
-
Cadmium (Cd) is a heavy metal widely spread in the environment[1, 2], which represents a significant water and food contaminant, and is among the most toxic metals in the environment. The long biological half-life (10–30 years) and the low excretion rate[3] of Cd have alerted researchers to its health risk potential[4-6]. Cd exerts toxic effects in various tissues[7-14] as a consequence of induced oxidative stress. Infiltration of immune cells in tissues[15-19] is also a recognized mechanism of Cd toxicity. The positive and negative effects of Cd on immune cell activities depend on cell type, Cd concentration, and exposure duration[20-23]. The variety of effects of Cd are mediated through activation of numerous transcription factors (either directly by the metal or by oxidative stress)[22, 24] of which nuclear factor kappaB and activator protein-1 are included in the regulation of immune cell activity.
The aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor residing in the cytoplasm in the form of a multiprotein complex. Following ligand binding, the receptor dissociates from the complex and translocates into the nucleus where it initiates transcription of various genes (in the first line of enzymes involved in drug metabolism, of which cytochrome P450/CYP is the most commonly analyzed). Various exogenous (dioxins, polycyclic aromatic hydrocarbons, and polychlorinated biphenyls) and endogenous ligands (indigoids, metabolites of arachidonic acid, heme, and tryptophan)[25] activate the receptor resulting in different cellular responses. In addition, stress factors, such as hyperoxia, hydrogen peroxide, and ozone[26] activate the AhR. Involvement of AhR and AhR-regulated genes has also been suggested as one of the effects caused by metals in various tissues, particularly toxic heavy metals[26]. Increased expression of AhR mRNA has been noted in a human keratinocyte cell line (HaCaT cells) in response to arsenic[27]. In contrast, Cd has no effect on the AhR level in a human lung adenocarcinoma cell line (H1355 cells)[28]. An increase of AhR-regulated genes, including cytochrome P450 (CYP) 1A1, CYP1A2, CYP1B1, and CYP2S1 in response to arsenic[27-30], mercury[30-32], lead[30-32], copper[30-32], and chromium[29, 30] has been shown. The majority of studies examining the effects of heavy metals on CYP450s are oriented toward CYP1A1 because of its role in bioactivation of environmental pollutants into toxic metabolites with carcinogenic potential. It remains controversial whether the effect of heavy metals on CYP1A1 is AhR-dependent or mediated through induced oxidative stress. The involvement of oxidative stress in the regulation of CYP1A1 expression has been shown in H1355[28] and HaCaT[27] cells stimulated with arsenic. In contrast, blocking CYP1A1 mRNA in H1355 cells co-treated with arsenic and an AhR antagonist[28] demonstrates that Cd directly affects the AhR. Data that mercury, lead, and copper induce de novo synthesis of CYP1A1 mRNA, which is AhR dependent[32], further confirm this finding. Changes in AhR and AhR-related genes induced by these metals affect reactivity to other exogenous[29, 31, 33] or endogenous ligands[27]. Cd also increases the expression of AhR-regulated genes (including CYP450 1A1, NAD(P)H: quinone oxidoreductase, and glutathione S-transferase Ya)[29]. In vitro Cd exposure results in an increase of CYP1A1 mRNA in human and mouse hepatoma cells[29, 30, 34]. Cd affects AhR pathways in the intestine and modulates AhR signaling (CYP1A1 expression) via estrogen receptors[35, 36]. Modulation of AhR-regulated genes by Cd could change reactivity to exogenous and endogenous AhR ligands by exerting an effect on the biological responses evoked by these compounds. Thus, inhibiting dioxin-induced AhR activation and subsequent activation of CYP1A1 by Cd in human hepatoma cells is considered to modulate the carcinogenic effects of dioxin and related compounds[29, 33, 37]. Similarly, higher cardiotoxicity (abnormalities in heart development) is observed in mice co-treated with Cd and the tryptophan metabolite formylindolo [3, 2-b] carbazole (FICZ) during the gestation period compared to the effect of FICZ alone[38]. Although the potential impact of Cd on AhR and AhR-related genes has been demonstrated in several cell types and tissues, there are no data concerning involvement of the AhR in the immunomodulatory effects of Cd.
Our previous study showed that Cd acquired via the oral route in rats gains access to the skin, causing tissue damage and inflammation[39], and that the metal deposition in the skin is associated with activation of the AhR[40]. Orally acquired Cd in this model reached the lungs as well, resulting in pulmonary inflammation[41]. To determine if the AhR is involved in the effects of Cd in the lungs, CYP1A1 and CYP1B1 expression was measured in lung leukocytes of rats orally exposed to Cd, and in lung leukocytes isolated from healthy (unexposed to Cd) animals following Cd exposure in vitro. As Cd affects proinflammatory cytokine production by leukocytes[42-47], which can be regulated by AhR ligands[48-50], the potential involvement of AhR signaling in the effects of Cd on the interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF) lung leukocyte response was investigated in vitro using the receptor antagonist CH-223191. CH-223191 has been shown to be a pure AhR antagonist (without agonistic effects even at high concentrations) that binds to the receptor and blocks AhR nuclear translocation and DNA binding[51].
-
Animal procedures were carried out in compliance with Directive 2010/63/EU on the protection of animals used for experimental and other scientific purposes and approved by the Ethical Committee of the Institute for Biological Research ‘Siniša Stanković’, University of Belgrade, Serbia.
Male Dark Agouti (DA) and Albino Oxford (AO) rat strains (age 8–12 weeks) were used for the experiments. They were conventionally housed in a controlled environment (12-h light/dark cycle, 22 ± 2 °C, and 60% relative humidity) with free access to standard rodent chow and water in the unit for experimental animals at the Institute for Biological Research ‘Siniša Stanković’, Belgrade, Serbia.
In the prolonged Cd exposure experiments, the DA rats were exposed for 30 d to Cd doses that included the environmentally relevant Cd concentrations to which humans are exposed[52-54]. Cadmium chloride (CdCl2) was prepared in distilled water at concentrations of 5 mg/L (5 ppm) and 50 mg/L (50 ppm) of the Cd (II) ion. Control rats were given distilled water. Eight rats were assigned per group in two independent experiments.
-
Prior to tissue collection, the animals were anesthetized with an intraperitoneal injection of 15 mg/kg body weight of Zoletil 100 (Virbac, Carros, France). The lungs were aseptically removed, cleared of blood, and finely minced. Lung leukocytes were isolated following incubation by gentle mixing for 30 min at 37 °C in RPMI-1640 culture medium supplemented with 2 mmol/L glutamine and 20 µg/mL gentamycin in the presence of 1 mg/mL collagenase type IV (Worthington Biochemical Corp., Lakewood, NJ, USA) and 30 µg/mL DNase I (Sigma Chemical Co., St. Louis, MO, USA). The cells were resuspended in culture medium supplemented with 5% (v/v) heat-inactivated fetal calf serum and voriconazole (5 µg/mL) (Pfizer PGM, Poce Sur Cisse, France). Total cell counts and viability were determined by trypan blue exclusion using a LUNA-IITM automated cell counter (Logos Biosystems, Anyang, South Korea).
Isolated leukocytes from healthy untreated animals (4 × 106 cells/well in a 96-well plate) were cultured for 48 h with various Cd concentrations (1, 5, 10, and 50 μmol/L) alone and in the presence of 3 µmol/L of the AhR antagonist CH-223191 (Sigma-Aldrich) to determine cytokine production and to isolate the RNA. The Cd doses used for cell stimulation were chosen to fit the most commonly used Cd doses in in vitro studies using cells from both human[21,44,45] and animal origins[29]. In a preliminary set of experiments, the cells were first pretreated for 1 h with CH-223191 and then Cd was added. No differences were noted between pretreated cell cultures and cells concomitantly treated with Cd and the antagonist, so the results obtained in cells co-treated with Cd and the antagonist are presented.
Leukocyte viability was measured by the MTT reduction assay following 48 h of culture. MTT (500 µg/mL) was added to each well of a 96-well plate and incubated for 3 h at 37 °C in a humidified atmosphere of 5% CO2. The formazan that formed was dissolved during an overnight incubation with 10% sodium dodecyl sulfate-0.01 N HCl, and absorbance of the extracted chromogen was read spectrophotometrically at 540 nm.
-
Lung leukocytes (1 × 106) were collected after a 48 h treatment with Cd and lysed in 10 mmol/L HCl. After precipitating the protein with 5% sulfosalicylic acid, the GSH level was quantified in the supernatant using 5,5'-dithio-bis-[2-nitrobenzoic acid] (DTNB) in Tris-HCl (pH 8.9) and reduced glutathione as the standard[55] spectrophotometrically at 412 nm. The GSH level was expressed as μmol of GSH/mg protein.
-
The dihydrorhodamine 123 assay (DHR 123; Life Technologies Corp. Carlsbad, CA, USA), based on the oxidation of DHR 123 to fluorescent rhodamine 123 by hydrogen peroxide, was used to measure ROS levels in lung leukocytes[56]. Lung leukocytes (1 × 106) treated with Cd for 48 h were incubated for 1 h in medium containing 4 μmol/L DHR 123. After the incubation, the cells were washed with PBS, fixed in 1% paraformaldehyde, and assayed for fluorescence intensity on the CyFLOW SPACE (Partec, Munich, Germany). A minimum of 10,000 events was acquired each time.
-
Cytokine concentrations were determined in lung leukocyte-conditioned medium using commercially available ELISA kits for rat IL-1β and IL-6 (R&D Systems, Minneapolis, MN, USA), and TNF (eBioscience Inc., San Diego, CA, USA) according to the manufacturer’s instructions. Cytokine titers were calculated with reference to a standard curve constructed using known amounts of the respective recombinant cytokine standards provided in the kits. The results are presented as relative change compared to the control (Cd 0 µmol/L, considered 1).
-
Lung leukocytes stimulated in vitro with Cd and leukocytes isolated from animals exposed for 30 d to Cd were used to isolate RNA. RNA (1 µg) was isolated using the mi-Total RNA Isolation Kit (Metabion, Martinsried, Germany) and reverse transcribed using random hexamer primers and MMLV (Moloney Murine Leukemia Virus) reverse transcriptase (Fermentas, Vilnius, Lithuania), following the manufacturer’s instructions. The prepared cDNAs were amplified using the Power SYBR® Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA) based on the manufacturer’s recommendations in a total volume of 20 μL in a Quant StudioTM 3 Real-Time PCR Instrument (96-well, 0.2-mL) (Applied Biosystems). The thermocycler conditions were: hold stage at 50 °C for 2 min, followed by 95 °C for 10 min; the PCR stage at 95 °C for 15 s followed by 40 cycles of 60 °C for 60 s each; and a melt curve stage at 95 °C for 15 s, followed by 60 °C for 60 s and 95 °C for 1 s. The PCR primers (forward/reverse) used in this study are listed in Table 1. The PCR products were detected in real-time and the results were analyzed with Quant StudioTM Design & Analysis Software v1.4.3 (Applied Biosystems) and calculated as 2−ΔCt where ΔCt is the difference between the threshold cycle (Ct) values of a specific gene and the endogenous control (β-actin).
Item Forward Reverse β-actin (housekeeping reporter gene) 5'-CCCTGGCTCCTAGCACCAT-3' 5-'GAGCCACCAATCCACACAGA-3' CYP1A1 5'-GGGGAGGTTACTGGTTCTGG-3' 5'-CGGATGTGGCCCTTCTCAAA-3' CYP1B1 5'-CTCATCCTCTTTACCAGATACCCG-3' 5'-GACGTATGGTAAGTTGGGTTGGTC-3' IL-6 5'-GCCCTTCAGGAACAGCTATGA-3' 5'-TGTCAACAACATCAGTCCCAAG-3' TNF-α 5'-TCGAGTGACAAGCCCGTAGC-3' 5'-CTCAGCCACTCCAGCTGCTC-3' IL-1β 5'-CACCTCTCAAGCAGAGCA-3' 5'-GGGTTCCATGGTGAAGTCAAC-3' NLRP3 5'-CAGAAGCTGGGGTTGGTGAA-3' 5'-CCCATGTCTCCAAGGGCATT-3' AhRR 5'-CAGCAACATGGCTTCTTTCA-3' 5'-GAAGCACTGCATTCCAGACA-3' AhR 5'-GCTGTGATGCCAAAGGGCAGC-3' 5'-TGAAGCATGTCAGCGGCGTGGAT-3' Table 1. List of the primers used in the study
-
The results were pooled from two independent experiments with four animals per group per experiment and presented as mean ± standard error. Data were analyzed by analysis of variance followed by Tukey’s test to examine differences between the groups. P-values < 0.05 were considered significant.
-
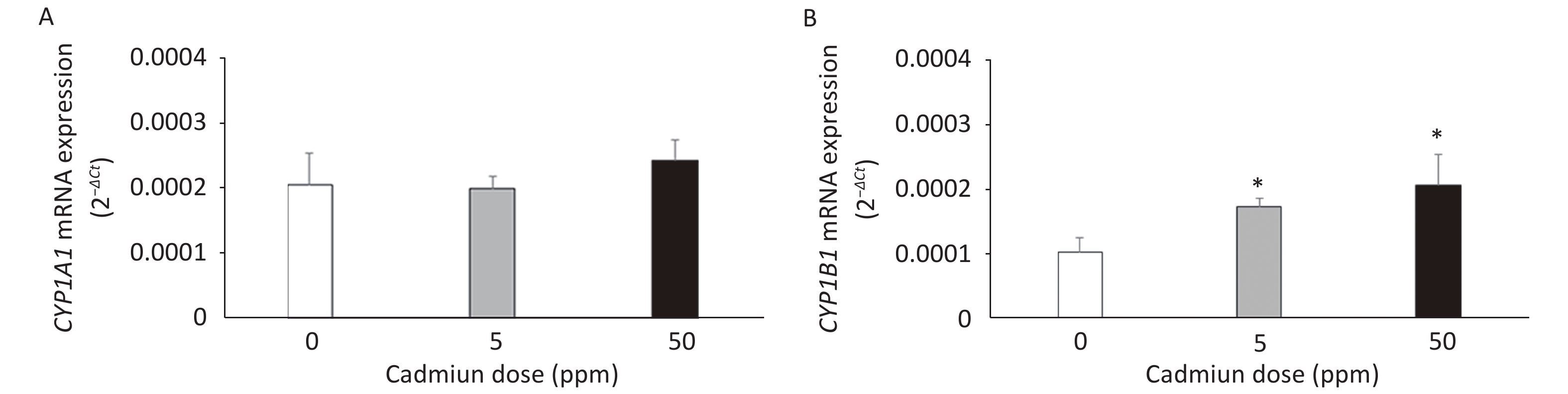
The expression of CYP mRNAs was measured using a DA rat model of prolonged oral metal exposure[39, 41], in which increased levels of Cd are noted in the lungs[41]. The CYP1A1 mRNA remained unchanged (Figure 1A) and CYP1B1 mRNA level increased (Figure 1B) in lung leukocytes of animals treated with either the low (5 ppm) or high (50 ppm) Cd dose compared to the controls (Cd 0 ppm). In addition, an increase in the AhR mRNA level was documented in lung leukocytes of Cd-exposed animals (0.0441 ± 0.0022 in response to the low, and 0.0458 ± 0.0039 in response to the high Cd dose compared to 0.0218 ± 0.0036 in the controls, P < 0.05).
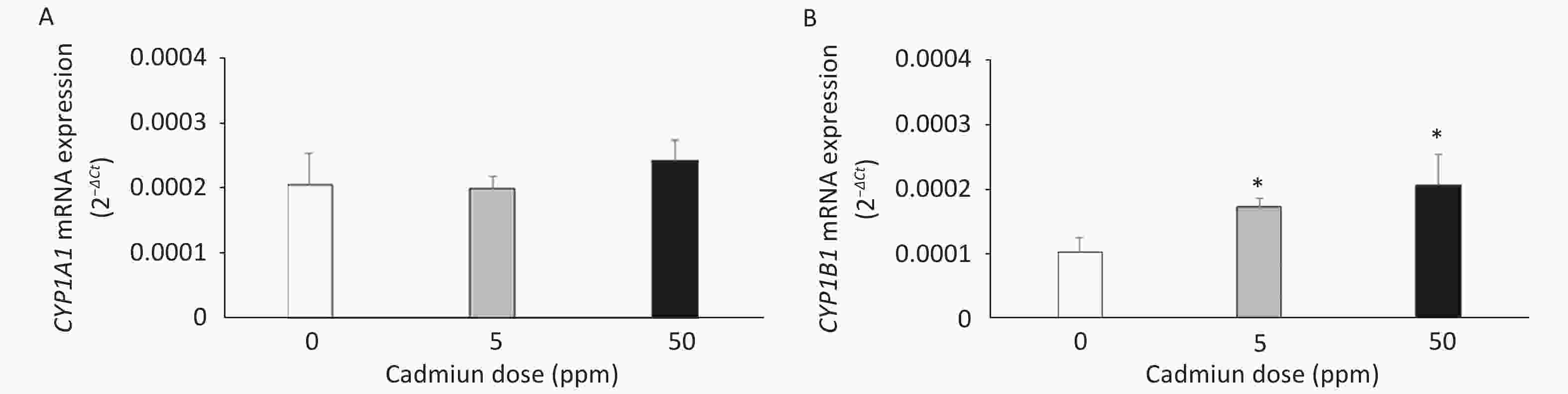
Figure 1. Expression of CYP1A1 (A) and CYP1B1 (B) mRNA in lung leukocytes isolated from animals orally exposed to Cd (5 and 50 ppm). Results are presented as mean ± standard error. Significance at: *P < 0.05 vs. control (Cd dose 0 ppm).
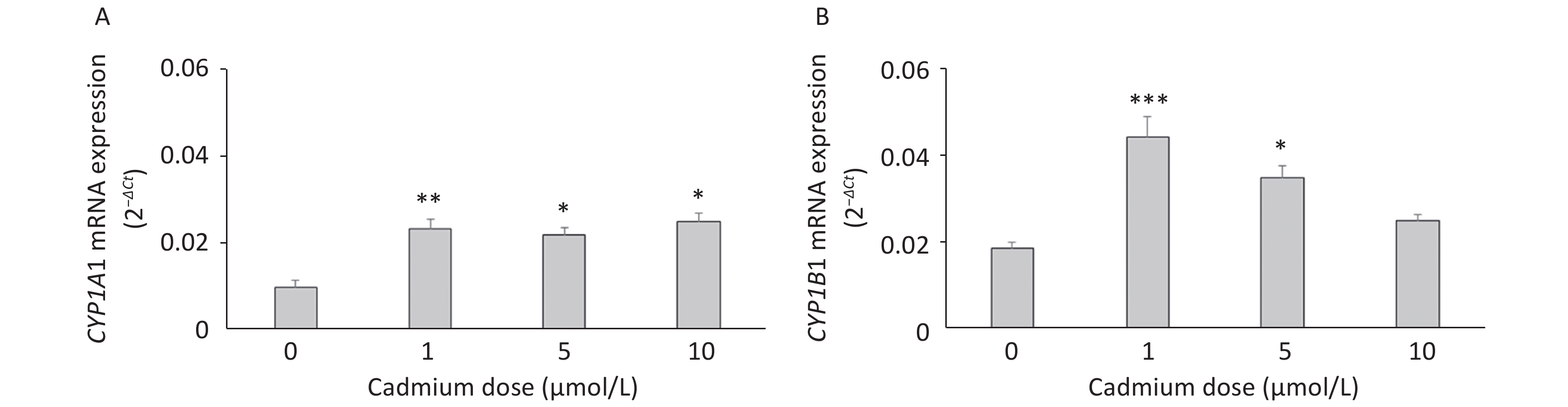
To verify these findings, we exposed lung leukocytes isolated from healthy untreated animals to increasing Cd concentrations in vitro. Cell viability following stimulation with Cd revealed that 50 µmol/L Cd resulted in the death of 15.6% of the cells in culture, so non-lethal doses (i.e., 1, 5, and 10 µmol/L) were tested further. Leukocytes responded to Cd exposure by increasing the expression of CYP1A1 mRNA at all doses tested (Figure 2A) and CYP1B1 at 1 and 5 µmol/L (Figure 2B).
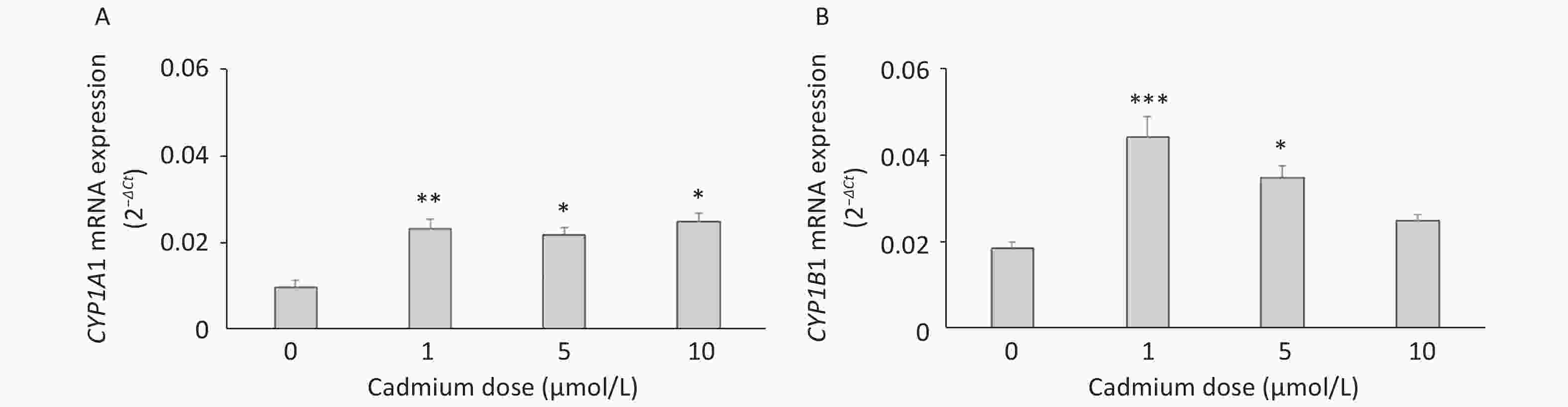
Figure 2. Expression of CYP1A1 (A) and CYP1B1 (B) mRNA in lung leukocytes isolated from healthy untreated animals following in vitro Cd exposure (1, 5, and 10 µmol/L). Results are presented as mean ± standard error. Significance at: *P < 0.05, **P < 0.01, and ***P < 0.001 vs. control (Cd dose 0 µmol/L).
Exposure to Cd was not accompanied by oxidative stress as judged by the lack of changes in the DHR assay and intracellular GSH (Table 2).
Parameters tested Cadmium dose (µmol/L) 0 1 5 10 DHR (mean fluorescence intensity) 1.72 ± 0.12 1.63 ± 0.06 1.63 ± 0.04 1.65 ± 0.05 Intracellular GSH (µmol/L per mg protein) 795.0 ± 88.7 846.7 ± 62.9 762.9 ± 81.4 738.6 ± 67.7 Table 2. Oxidative stress in lung leukocytes following in vitro Cd stimulation
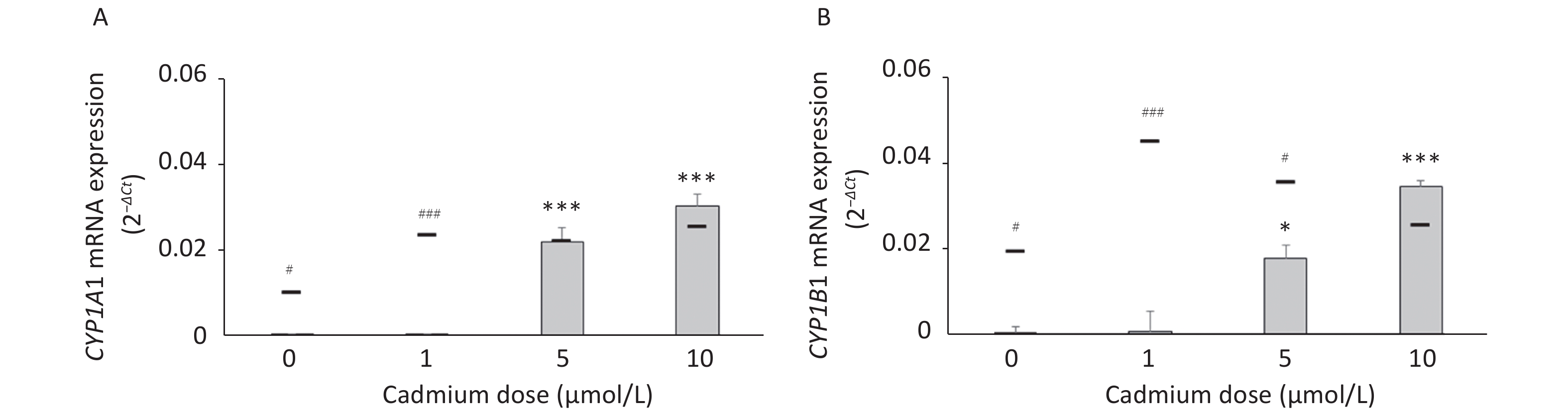
To determine if increased expression of CYPs in response to Cd is a consequence of AhR activation, we measured CYP1A1 and CYP1B1 mRNA levels in the presence of the AhR antagonist CH-223191. The presence of CH-223191 in the culture decreased CYP1A1 mRNA in cells treated with 0 and 1 µmol/L Cd and CYP1B1 mRNA at 1 and 5 µmol/L Cd (Figure 3). As the effect of the AhR inhibitor was observed at 1 and 5 µmol/L Cd, these Cd doses were used for the remaining experiments.
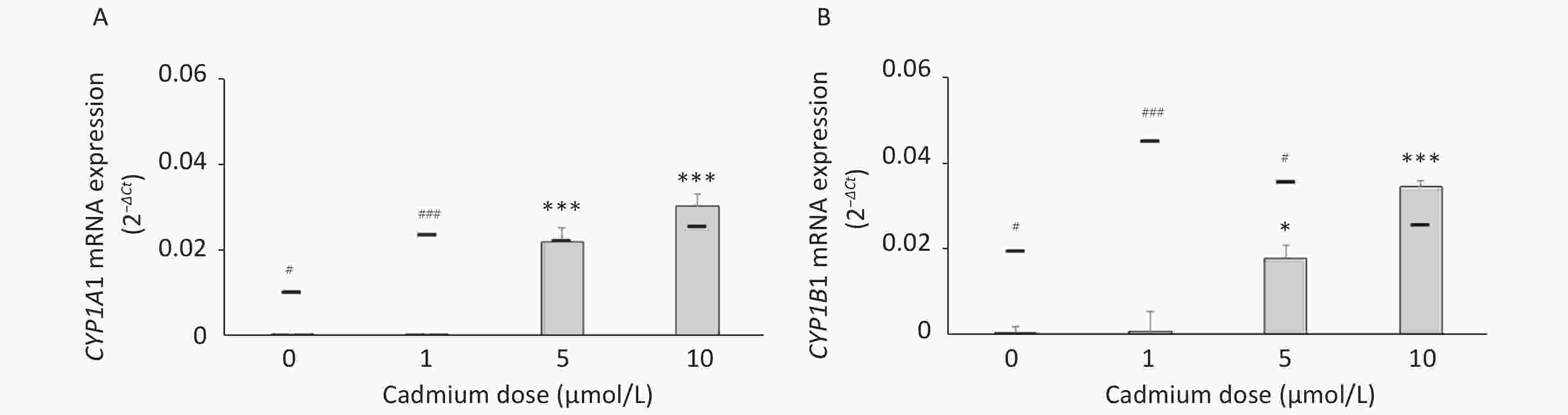
Figure 3. Expression of CYP1A1 (A) and CYP1B1 (B) mRNA in lung leukocytes co-treated with Cd and an AhR antagonist. Lines represent expression levels in cells treated with Cd alone (−CH-223191). Results are presented as mean ± standard error. Significance at: *P < 0.05 and ***P < 0.001 vs. control (Cd dose 0 µmol/L), #P < 0.05, and ###P < 0.01 for CH-223191 vs. −CH-223191.
-
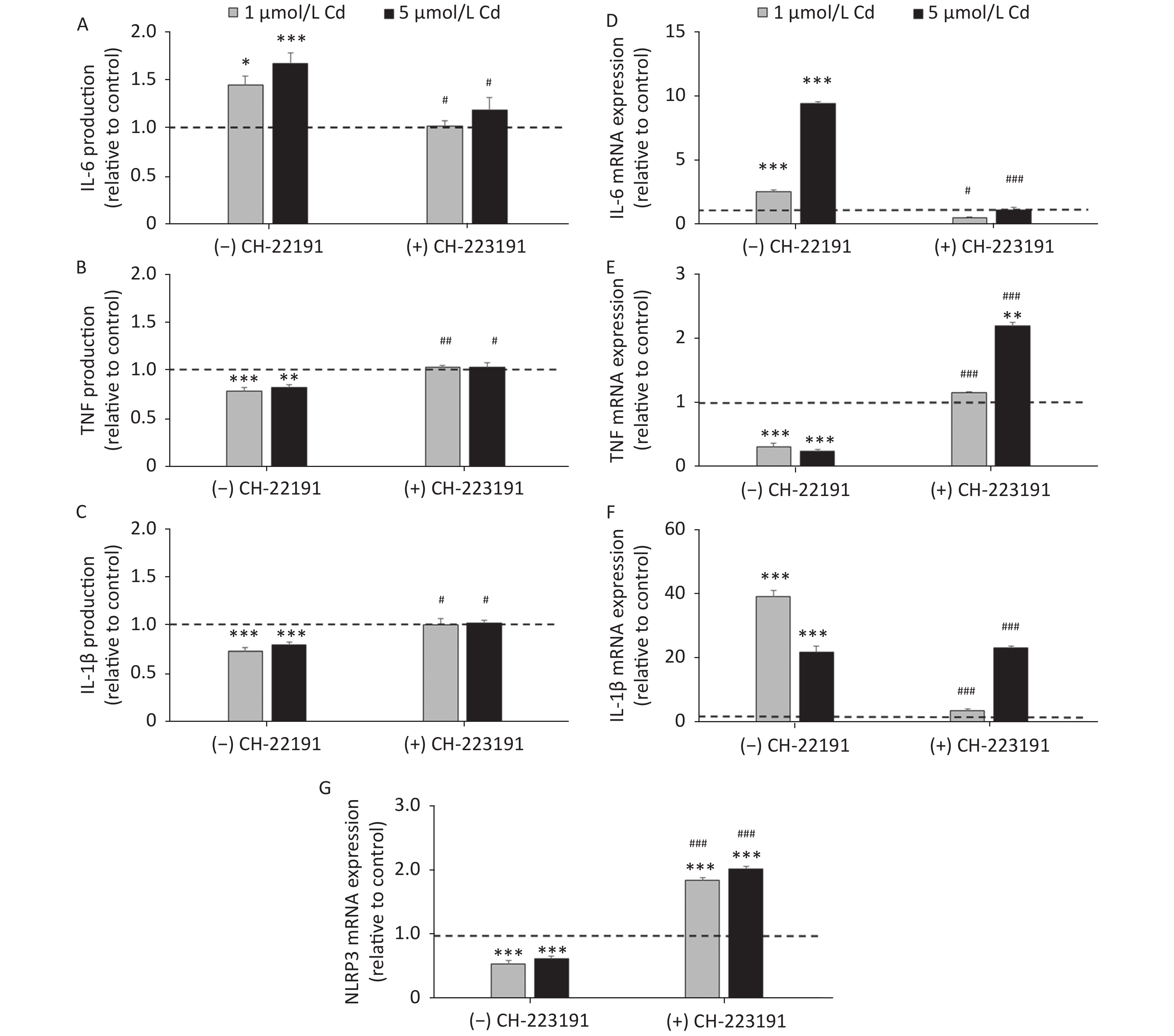
To determine if the effect of Cd on inflammatory cytokine production involves the AhR, IL-1β, IL-6, and TNF production and mRNA expression were measured in cells cultured with Cd alone and in the presence of the AhR antagonist CH-223191 (Figure 4). Lung leukocytes responded to Cd with increased IL-6 (Figure 4A) and decreased TNF (Figure 4B) and IL-1β (Figure 4C) production. The AhR antagonist diminished the effect of Cd and reversed cytokine production to the levels noted in cell cultures without Cd. Similarly, increased IL-6 (Figure 4D) and decreased TNF (Figure 4E) mRNA were noted in response to Cd stimulation. CH-223191 generally returned the mRNA levels to those comparable to the controls (except TNF where a higher mRNA level was noted in cells treated with 5 µmol/L Cd). However, in contrast to decreased production of IL-1β, mRNA of IL-1β increased following stimulation with Cd (Figure 4F). The AhR antagonist suppressed mRNA expression in cells treated with 1 µmol/L Cd, but had no effect on mRNA expression in response to 5 µmol/L Cd.
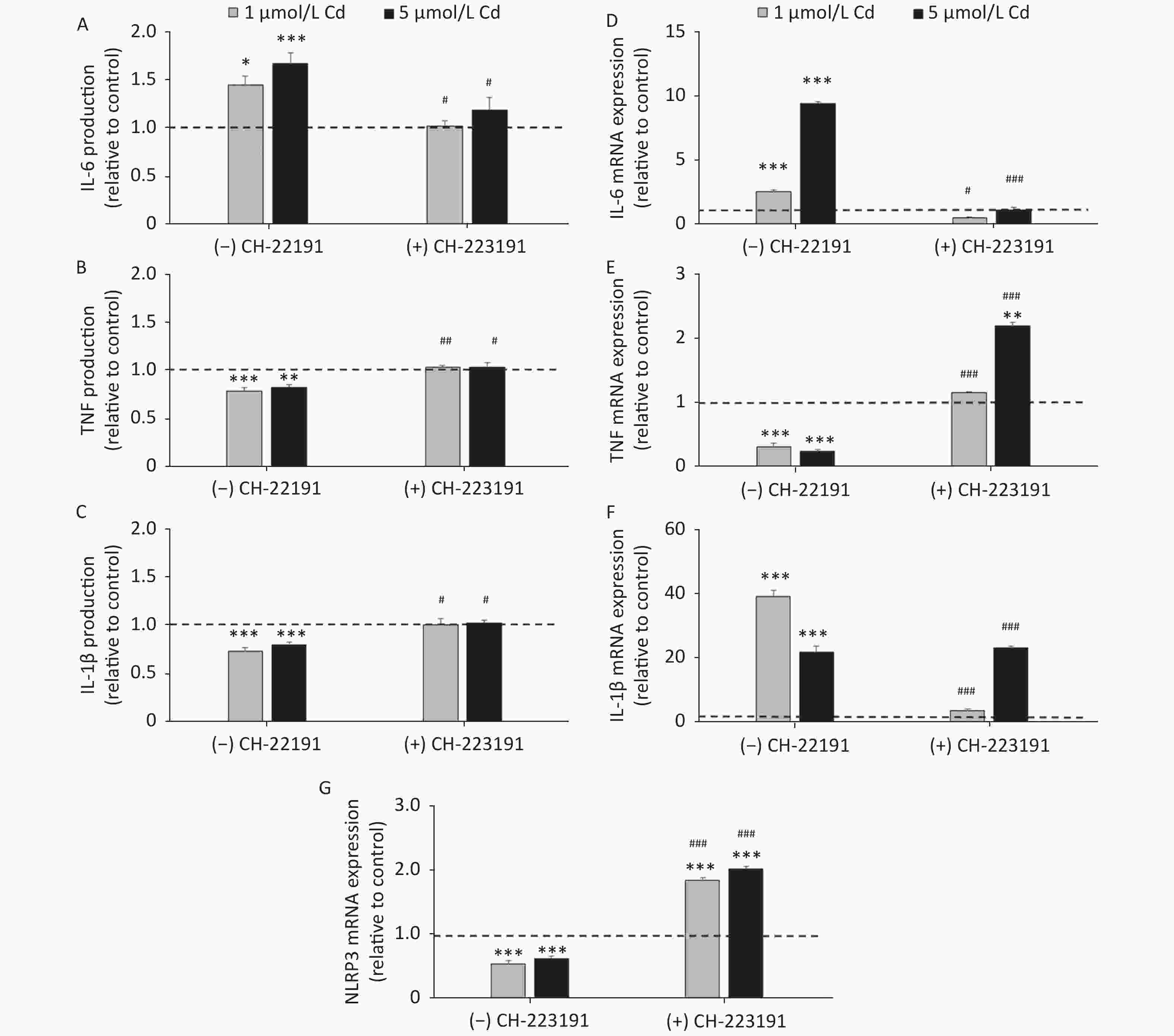
Figure 4. Cytokine production and gene expression by lung leukocytes isolated from healthy untreated animals following in vitro Cd exposure (1 and 5 µmol/L) in the absence or presence of CH-223191. IL-6 production (A) and mRNA expression (D). TNF production (B) and mRNA expression (E). IL-1β production (C) and mRNA expression (F). NLRP3 mRNA expression (G). Results are expressed as relative values compared to the control (considered 1, presented as a line on the graphs) and presented as mean ± standard error. Significance at: *P < 0.05, **P < 0.01 and ***P < 0.001 vs. control and #P < 0.05, ##P < 0.01, and ###P < 0.01 for CH-223191 vs.−CH-223191.
As activation of the AhR causes a decrease in IL-1β production by reducing the mRNA level of NLRP3, an inflammasome component[57], we next determined the NLRP3 level in cells treated with Cd (Figure 4G). As a result, the NLRP3 mRNA level decreased in response to Cd alone. Co-treatment of lung leukocytes with Cd and CH-223191 resulted in increased NLRP3 expression compared to the controls.
-
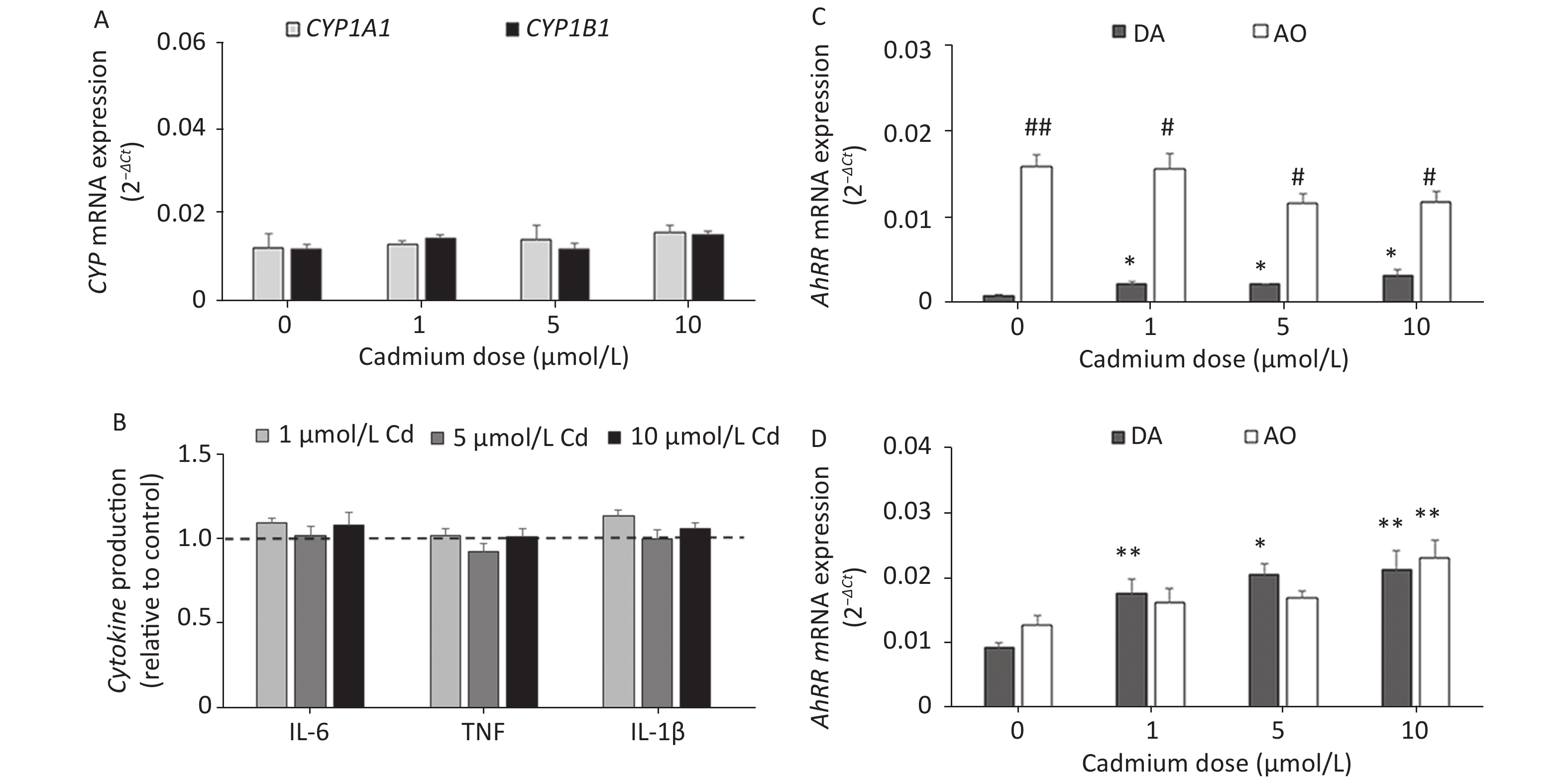
Given that Cd toxicity is rat strain-dependent, the effect of Cd exposure on lung leukocytes from AO rats, a strain that is less sensitive to Cd immunotoxicity[58,59], was examined. In contrast to lung leukocytes from DA rats, CYP1A1 or CYP1B1 mRNA expression (Figure 5A) and proinflammatory cytokine production (Figure 5B) did not change in cells from the lungs of AO rats in response to Cd.
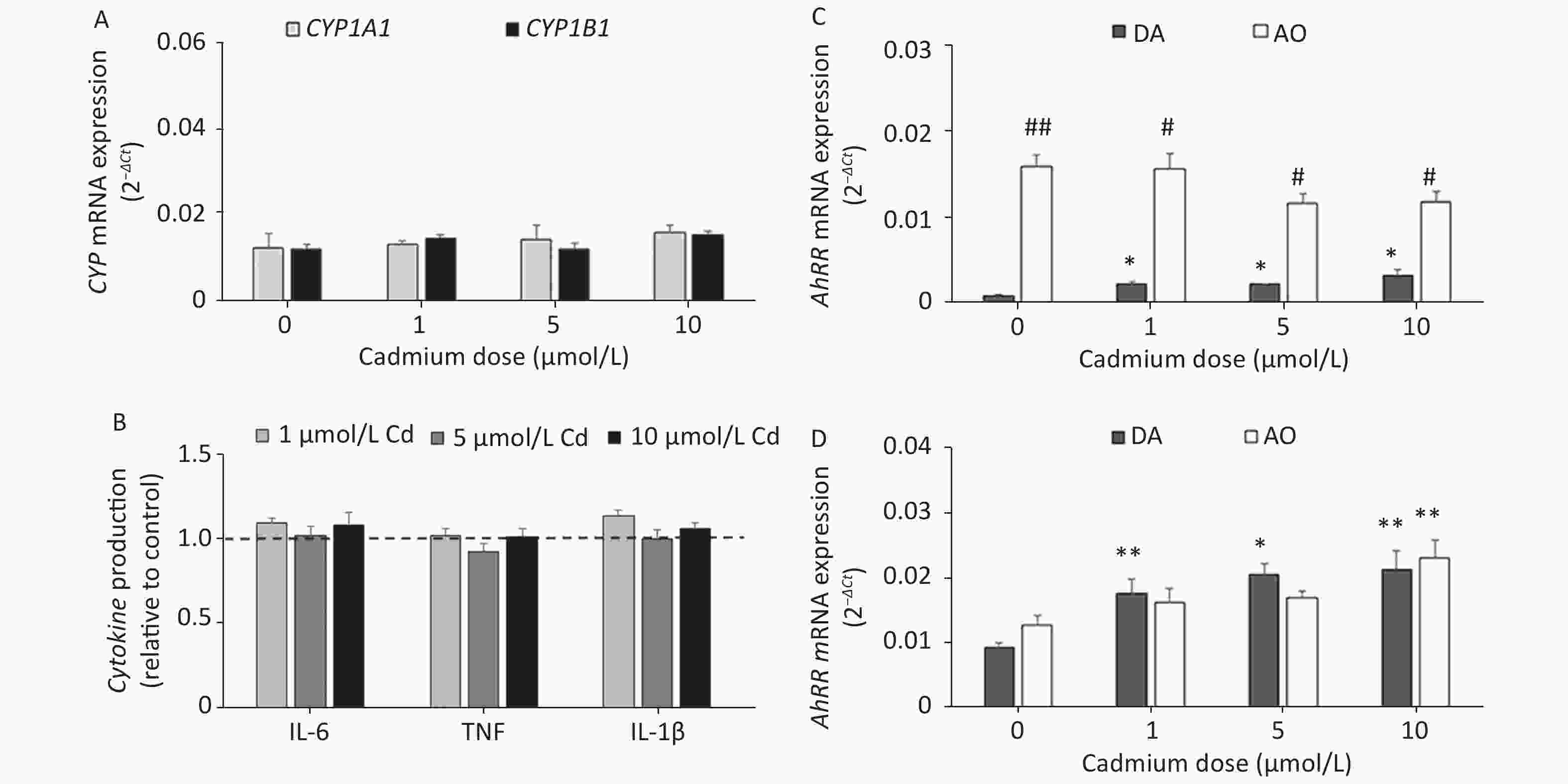
Figure 5. The effect of Cd on lung leukocytes isolated from healthy untreated AO rats. (A) Expression of CYP1A1 and CYP1B1 mRNA in lung leukocytes. (B) Relative cytokine production (IL-6, TNF, and IL-1 β) compared to the control (considered 1, presented as a line on the graphs). AhRR (C) and AhR (D) mRNA expression in lung leukocytes isolated from AO and DA rats. Results are presented as mean ± standard error. Significance at: *P < 0.05 and **P < 0.01 vs. control (Cd dose 0 µmol/L) and #P < 0.05, and ##P < 0.01 for AO vs. DA rats.
To determine what might have contributed to the lack of a Cd effect in AO rats, we measured the AhR repressor (AhRR) expression level, which is a natural regulator of the AhR, in Cd-treated leukocytes of both rat strains (Figure 5C). Generally higher levels of AhRR mRNA were noted in AO compared to DA rats. AhRR mRNA expression increased in DA rats at all Cd doses tested, but there were no changes in AO rats. The increased AhRR expression noted in DA rats may have been related to the increased AhR expression noted in leukocytes of this strain (Figure 5D).
-
In the present study, the potential involvement of the AhR in the immunomodulatory effects of Cd on lung leukocytes was investigated in rats. Cd is a known modulator (stimulator/inhibitor) of several signaling pathways in vivo[60, 61]. Both inhibition[62, 63], as well as stimulation of CYP450[64, 65] have been shown in rodent liver and intestinal tissues[66]. The increased levels of CYP1B1 mRNA in the leukocytes from the lungs of Cd-treated rats in this study showed that Cd affects this AhR-regulated gene in other tissues as well. The increase of CYP1B1 (but not CYP1A1) is in accordance with data showing higher responsiveness of CYP1B1 genes to AhR ligands in several cell lines[67]. In addition, the expression of CYP1A1 and CYP1B1 genes differs between cell sources (higher CYP1B1 mRNA levels in human renal adenocarcinoma compared to liver hepatoblastoma cells)[68]. Nevertheless, in vitro Cd exposure stimulated genes of both CYPs. The increased levels of CYP1A1 mRNA in lung leukocytes following in vitro Cd exposure are in line with data showing higher levels of this AhR-regulated gene mRNA in human HepG2 hepatoma cells[34] and murine Hepa1c1c7 hepatoma cells[29]. A decrease in CYP1A1 mRNA (at 1 μmol/L of Cd) and CYP1B1 mRNA (at 1 and 5 μmol/L of Cd) levels in response to the AhR antagonist indicates involvement of the AhR in the observed increases of the CYPs at these Cd doses. The lack of an effect at a higher Cd dose (10 μmol/L) suggests the effect of high Cd doses on the activity of other transcriptional factors, as the AhR is not the only factor regulating CYP expression[69, 70].
ROS are often related to Cd toxicity[71]. Increases in lung leukocyte CYP1B1 from rats that consumed Cd might be related to the observed oxidative stress[41]. Oxidative stress seems not to be the potential reason for the increase in CYP mRNAs in lung leukocytes exposed to Cd in vitro.
Stimulation of lung leukocytes with Cd resulted in increased IL-6 protein and gene expression levels, and applying CH-223191 abolished this increase, suggesting involvement of the AhR in the IL-6 response to Cd. Data regarding a role for AhR activation in the IL-6 response vary from suppression, as noted in mouse peritoneal macrophages following stimulation with 2, 3, 7, 8 -tetrachlorodibenzo-p-dioxin (TCDD) and FICZ[57] and in a bone marrow stromal cell line in response to TCDD[72], to stimulation measured in response to FICZ in HaCaT cells[73] and in human and mouse mast cells[74]. The different effect of AhR activation on IL-6 probably depends on the type of cells and/or the AhR agonist used. Our results suggest that activation of the AhR might result in a greater IL-6 response in lung leukocytes. Although the cytokine response to FICZ stimulation in HaCaT cells is mediated by oxidative stress[73], no effect of Cd on the production of ROS or GSH was noted in our experimental setting, suggesting a direct role of the AhR in the regulation of the IL-6 response to Cd.
Stimulation of lung leukocytes with Cd in culture resulted in a decrease in the TNF response. Both TNF protein and mRNA reached levels comparable to controls (cells unexposed to Cd) in the presence of CH-223191, suggesting a role of the AhR in the Cd-induced TNF response to Cd. Data regarding the role of the AhR in the TNF response are conflicting. Activation of the AhR with TCDD and FICZ suppresses TNF in mouse peritoneal macrophages[57], while no effect of FICZ was noted on the cytokine response in HaCaT cells[73]. In contrast, an increase of TNF has been observed in response to TCDD in differentiated THP-1 cells[75] and in the lungs of C57BL mice[76]. Increased TNF expression in response to 5 µmol/L Cd in the presence of the AhR antagonist, indicates that higher Cd doses affect mRNA transcription by additional mechanisms. However, the absence of changes at the protein level suggest a role of the AhR in post-transcriptional processing of TNF mRNA or in mRNA stability.
The effect of the AhR inhibitor on IL-1β production suggests involvement of the AhR in the cytokine response to Cd as well. Decreased IL-1β production due to activation of the AhR has been documented in mouse peritoneal macrophages following stimulation with TCDD and FICZ[57]. In accordance with that study, our data show that decreased IL-1β production was not due to decreased expression of IL-1β mRNA, as higher levels were noted in cells treated with Cd compared to controls. The involvement of AhR activation in the stimulation of IL-1β transcription has been shown in HaCaT cells treated with FICZ as well[73], although those authors did not observe any changes in cytokine production. The absence of IL-1β mRNA expression (compared to cells not exposed to Cd) in cells co-treated with a low Cd dose and the AhR antagonist is opposite to the increased expression noted in cells treated with Cd alone, suggesting involvement of the AhR in the synthesis of IL-1β mRNA. However, similar IL-1β transcription in response to a higher Cd dose regardless of the presence of the AhR antagonist proposes some additional mechanisms of cytokine transcriptional regulation by Cd, and suggests that the effect of the AhR on IL-1β transcription depends on the metal dose applied.
A discrepancy between the IL-1β production and gene expression data in response to Cd in our study might be explained by decreased expression of NLRP3. NLRP3 is a component of the inflammasome, a protein complex which is included in the processing of pro-IL-1β to secretory IL-1β. The increase of NLRP3 mRNA expression in cells co-treated with Cd and CH-223191 was opposite to the decreased expression in cells treated with Cd alone, suggesting that the observed effect is a consequence of AhR activation. In support of this assumption are data obtained in mouse peritoneal macrophages treated with TCDD and FICZ that show a decrease in NLRP3 mRNA following receptor activation[57].
Increased levels of AhR repressor mRNA in AO rats seemed responsible for the lack of the effect of Cd on CYP mRNA levels. In addition, high AhRR levels in leukocytes of AO rats could be responsible for the lack of an effect of Cd on cytokine production, as inhibited IL-1β production is seen in AhRR-transgenic mice[77]. The lack of a change in the AhRR mRNA levels in response to Cd in AO rats is in line with data that animals over-expressing AhRR do not increase expression of AhRR in the lungs when treated with TCDD in contrast to wild-type animals in which increased AhRR expression has been reported[50]. Increased levels of AhRR in leukocytes of DA rats exposed to Cd seemed to be related to an increase in the AhR in line with data that activation of the AhR results in increased repressor expression[50]. The strong positive correlation between the AhR and AhRR (AhRR = 0.0004 + 0.073 × AhR, r = 0.733, P = 0.0012) noted in lung leukocytes from DA rats supports such an assumption. The relevance of the AhR for animal strain differences in chemical toxicity has been recognized in terms of the different toxicities of various AhR ligands in inbred mice and rats (as a consequence of gene polymorphism within the receptor)[78, 79], the structure of the AhR receptor in rats[80], or due to different levels of CYP450, AhR, and the AhR nuclear translocator[81]. Our data suggest that differences in the AhR repressor may also be responsible for strain differences in chemical toxicity.
In conclusion, the results presented in this study show that Cd caused an increase in expression of AhR-regulated genes in vitro and in vivo. AhR signaling was involved in the lung leukocyte proinflammatory cytokine response, which is a novel finding. Strain differences in AhR activity in the lung leukocyte response to Cd were, at least partially, mediated by different AhR repressor expression levels. Involvement of the AhR in the leukocyte cytokine response to Cd in inflammation-prone rats provides new insight into the mechanisms of Cd immunotoxicity.
-
The authors declare no conflicts of interest.
-
The authors thank Marta Despotovic for technical assistance during part of the investigation.
Aryl Hydrocarbon Receptor is Involved in the Proinflammatory Cytokine Response to Cadmium
doi: 10.3967/bes2021.025
Funds:
This study was supported by the Ministry of Education, Science, and Technological Development of the Republic of Serbia [451-03-68/2020-14/200007]
- Received Date: 2020-03-18
- Accepted Date: 2020-06-02
-
Key words:
- Cadmium /
- Lung leukocytes /
- Aryl hydrocarbon receptor /
- Cytokine (IL-6, TNF, IL-1β) response
Abstract:
| Citation: | KULAS Jelena, TUCOVIC Dina, ZELJKOVIC Milica, POPOVIC Dusanka, POPOV ALEKSANDROV Aleksandra, KATARANOVSKI Milena, MIRKOV Ivana. Aryl Hydrocarbon Receptor is Involved in the Proinflammatory Cytokine Response to Cadmium[J]. Biomedical and Environmental Sciences, 2021, 34(3): 192-202. doi: 10.3967/bes2021.025

|

 Quick Links
Quick Links
 DownLoad:
DownLoad: