



-
PM2.5 are fine inhalable particles with diameters that are generally 2.5 micrometers and smaller and are characterized by a small particle size, a large surface area, and a strong toxin absorption ability[1-3]. PM2.5 contains heavy metals and organic pollutants that are harmful to human health and exert carcinogenic, teratogenic, and mutagenic effects[4-7]. PM2.5 has been classified as a human carcinogen by the International Agency for Research on Cancer (IARC)[2]. The economy has developed rapidly making air pollution a serious environmental health problem in China. The purpose of this study was to explore the differentially expressed genes and pathways after a PM2.5 exposure in the human bronchial epithelial (HBE) cells via gene chip technology and bioinformatics analysis.
Gene chip technology was proposed in the mid-1980s for the large-scale examination of gene expression. In this study, we used the gene chip technology to perform differential gene screening of the HBE cells exposed to PM2.5. Incorporating bioinformatics approaches, we also screened the selected differential genes through clustering and functional enrichment analyses, determined the core genes, and constructed a network map of protein-protein interactions to investigate the PM2.5-induced changes in the gene expression levels of the HBE cells. The results provide a scientific basis for the mechanism of PM2.5-induced carcinogenesis, mutagenesis, and for the exploration of new research directions.
PM2.5 samples were collected in Taiyuan during 2017–2018, at sampling sites where there were no visible pollution sources. An intelligent medium-volume total suspended particle air sampler, and a PM2.5 cutter was used to collect PM2.5 from the air. The sampling filter was a quartz fiber filter with a sampling flow of 100 L/min. Each site was sampled once every quarter for 3 days, and samples were continuously collected for 24 h every day during 2017–2018. The concentration of PM2.5 samples was determined by weighing the sampling filters using an electronic balance and calculated from the total collected air volume.
The HBE cells were purchased from the Shanghai Cell Resource Center of the Chinese Academy of Sciences. The cells were cultured in a constant temperature incubator (37 °C) with a volume fraction of 5% CO2. The cells were allowed to reach 70% to 80% confluency at the bottom surface of the culture flask. The PM2.5 treatment was administered when the cells reached 80% confluency at the bottom of the culture bottle. The IC50 value of PM2.5 was 70.12 μg/mL, based upon our previous experimental results; therefore, the PM2.5 concentration used in this study was 50 μg/mL, and the duration of treatment was 24 h. The total RNA was extracted from the HBE cells and purified using a QIAGEN RNeasy® Mini Kit. The concentration and purity of the RNA were measured using a NanoDrop ND-1000 kit (Thermo Scientific), and RNA integrity was determined from the RNA integrity number (RIN) value determined using the Agilent RNA 6000 nano assay.
The gene chip used in this study was the Human OneArrayTM (HOA) provided by Shenzhen Huada Gene Technology Co. Ltd. Based on the latest database of UniGene and via the IMPORT-patented technology, highly specialized 60-mer oligonucleotide probes were developed. More than 30,000 probes were placed on each chip, covering more than 30,000 genes and several control probes.
The pre-processing of the gene expression profile chip data (including screening and calibration) and statistical analysis were performed by the Rosetta Resolver® system (Rosetta Biosoftware), with the differentially expressed genes determined by the gene expression fold-change. The screening conditions were: log2[fold change (FC)] ≥ 1 and P < 0.05; the probe signal of at least one sample in the comparison group was ≥ 200. We further determined the top 10 up-regulated genes and the top 10 down-regulated genes.
Differentially expressed genes were annotated according to gene ontology (GO) using the clusterProfiler software, and the Kyoto Encyclopedia of Genes and Genomes (KEGG) functional annotation analysis was performed to determine the biological functions and pathways mainly affected by the differential genes. A P-value < 0.05 indicated a statistically significant difference.
The STRING protein interaction database was used to analyze the protein interactions of differentially expressed genes, and the protein interaction network map was constructed. The results of the STRING database analysis were imported into the Cytoscape software, with the degree of the nodes (the number of interconnections) calculated using the network analysis plug-in cytoHubba. The hub node was determined, and the protein corresponding to the hub node was identified as the hub protein.
According to the differential expression screening conditions, a total of 245 differentially expressed genes were determined, including 27 up-regulated and 218 down-regulated genes. Figure 1 shows the volcano map for the differentially expressed genes. The genes with log2FC < -1 were the down-regulated genes, and those genes with log2FC > 1 were up-regulated. We further narrowed the screening range and screened out the top ten up-regulated genes and down-regulated genes as the hub genes. Heat maps were generated using cluster analysis of visualized repeats and similarities between samples. Each row in the figure indicated a chip, and each column indicated a gene probe. The red-green level indicates the magnitude of the change in the up or down direction of the probe signal. The length of the line represents the distance index, and the similarity of the sample or genes within the same cluster was found to be high.
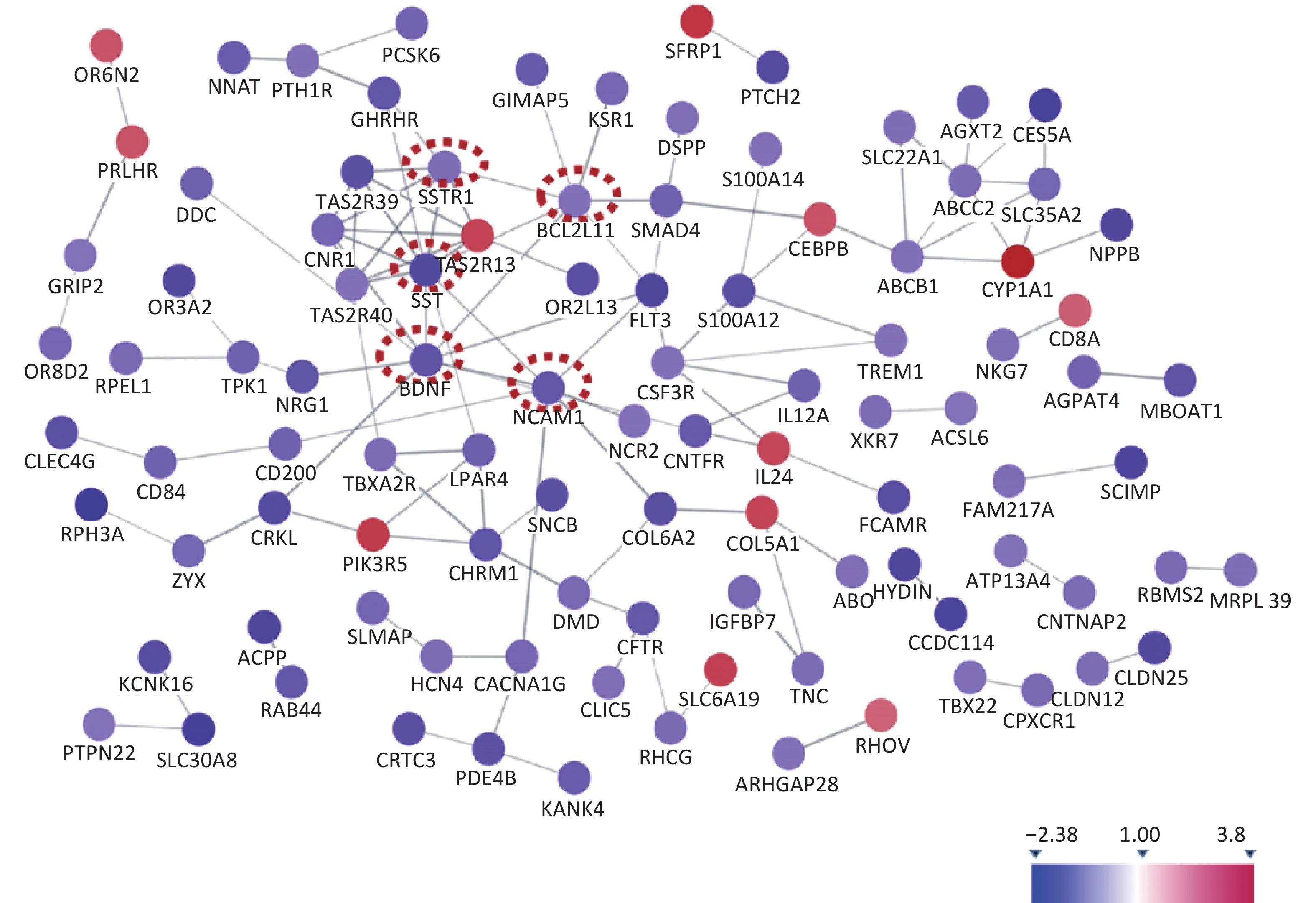
Figure 1. Protein interaction network diagram.
The selected differentially expressed genes were subjected to the GO functional annotation enrichment analysis. According to the results of the significant enrichment of GO, the differentially expressed genes primarily represented ‘cell compositions’, including the intrinsic components of the membrane, membrane components, and cell periphery, and were enriched in ‘molecular functions’, such as transmembrane receptor activity, receptor activity, signal transducer activity, and molecular transducer activity (Table 1).
Gene symbols logFC The scale of up-regulation or down-regulation Up-regulation or down-regulation Gene products PHACTR2 3.722545 13.200720 up Phosphatase and actin regulator 2 CYP1A1 2.156299 4.457698 up Cytochrome P450 family 1 subfamily a member 1 TMEM156 2.081564 4.232658 up Transmembrane protein 156 IL1RAPL2 1.793822 3.467323 up Interleukin-1 receptor accessory protein like-2 SIRPG 1.730599 3.318656 up Signal regulatory protein gamma SFRP1 1.618474 3.070501 up Secreted frizzled-related protein 1 PIK3R5 1.519071 2.866064 up Phosphoinositide-3-kinase regulatory subunit 5 SLC6A19 1.434983 2.703791 up Solute carrier family 6 member 19 TAS2R13 1.399616 2.638313 up Taste 2 receptor member 13 TSPAN18 1.390634 2.621938 up Tetraspanin 18 BDH2 −2.28103 4.860234 down 3-hydroxybutyrate dehydrogenase 2 RPH3A −1.93591 3.826191 down Rabphilin 3A WFDC1 −1.80125 3.485228 down WAP four-disulfide core domain 1 SLC30A8 −1.78310 3.441645 down Solute carrier family 30 member 8 CES5A −1.75611 3.377869 down Carboxylesterase 5A CCDC114 −1.67944 3.203035 down Coiled-coil domain containing 114 ERG −1.65794 3.155653 down ETS transcription factor ERG SCIMP −1.64625 3.130183 down SLP adaptor and CSK interacting membrane protein PPP1R42 −1.64355 3.124335 down Protein phosphatase 1 regulatory subunit 42 FLT3 −1.61497 3.063059 down Fms-like tyrosine kinase 3 Table 1. The core gene expression after PM2.5 exposure
The KEGG enrichment analysis showed the top seven most significant enrichment pathways (Table 2), which mainly involved tumor cell migration, signal transduction, bile metabolism, and genotoxicity. The enrichment analysis included the top seven biological pathways. We used the STRING protein interaction database to analyze the differential genes and construct a protein interaction network map (Figure 1). We screened out five of the most relevant hub proteins according to the Cytoscape software analysis, and the corresponding genes were SST, BDNF, NCAM1, SSTR1, and Bcl2L11, respectively.
Pathway annotation Number of genes Gene symbols Focal adhesion 7 PIK3R5, COL5A1, ZYX, PARVG, TNC, COL6A2, CRKL Bile secretion 4 ABCB1, SLC22A1, ABCC2, CFTR ABC transporters 3 ABCB1, ABCC2, CFTR Neuroactive ligand-receptor interaction 8 PRLHR, LPAR4, CNR1, SSTR1, CHRM1, TBXA2R, PTH1R, GHRHR Type II diabetes mellitus 3 PIK3R5, HK2, CACNA1G Taste transduction 3 TAS2R13, TAS2R39, TAS2R40 JAK-STAT signaling pathway 5 PIK3R5, IL24, IL12A, CNTFR, CSF3R Table 2. Analysis of the top seven enriched biological pathways
Gene chip technology, also known as DNA microchip or DNA microarray technology, is based on the in situ hybridization of nucleic acids. DNA fragments are fixed to the medium, and the fluorescently labeled samples are hybridized with the DNA fragments for the large-scale examination of the genes in the sample. The GO functional annotation and the KEGG functional analysis can reveal the potential pathogenesis and core genes in liver cancer, providing a basis for the diagnosis and treatment of the disease.
In this study, the HBE cells were acutely exposed to PM2.5 for 24 h, and bioinformatic approaches were used to analyze the gene chip results. According to the screening criteria, 27 genes were up-regulated, and 218 genes were down-regulated. These results suggest that PM2.5 may affect the gene transcription process of the HBE cells. By focusing on the GO functional annotation and aggregation analysis of the differential genes and the KEGG biological pathway enrichment analysis, we showed that the differential gene expression was mainly enriched in molecular conduction and other molecular functions, including the transmembrane receptor activity and cell signaling. In addition, the differential gene expression was enriched in biological processes such as the ion transmembrane transport and the cell nutrient transport. KEGG enrichment identified pathways related to differential gene expression, including those relevant to the focal adhesion associated with a tumor cell migration, bile secretion associated with bile metabolism, and neuroactive ligand-receptor interactions associated with neurotoxicity and genotoxicity, as well as the JAK-STAT pathway, a signaling pathway involved in cell differentiation, immunity, and apoptosis. These data provide a reference for the mechanism of PM2.5-induced apoptosis and carcinogenesis.
By analyzing the protein network map, we determined five differentially expressed genes with the most interactions and they were SST, BDNF, NCAM1, SSTR1, and Bcl2L11. Some studies have shown that the SST protein can significantly promote the proliferation, migration, and invasion of cancer cells[8]. SSTR1 is one of its subtypes. BDNF is the brain−derived neurotrophic factor−a protein synthesized in the brain and is widely distributed in the central nervous system. BNDF plays an important role in the survival, differentiation, and growth of neurons during the development of the central nervous system[9]. Bcl2L11 belongs to the BCL-2 family of proteins and contains a Bcl-2 homology domain 3 (BH3), which has been shown to interact with other members of the BCL-2 family and activate apoptosis[10].
In summary, after PM2.5 exposure, the HBE cells exhibited various changes in the gene profile that involved many biological processes and pathways. These multigene and multichannel changes will shed light on future in-depth studies of the mechanisms underlying PM2.5 toxicity.
PM2.5-induced Alterations of Gene Expression in HBE Cells Revealed by Gene Chip Analysis
doi: 10.3967/bes2020.030
Funds:
This project was supported by the basic research programs of Shenzhen Science and Technology Innovation Committee to XU Xin Yun [JCYJ20170413101713324]
- Received Date: 2019-09-05
- Accepted Date: 2020-01-03
| Citation: | WANG Bing Yu, CAI Ying, ZHENG Kai, HUANG Hai Yan, QIN Xiao Yun, XU Xin Yun. PM2.5-induced Alterations of Gene Expression in HBE Cells Revealed by Gene Chip Analysis[J]. Biomedical and Environmental Sciences, 2020, 33(3): 213-216. doi: 10.3967/bes2020.030

|

 Quick Links
Quick Links
 DownLoad:
DownLoad: