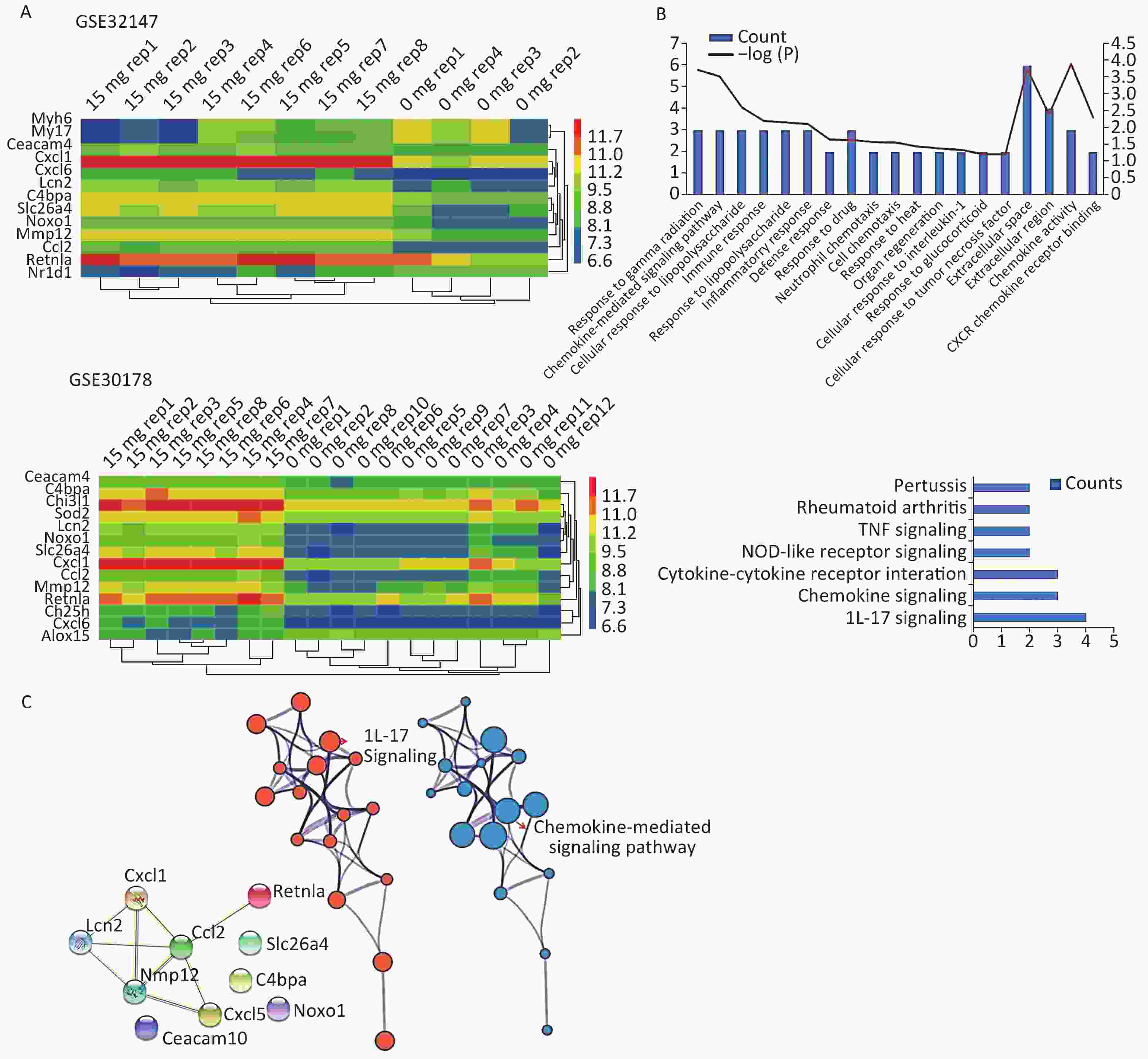
Figure 1. Bioinformatics analyses used to screen for mutual DEGs in silicosis. (A) DEG selection and hierarchical clustering analysis by heatmap. The vertical axis represents the sample. The horizontal axis represents the DEGs. (B) GO and KEGG analysis of the biological function and pathway of the co-expressed DEGs. (C) Construction of a PPI network for the 10 DEGs.
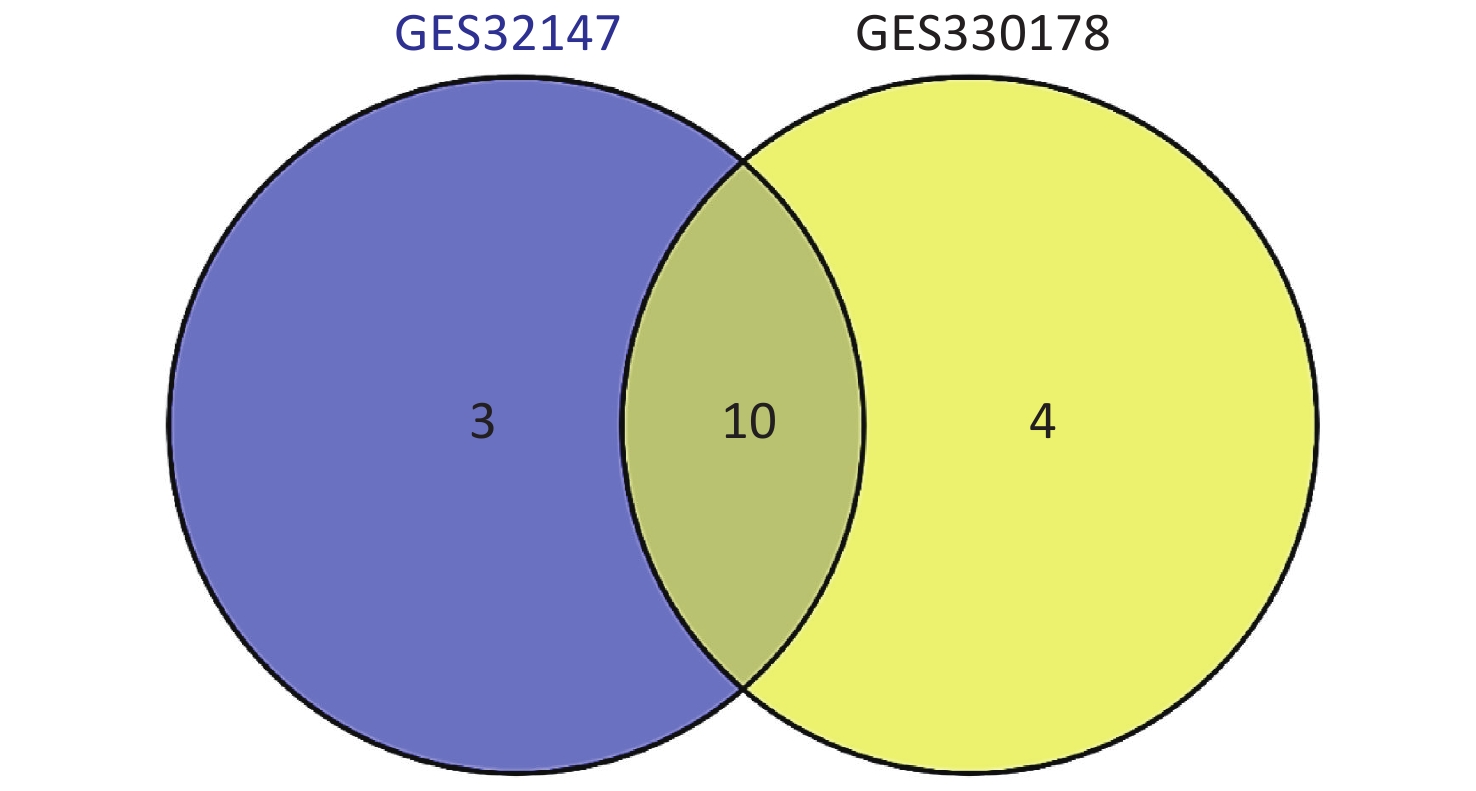
Subsequently, we searched for overlapping DEGs using a Venn diagram (Supplementary Figure S2 available in www.besjournal.com) and listed in Supplementary Table S1 (available in www.besjournal.com) with their full names. We identified 10 common DEGs from both datasets that were markedly up-regulated in SE.
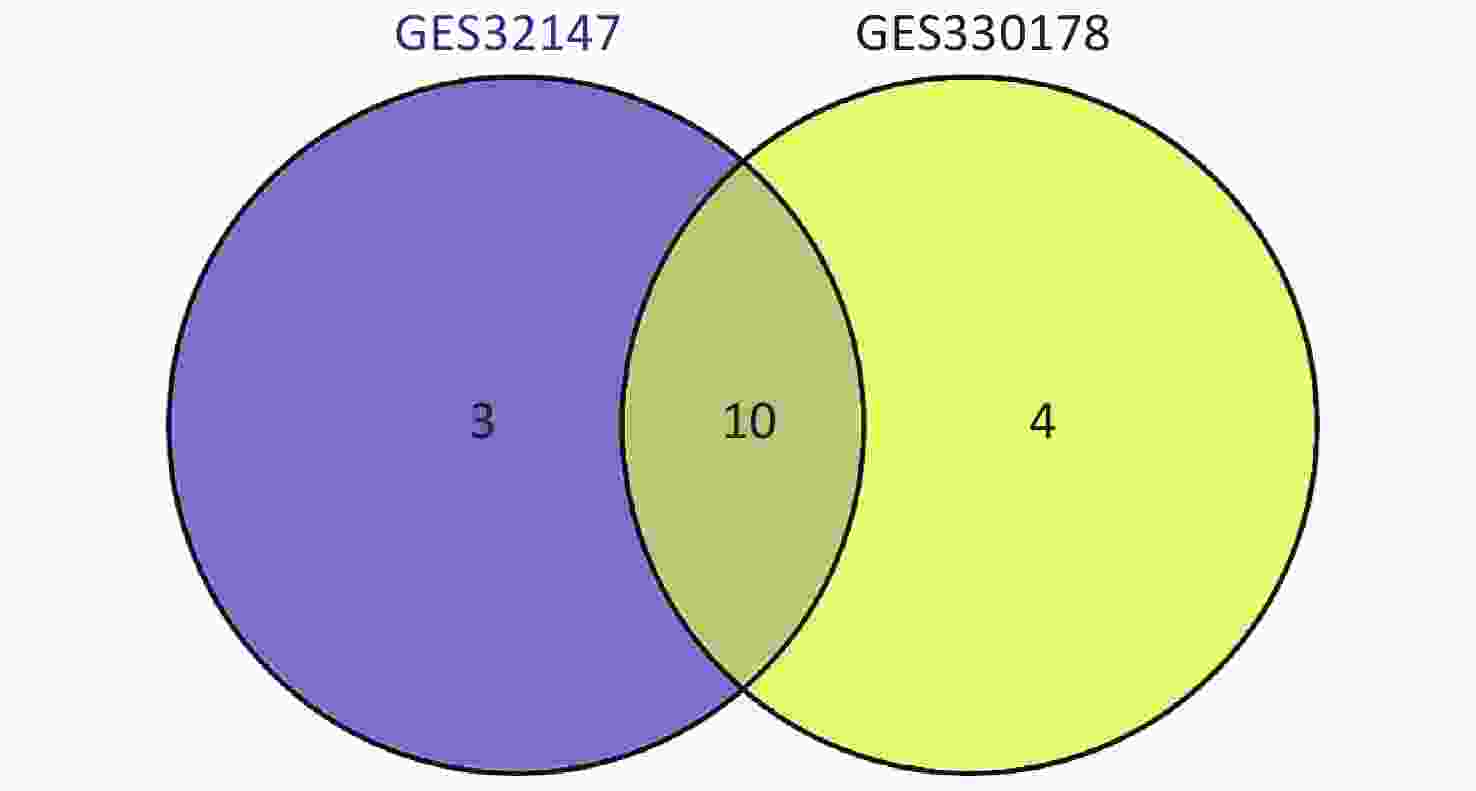
Figure S2. Intersection of the screened DEGs from GSE32147 and GSE30178 represented by a VENN diagram.
Gene symbol Gene title Ceacam4 carcinoembryonic antigen-related cell adhesion molecule 4 C4bpa complement component 4 binding protein, alpha Cxcl1 C-X-C motif chemokine ligand 1 Ccl2 C-C motif chemokine ligand 2 Mmp12 matrix metallopeptidase 12 Retnla resistin like alpha Lcn2 lipocalin 2 Noxo1 NADPH oxidase organizer 1 Cxcl6 C-X-C motif chemokine ligand 6 Slc26a4 solute carrier family 26 member 4 Table S1. Common DEGs with their corresponding full name
To gain insights into the biological roles of these DEGs, we performed a GO categories enrichment analysis. In this study, all DEGs were uploaded to the online software DAVID in order to identify over-represented GO categories. GO enrichment analysis results varied based on their GO classification and change in expression. Using a cutoff of P < 0.05, ‘chemokine activity’ for MF (P = 1.26 × 10−4), ‘extracellular space’ for CC (P = 1.78 × 10−4), and ‘response to gamma radiation’ for biological process (BP) (P = 1.87 × 10−4) exhibited the most significant enrichment (Figure 1B. Upper). KEGG pathway enrichment analysis was then used to understand the signaling pathway enrichment of the common DEGs. With a P < 0.05 cutoff, we were able to identify the most enriched biological pathways associated with silica exposure. These included the ‘Chemokine signaling pathway’ and ‘Pertussis’ (Figure 1B. Lower). Following this, a PPI network with 10 nodes and 9 PPI pairs was constructed (Figure 1C). In this network, C-C motif chemokine ligand 2 (CCL2) (degree = 5), matrix metallopeptidase 12 (MMP12) (degree = 4), which both had more than 3 interactions, were regarded as hub nodes (Supplementary Table S2 available in www.besjournal.com). Finally, the protein network containing the 10 common DEGs was generated using STRING and shown in Figure 1C (Left). To further understand and capture the relationships between the GO terms, a subset of enriched terms were selected and rendered as a network plot using Metascape (http://metascape.org), where terms with a similarity > 0.3 are connected by edges. The network was visualized using Cytoscape 3.5, where each node represents an enriched term and the size of the node was determined by the -logP value (Figure 1C. Central) and its cluster enrichment (Figure 1C. Right). As a result, we were able to show that IL-17 signaling, and the Chemokine-mediated signaling pathway was the most enriched terms.
Hub gene Degree score Ccl2 5 Mmp12 4 Cxcl1 3 Lcn2 3 Cxcl5 2 Retnla 1 Table S2. The hub genes with corresponding degree scores
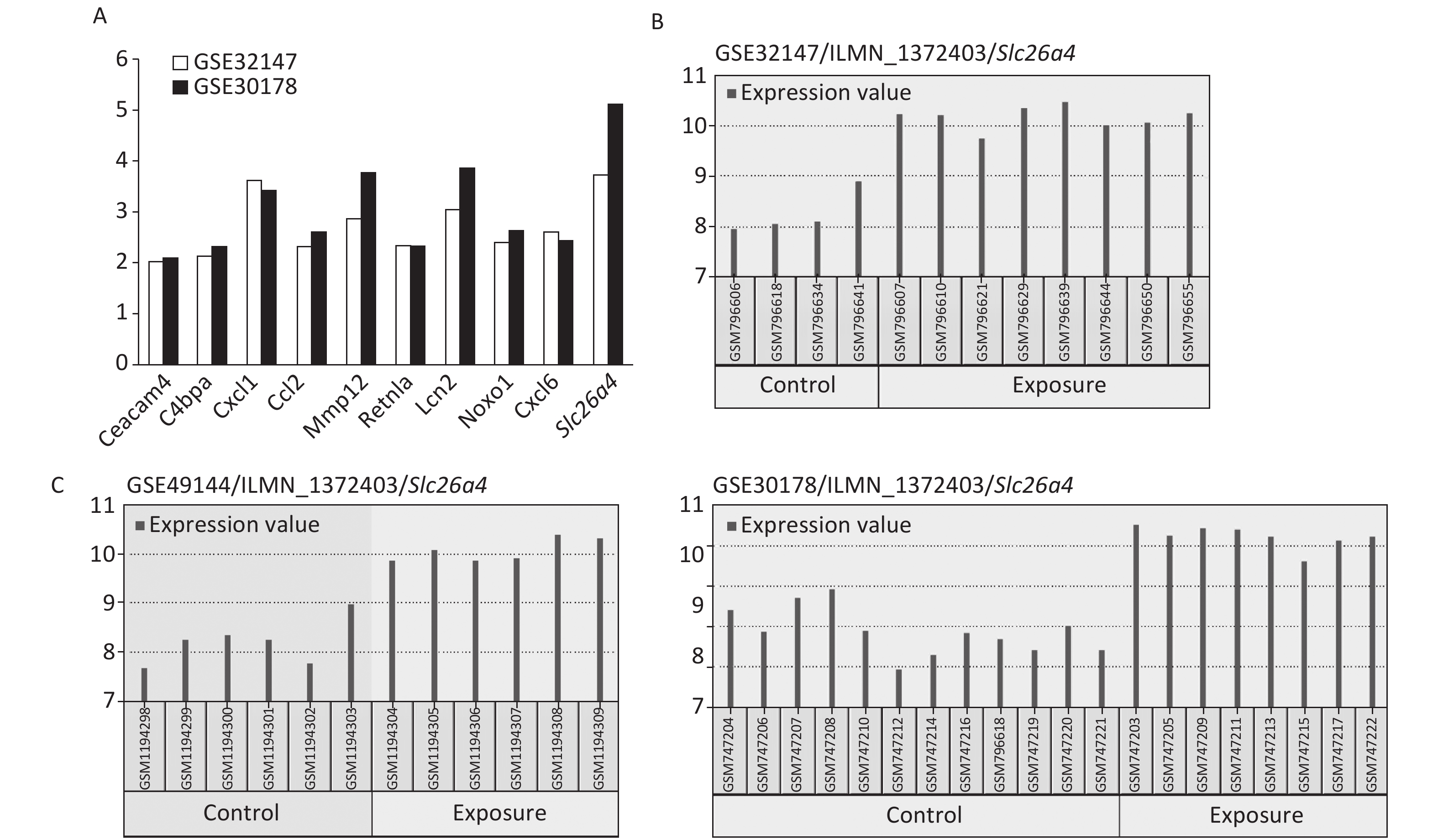
The most significant changes were observed in the Slc26a4 (Figure 2A) gene, which is also known as Pendrin or Dfnb4. This gene functions as sodium-independent transporter of chloride and iodide. The expression level of Slc26a4 was determined in the GES49144 validation dataset, which was made up of 6 rats exposed to crystalline silica for 3 weeks and 6 unexposed rats as controls. Slc26a4 expression was still significantly increased even after 3 weeks’ exposure to silica when compared with healthy controls (Figure 2C).
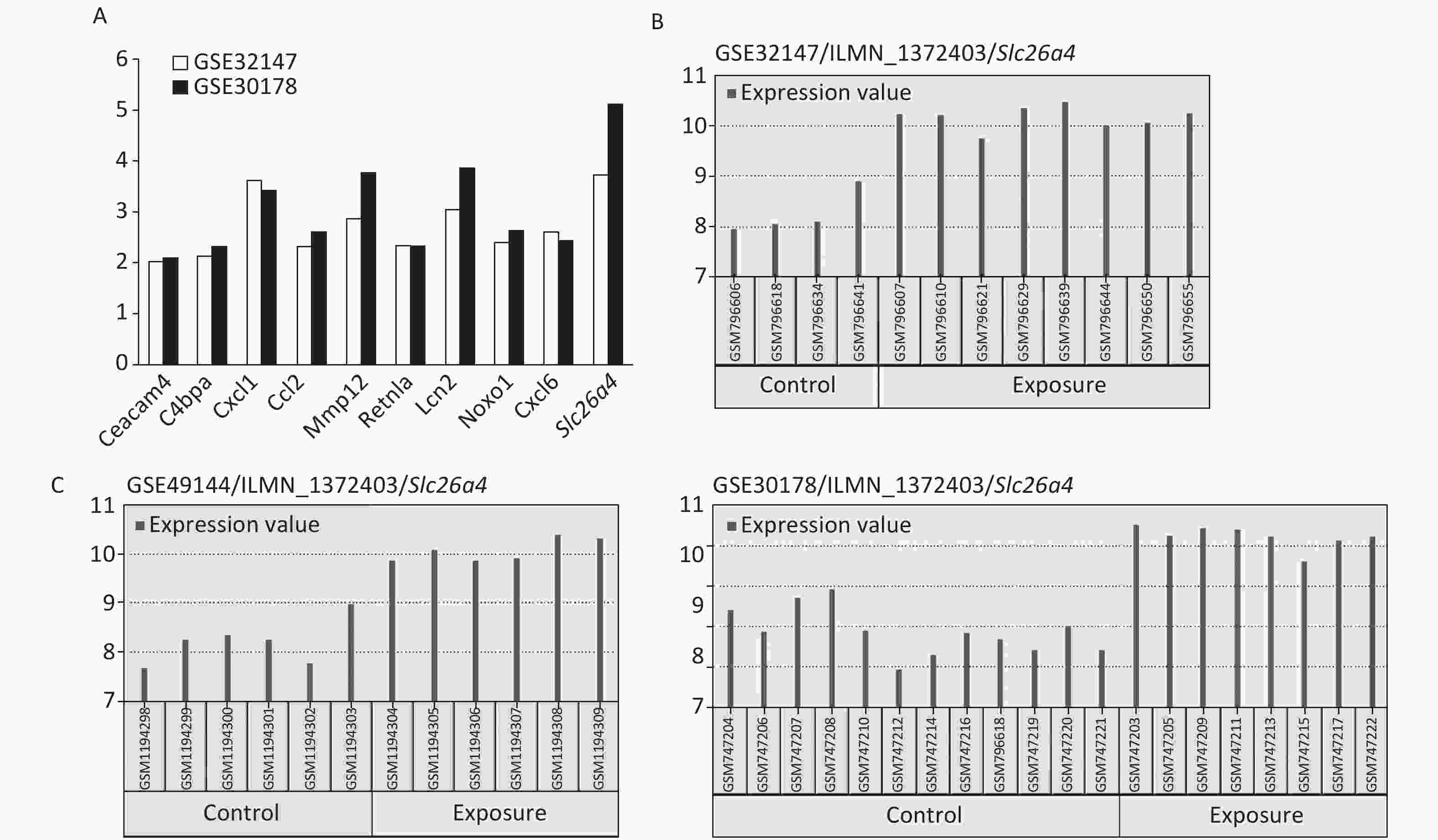
Figure 2. Selection of Slc26a4 for further verification. (A) Fold changes of co-expressed DEGs in each dataset. (B) The expression values of Slc26a4 across samples in each dataset. (C) Slc26a4 expression values across samples in the validation group.
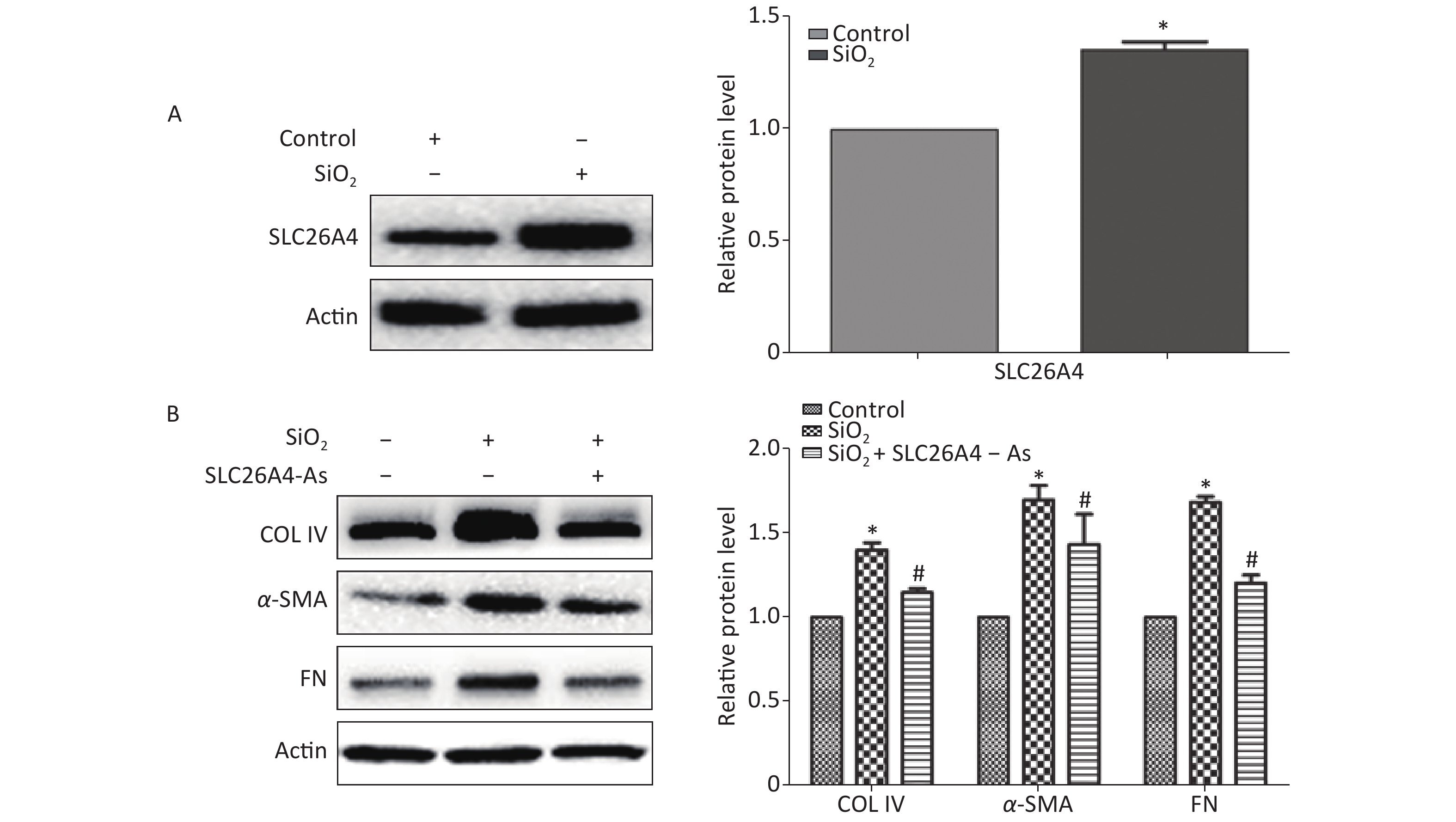
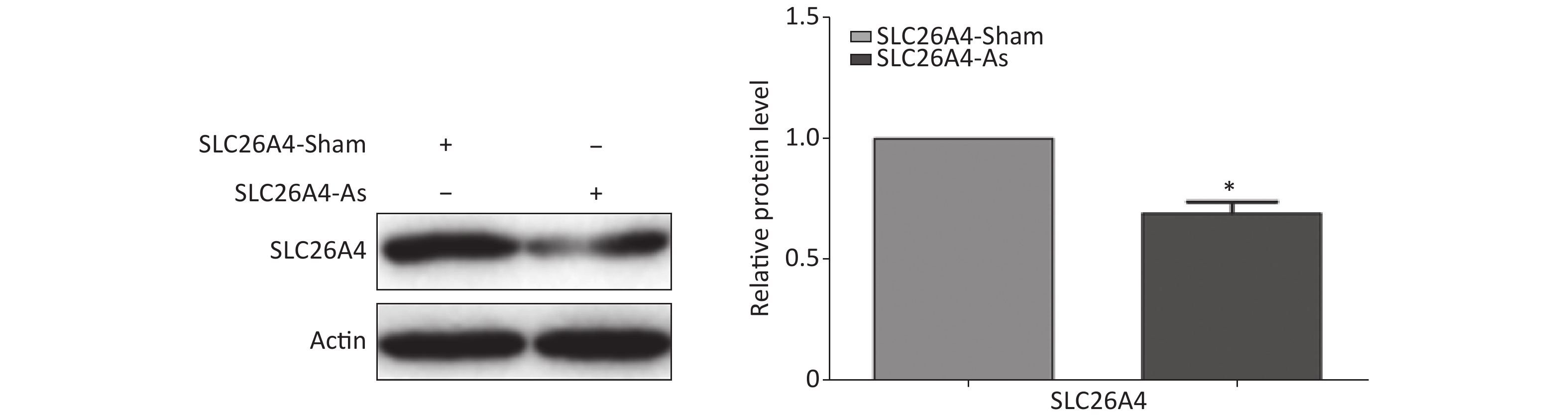
To establish the role of SLC26A4 in humans, we first validated the expression profile of SLC26A4 in human lung epithelial A549 cells treated with SiO2. We found that SiO2 exposure led to an increase in SLC26A4 protein expression (Figure 3A). Given the potential link between SLC26A4 and silicosis induced fibrosis, we also looked at the effect of increased SLC26A4 expression on the well-known ECM markers. Expression of COL IV, α-SMA and FN were all significantly downregulated in A549 cells transfected with SLC26A4 siRNA after pretreatment with SiO2 (Figure 3B, Supplementary Figure S3 available in www.besjournal.com). That means that, SiO2-induced ECM generation was dependent on SLC26A4 induction.
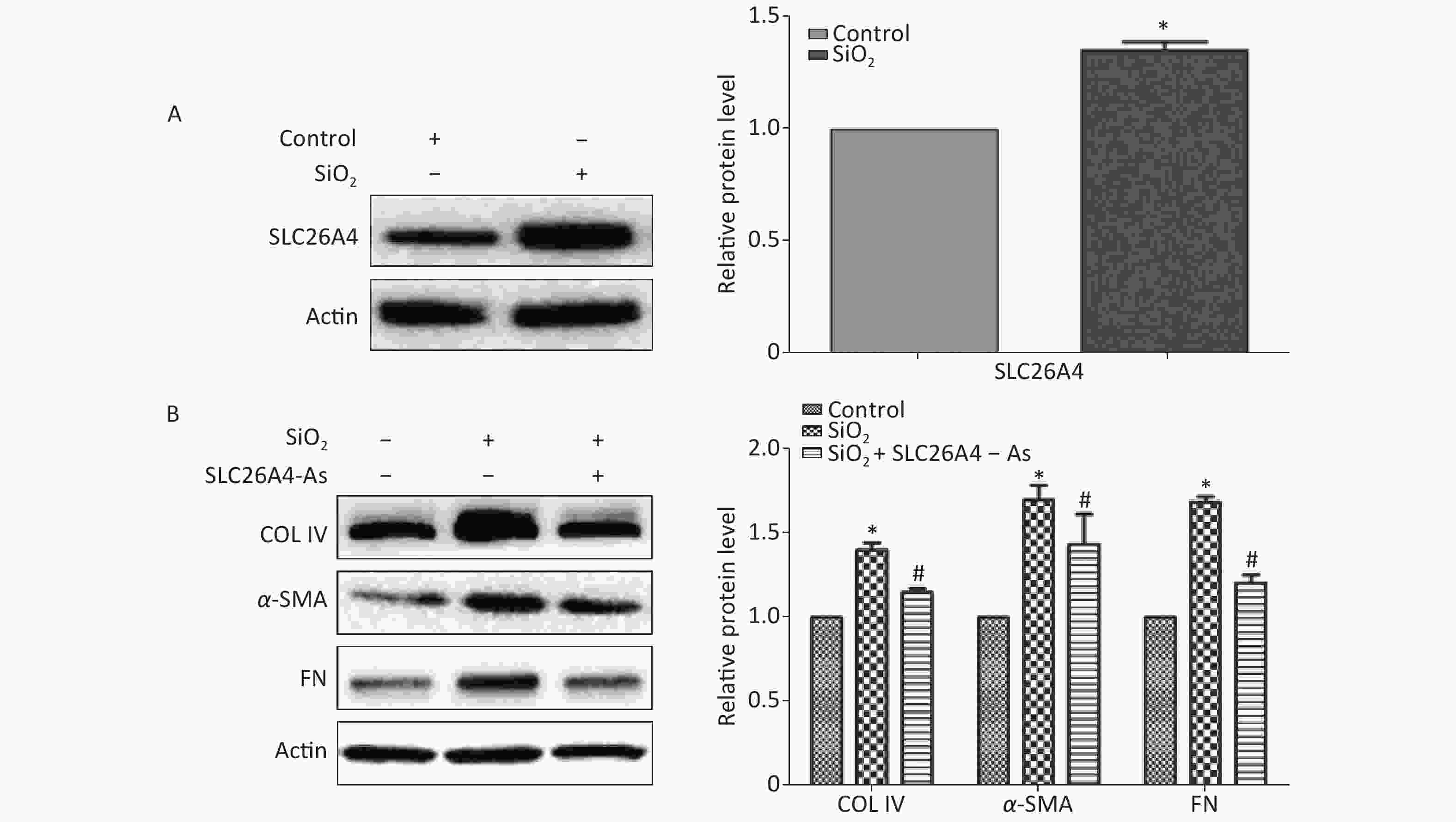
Figure 3. The influence of SLC26A4 in silica treated lung epithelial cells. (A) The expression of SLC26A4 in A549 cells exposed to silica; (B) siRNA mediated inhibition of SLC26A4 mitigated silica-induced ECM production. Quantitative results are presented as mean ± SD of the IntDen from three independent experiments (right). *P < 0.01 compared with the control group; #P < 0.05 compared with the SiO2 group.
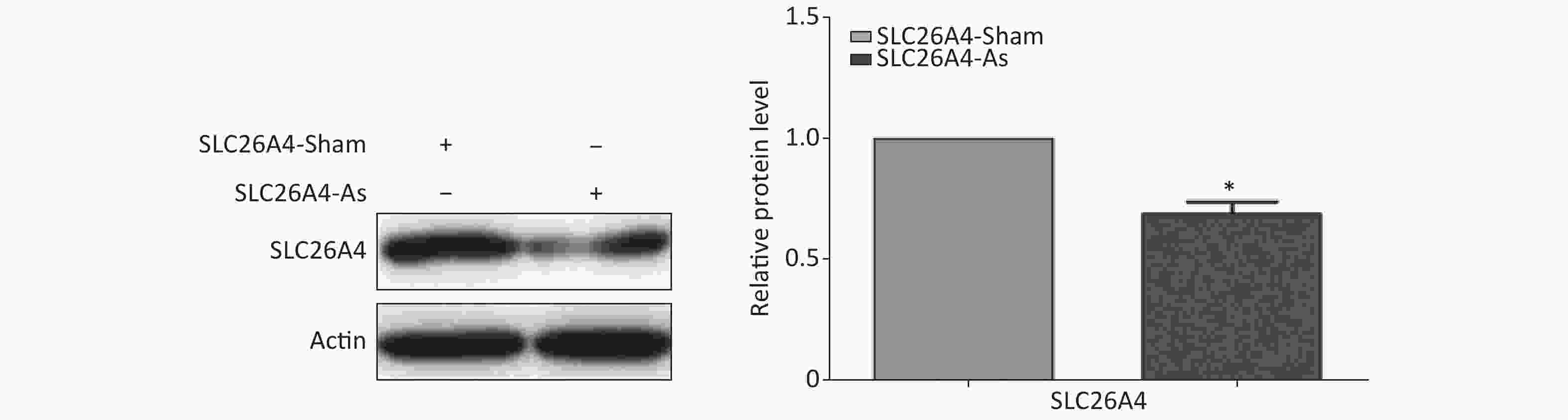
Figure S3. Effect of SLC26A4 siRNA in A549 lung epithelial cells. Quantitative results are presented as mean ± SD of the IntDen from three independent experiments (right). *P < 0.01 compared with the control group.
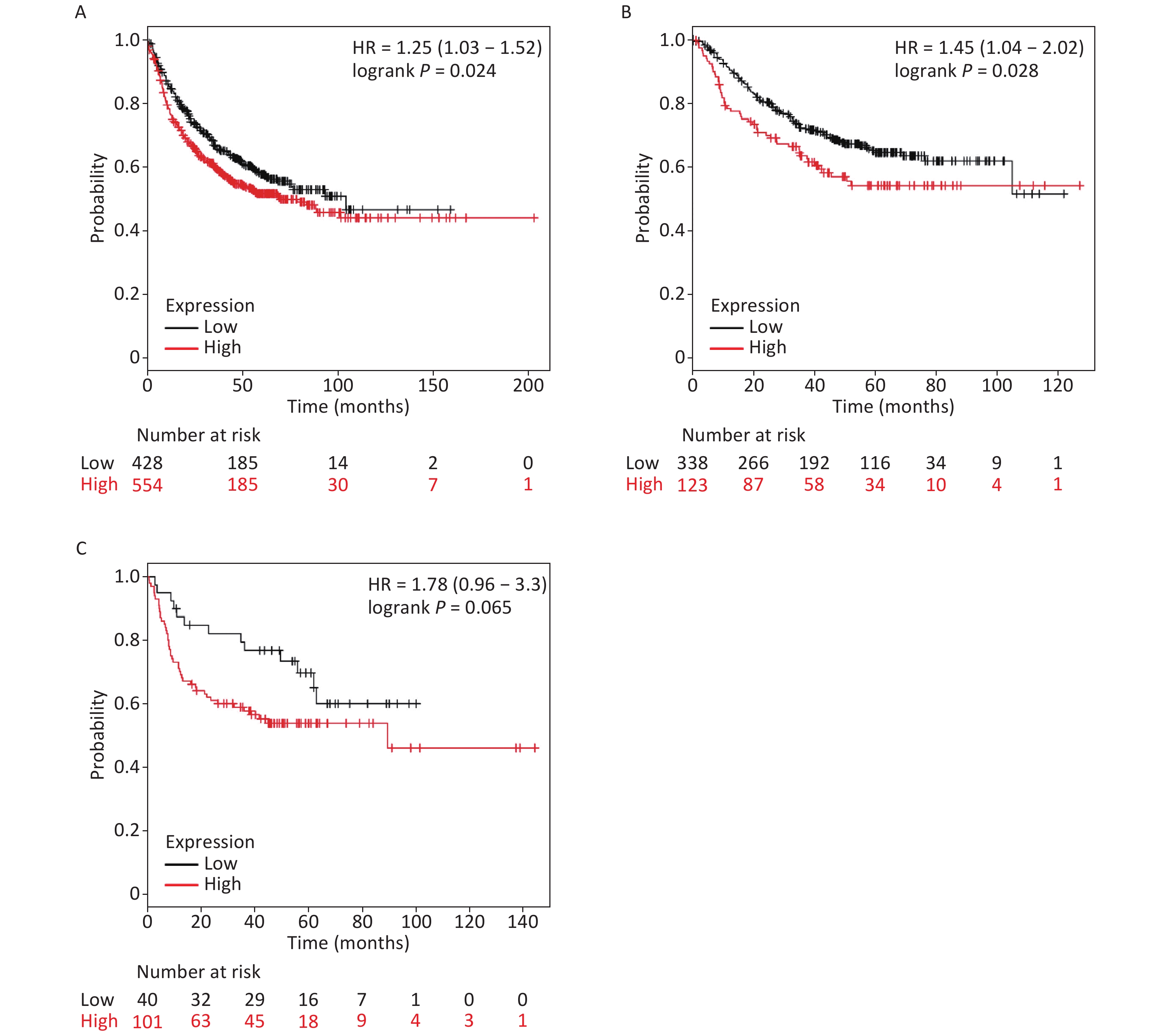
It is well established that silicosis is closely associated with the development of LC, especially LA. Therefore, we studied the relationship between SLC26A4 expression and clinical outcome using an online tool, KM-plotter. First, we were able to show that increased SLC26A4 transcript was correlated to reduced OS for all LC patients [HR: 1.25 (1.03–1.52), P = 0.024] (Figure 3B, Supplementary Figure S4A available in www.besjournal.com). In addition, elevated SLC26A4 mRNA levels were also shown to be an indicator of unfavorable OS outcomes in LA [HR: 1.45 (1.04–2.02), P = 0.028] (Figure 3B, Supplementary Figure S4B), but not in LSCC patients [HR: 1.78 (0.96–3.30), P = 0.065] (Figure 3B, Supplementary Figure S4C).
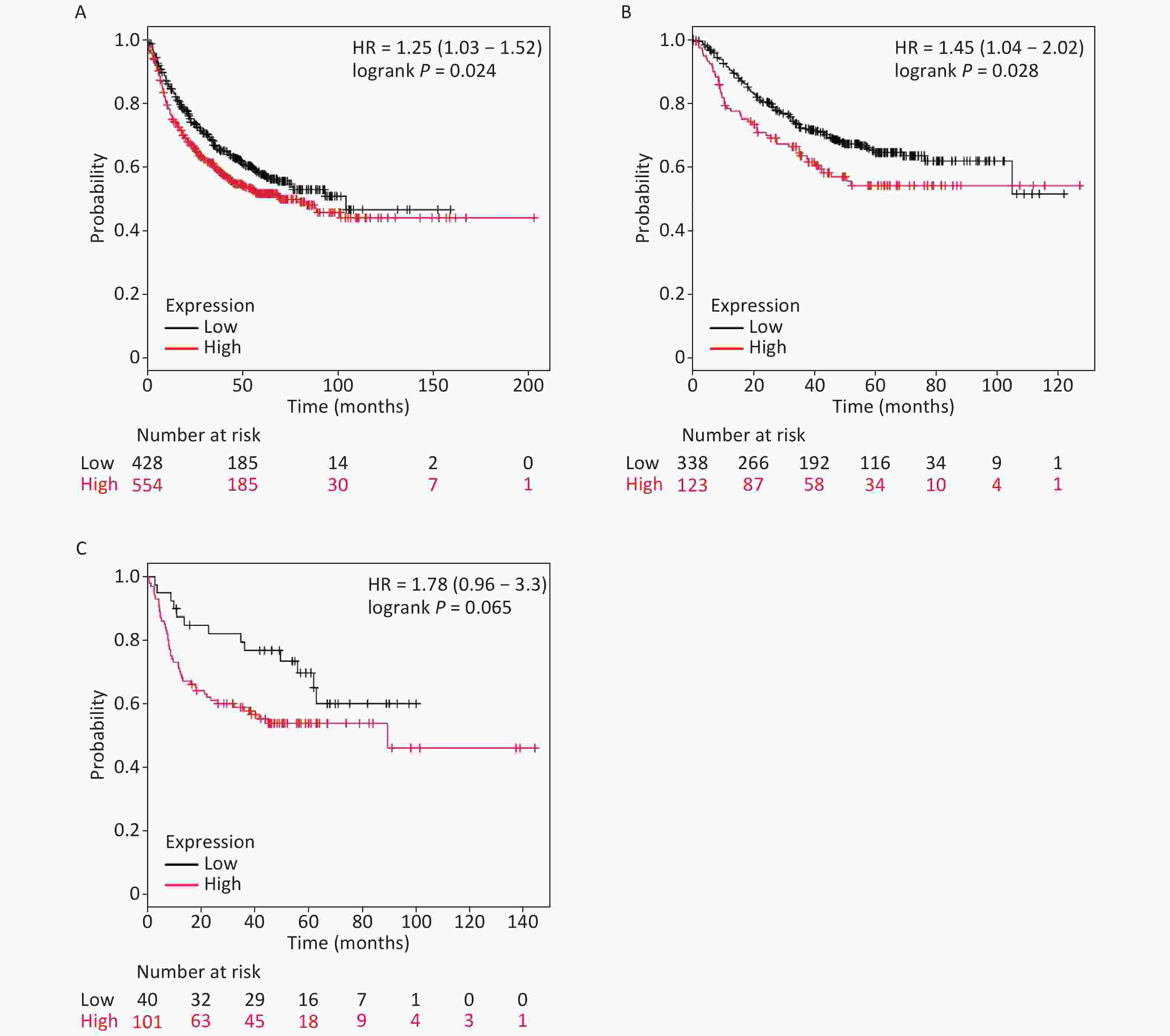
Figure S4. The prognostic value of SLC26A4 mRNA expression after analysis in the KM-plotter tool. Overall survival curves are plotted for (A) all patients with lung cancer (n = 982) and patients with (B) lung adenocarcinoma (n = 461), (C) lung squamous cell carcinoma (n = 141).
Despite the rapid advances in the field of pneumoconiosis research, effective intervention and treatment for silicosis are still not available[6]. Thus, easily evaluated molecular markers are urgently needed to reduce the occurrence and development of fibrotic disease. With early intervention being pivotal to enhanced survival. Here, we used a bioinformatics approach to explore key genes linked to silicosis onset and progression in in vivo models. We identified 10 common DEGs, that were consistently upregulated in lung tissue samples from rats treated with SiO2.
We performed GO analysis of the 10 DEGs, and the most enriched GO terms were associated with cellular stress responses and cytokine activity in the SE group. KEGG pathway and PPI analysis revealed that the common DEGs were predominantly enriched for the chemokine signaling pathway and inflammation. These findings fit into the inflammatory phenotype associated with the initiation of silicosis. In addition, terms associated with ‘Extracellular space’ and ‘Pertussis’ were found to be tightly correlated with silicosis because in both pertussis and silicosis, an increased deposition of ECM proteins is observed in the extracellular space.
We selected gene Slc26a4 due to its high FC. This gene encodes a protein which belongs to the SLC26 anion/bicarbonate transporter family. Mutations in this gene are commonly associated with Pendred syndrome, the most common form of syndromic deafness[7]. Pendrin is another common name for Slc26a4[8]. SLC26A4A is an anion transporter located at the apical side of airway epithelial cells, and it works as a downstream effector of the IL-4/IL-13 signaling pathway, which is implicated in asthma pathogenesis. Kenji et al. described the significance of the Pendrin/DUOX/peroxidase pathway in PENDRIN-induced airway inflammation[9]. SLC26A4 also plays an important role in vascular volume homeostasis. Additionally, PENDRIN is associated with the development of metabolic alkalosis in cystic fibrosis patients. Moreover, Slc26a4 expression was still clearly elevated even after 3 weeks’ exposure to silica in a validation group, which is consistent with the data in this study. This supports the hypothesis that SLC26A4 plays an important and sustained role in silicosis.
To date no evaluation of SLC26A4 expression in the early phases of silicosis has been published. To investigate the potential role of SLC26A4 in SiO2-induced pulmonary fibrosis in vitro, we first incubated A549 cells with SiO2. This experiment showed that SiO2 in A549 cells induced an increase in SLC26A4 expression. This indicates that SLC26A4 could be a key effector in the fibrogenic reaction associated with silicosis. We next analyzed the interventional potential of SLC26A4 siRNA against SiO2-stimulated production of representative ECM proteins, including COL IV, α-SMA and FN, whose expression is upregulated during inflammation and fibrosis. It was evident from this study that inhibition of SLC26A4 alleviated SiO2- evoked ECM induction.
Mounting evidence supports the hypothesis that patients with silicosis are more vulnerable to LC[10]. Certain factors may affect the increased cancer susceptibility due to continuous silica exposure. Consequently, we employed the KM-Plotter tool from the LC-database to establish a correlation between survival time and SLC26A4 expression. This analysis showed that increased SLC26A4 expression was correlated with reduced survival in LC patients. This was particularly true for LA patients but not for LSCC patients. This suggests that SLC26A4 could be a critical node in the pathogenic evolution of silicosis induced tumor development.
Taken together, our analysis of the biological functions and pathways shared between these datasets may allow for further insights regarding silicosis development at the molecular level and pave the way toward understanding its pathological mechanism. Here, we were able to confirm that SLC26A4 closely correlates with the onset and the prognosis of silicosis. Further investigation is required to establish the clinical relevance of these findings.
Funds:
This work was supported by Jiangsu Province’s Outstanding Medical Academic Leader program [CXTDA2017029]; Jiangsu Provincial 333 Project [BRA2018398]; and Scientific research project of Jiangsu Commission of Health [Y2018076]
Reference 
 Quick Links
Quick Links
 DownLoad:
DownLoad: