-
Beryllium (Be) is a non-radioactive element with carcinogenic properties, and presents serious occupational and environmental hazards from its resulting toxicants, which could significantly impact the health of the occupational population[1-3]. Beryllium and its compounds can cause diseases such as acute chemical pneumonia, lung cancer, and chronic beryllium disease, which predominantly occurs alongside either lung granuloma or pulmonary fibrosis[4,5]. Furthermore, the mechanism by which beryllium is toxic has not been fully elucidated; therefore, studies on its health hazards as well of its compounds are important. An in-depth study of the mechanism of beryllium toxicity is especially essential to prevent and control its associated health hazards. Tandem mass tag (TMT) technology is a reliable technology in quantitative proteomics, which can be implemented to perform relative quantification and identification analysis of proteins, peptides, nucleic acids, and other biological macromolecules.
In this study, TMT-labeled quantitative proteomics technology and bioinformatics analyses were used to evaluate the differential protein expression profiles in 16HBE cells treated with BeSO4. In addition, we screened selected key proteins that have important reference values for potential follow-up research, thereby providing a new direction for exploring the mechanism of BeSO4-treated 16HBE cytotoxicity and screening for potential biomarkers.
The human bronchial epithelial (16HBE) cells were cultured in DMEM containing 10% FBS at 37 °C in a 5% CO2 incubator and treated with BeSO4 at 150 μmol/mL for 48 h. Protein quantification was performed according to the instructions of the BCA Protein Quantification Kit. One hundred and fifty μg of total protein was used for each sample in an ultrafiltration tube to which 10 mmol/L DTT (DL-dithiothreitol) was added to 500 μL at 4 °C, then the protein sample was centrifuged at 10,464 ×g for 15 min, and incubated at room temperature for 1 h. Subsequently, 400 μL of 20 mmol/L IAA (Indole-3-acetic acid) was added at 4 °C, centrifuged at 14,243 ×g for 15 min, after which the filtrate was discarded in the collection tube. Later 400 μL of 100 mmol/L triethylamine borane (TEAB) was added and the mixture was centrifuged at 14,243 ×g for 15 min at 4 °C, and the filtrate was discarded. TMT labeling was performed according to instructions by Thermo ScientificTM TMTTM, and the TMT-labeled samples were combined and transferred into a 1.5 mL tube. The evaporated samples were dissolved in 0.1% formic acid (FA). The samples were divided into groups using Ultimate 3000 and APC-3000 instruments (Thermo, USA). Each component was evaporated to dryness using a PVC 2–18 centrifugal concentrator, and the components of the spin-dried samples were dissolved in 20 μL of 0.1% formic acid (FA) (mixed by vortexing), and tested on a Q Exactive mass spectrometer (Thermo, USA). The original data obtained by searching the UniProt human database were exported to Microsoft Excel software and then normalized using Persue software for subsequent analyses.
Based on the R language “clusterProfiler” and “org.Hs.eg.db” software packages, Gene Ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) enrichment analysis of DEPs were carried out, whose biological effect is analyzed from three aspects: biological function, cell composition and molecular function. Simultaneously, the related signaling pathways involved in the differential proteins of 16HBE cells treated with BeSO4 were analyzed. The protein-protein interaction network (PPI) is a visual network diagram of the interaction between proteins, highlighting the key proteins from the interconnected nodes and the number. After importing DEPs into the STRINIG online database (
https://string-db.org/ ), they were combined with Cytoscape software to visually analyze the TSV files from STRINIG. The MCODE plug-in in Cytoscape, based on the calculation method of node information in the network graph. This was done to further determine the more closely connected protein modules. R language was used to perform GO enrichment analysis on the protein modules selected in PPI and to screen key proteins.A total of 4,054 proteins were obtained, and the subsequent analysis illustrated that a total of 883 proteins were (significantly) differentially expressed between the groups (P < 0.05). The volcano and heat maps more intuitively demonstrate the distribution of differential protein expression between the experimental and control groups. Criteria of FC ≥ 1.2, FC ≤ 0.83, and P < 0.05, were established to identify the initial 15 proteins exhibiting significant differential expression (Table 1).
Protein accession number Protein name Gene FC Up/Down P value P68431 Histone H3.1 H3C1 1.33 up 0.05 P04259 Keratin, type II cytoskeletal 6B KRT6B 1.29 up 0.01 Q9BVS4 Serine/threonine-protein kinase RIO2 RIOK2 1.21 up < 0.01 Q8WXF1 Paraspeckle component 1 PSPC1 1.20 up < 0.01 P08263 Glutathione S-transferase A1 GSTA1 0.71 down 0.02 O60499 Syntaxin-10 STX10 0.71 down 0.05 Q9P1F3 Costars family protein ABRACL ABRACL 0.72 down 0.03 Q96FK6 WD repeat-containing protein 89 WDR89 0.73 down 0.05 P02795 Metallothionein-2 MT2A 0.74 down < 0.01 P17066 Heat shock 70 kD protein 6 HSPA6 0.77 down 0.02 Q9Y4G6 Talin-2 TLN2 0.78 down 0.03 P54868 Hydroxymethylglutaryl-CoA synthase, mitochondrial HMGCS2 0.80 down < 0.01 Q92604 Acyl-CoA:lysophosphatidylglycerol acyltransferase 1 LPGAT1 0.81 down < 0.01 Q15651 High mobility group nucleosome-binding domain-containing protein 3 HMGN3 0.81 down 0.02 P07477 Trypsin-1 PRSS1 0.82 down 0.01 Table 1. Top 15 DEPs screened in the 16HBE cells treated with beryllium sulfate
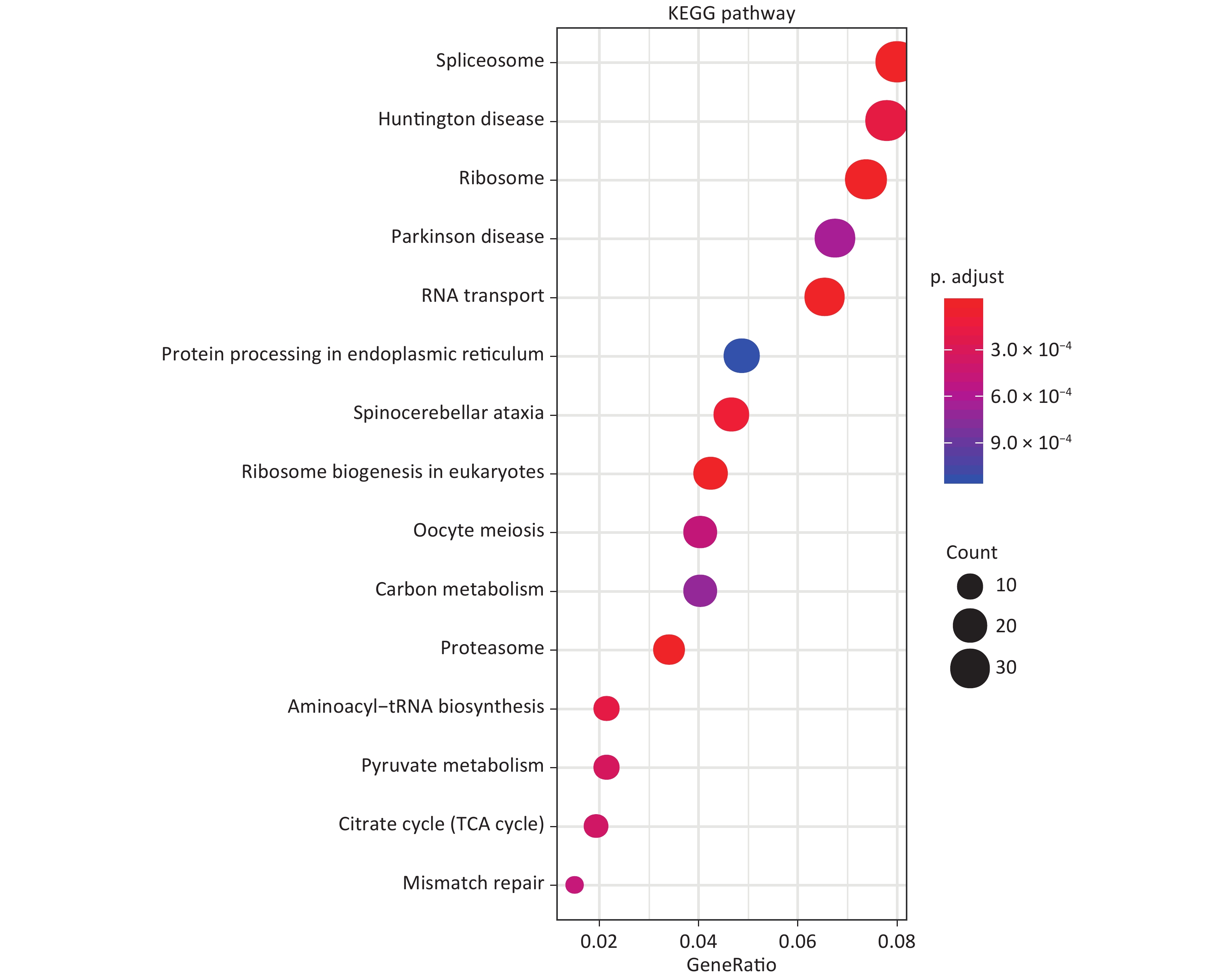
GO analysis showed that in terms of biological processes, the identified DEPs were mainly involved in ribonucleoprotein complex biogenesis, ribosome biogenesis, and RNA catabolism and processing. Cell component analysis showed that differentially expressed proteins are involved in cell-substrate junctions, focal adhesions, cell-substrate adherens junctions, etc. According to molecular function, the most differentially expressed proteins are related to cadherin binding, cell adhesion molecule binding, and structural constituents of ribosomes (Figure 1). KEGG analysis was used to explore the signaling pathways involved in the regulation of differentially expressed proteins. It was found that these differential proteins are mainly involved in signaling pathways involving the spliceosome, Huntington’s disease, and ribosomes (Figure 2).
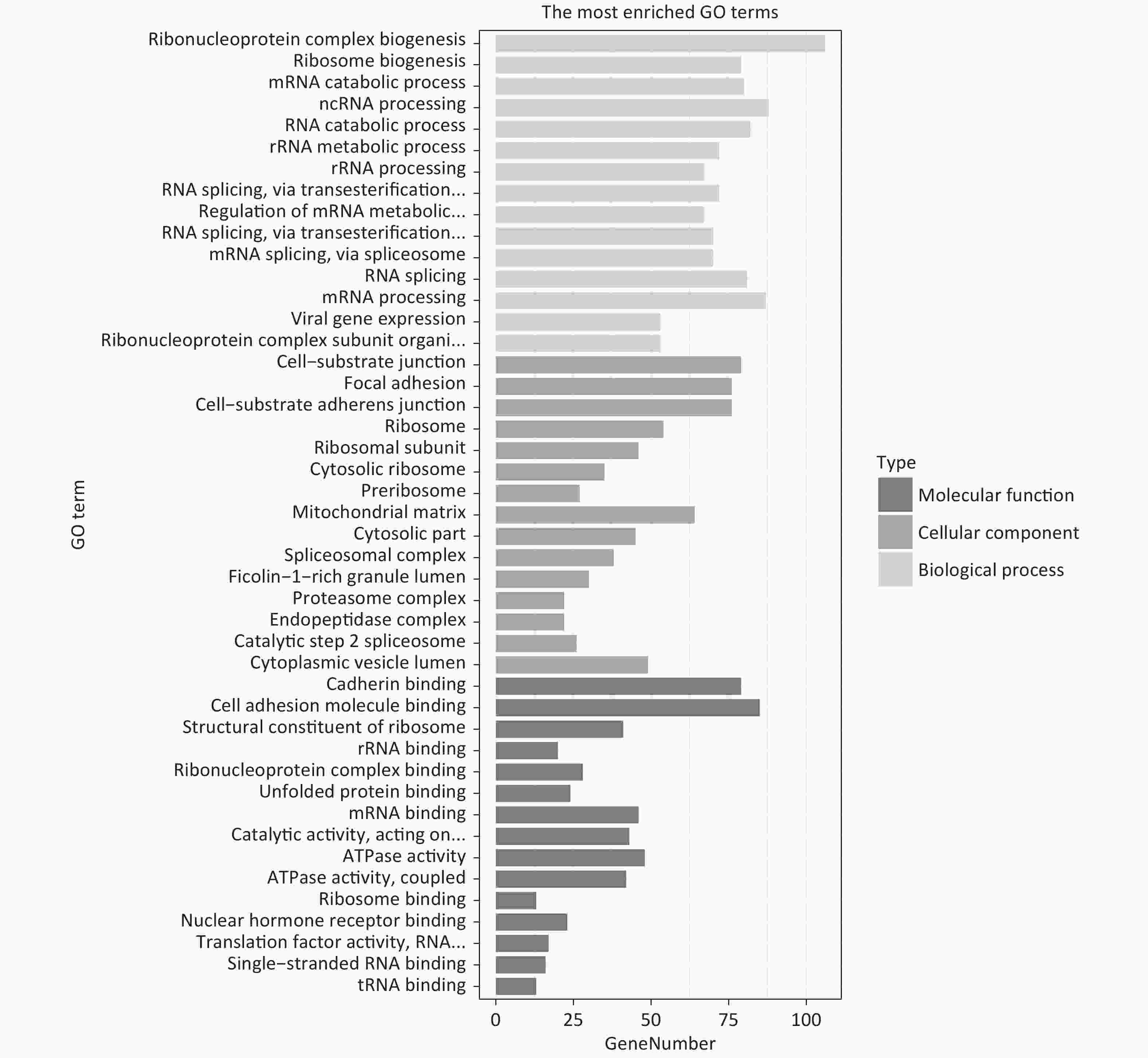
Figure 1. GO enrichment analysis of DEPs
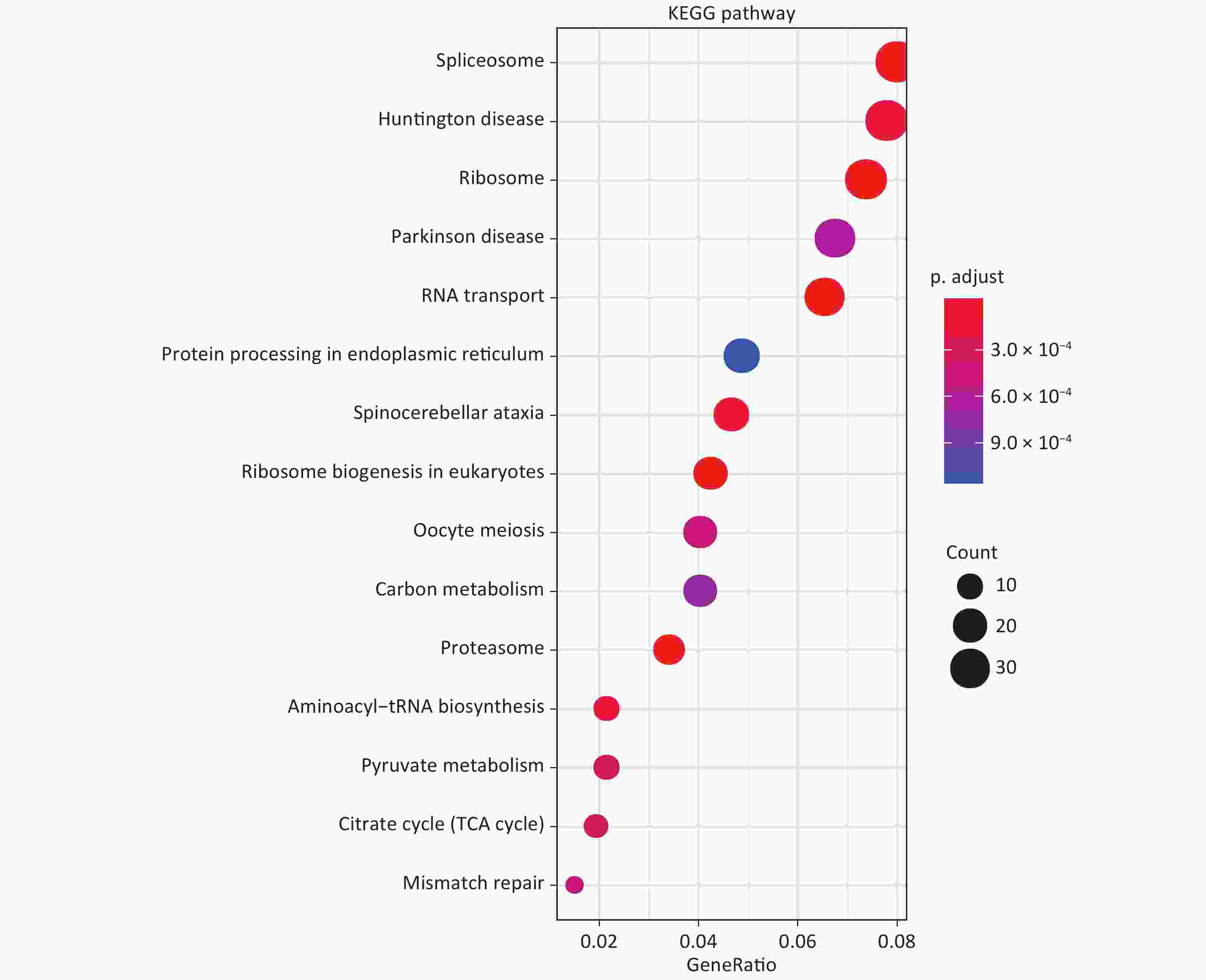
Figure 2. KEGG enrichment analysis of DEPs.
Cytoscape software was used to visually display the protein interaction- relationship data obtained from the STRING online database. The network graph composed of upregulated proteins consists of 421 nodes and 4,566 edges, whereas the downregulated protein network consists of 284 nodes and 1,756 edges. Thereafter, MCODE was used to filter out the key protein modules associated with the upregulation and downregulation of network graphs. The key protein modules are more intuitive. The key protein modules in the up- and down-regulated protein network graphs were 74 nodes, 1,574 connections, and 20 nodes and 47 connections, respectively.
Further GO analysis of the selected key protein modules revealed that the upregulated modules were mainly related to biological processes such as rRNA processing, metabolism, and protein localization. These differential proteins are mainly distributed in the cytoplasm and ribosomes and are involved in molecular functions, such as rRNA binding and translation factor activity. The biological processes involved in the downregulation of key protein modules are mainly related to oxidative stress and interleukin-12. Cell component analysis highlights that the proteins in this module are mainly distributed intercellularly, in the melanosome, or in the myelin. However, they are also related to molecular functions, such as oxidoreductase activity. The identified proteins from the upregulated and downregulated key protein modules were further screened to reveal that RIOK2, CSNK1D, and HEATR1 are upregulated, all of which are involved in rRNA processing and metabolism. Nine were downregulated, including PRDX5, P4HB, GLRX3, PDIA3, PRDX2, TXN, SOD2, CTNNB1, and HMGB1, all of which are related to oxidative stress. Additionally, HMGB1, P4HB, and TXN are involved in the interleukin-12-mediated signaling pathways. After importing these 12 key proteins into the STRING database, the data illustrate an interaction relationship between them. This suggests that these 12 key proteins form a regulatory network, and may play a regulatory role in the biological processes of oxidative stress and those of interleukin-12 in 16HBE cells treated with BeSO4.
In the present study, proteomics and bioinformatics were used to investigate DEPs before and after BeSO4 treatment in 16HBE cells, and to further explore any relevant signaling pathways. A total of 883 DEPs were identified, of which 520 were upregulated and 363 were downregulated. Additionally, GO and KEGG enrichment analyses were performed on the DEPs to explore the biological processes and signaling pathways involved. Visualization software was used to display the PPI interaction relationship of DEPs and to further identify key proteins. These analyses provide valuable mechanistic information in the study of BeSO4- treated 16HBE cell damage.
To predict the function of the key protein modules in the PPI network, we screened three key proteins from the upregulated modules: RIOK2, CSNK1D, and HEATR1, all of which are related to rRNA processing and metabolism. Additionally, nine key downregulated proteins related to oxidative stress: PRDX5, P4HB, GLRX3, PDIA3, PRDX2, TXN, SOD2, CTNNB1, and HMGB1 were screened. Among these, HMGB1, P4HB, and TXN are also involved in biological processes related to interleukin-12. Interleukin-12 is a multifunctional cytokine that is linked to effective anti-tumor and anti-infection immunomodulatory activities both, in vivo and in vitro[6]. Oxidative stress refers to the imbalance between the production of free radicals and reactive metabolites, and the elimination of antioxidants. This imbalance can promote damage to important biological molecules and cells, potentially affecting the entire organism[7]. In addition, oxidative stress is related to the pathogenesis of many diseases, such as chronic lung disease, chronic disease including chronic obstructive pulmonary disease, hypertension, and Alzheimer's disease[8, 9]. Studies have shown that BeSO4 stimulates the formation of reactive oxygen species (ROS) in mouse macrophages and plays an important role in apoptosis[10].
In summary, the results of this study identified three key potential differential proteins related to rRNA processing and metabolism, and nine were involved in oxidative stress signaling pathways. The DEPs demonstrate specific regulatory effects, and could thus be the basis of novel research directions and ideas for subsequent follow-up studies pertaining to the molecular mechanism of BeSO4-induced 16HBE cell damage. Our findings provide valuable references for future research on potential biomarkers and metabolic pathways of BeSO4 toxicity mechanisms.
Proteomics Study on the Differentially Expressed Proteins in 16HBE Cells Exposed to Beryllium Sulfate
doi: 10.3967/bes2021.128
Funds:
This study was supported by the National Natural Science Foundation of China [81573193] and the Natural Science Foundation of Hunan Province [2020JJ4082]
- Received Date: 2021-01-25
- Accepted Date: 2021-03-17
| Citation: | CAI Ying, LIU Yan Ping, ZHENG Kai, LEI Yuan Di, WANG Ye, XU Xin Yun, ZHANG Zhao Hui. Proteomics Study on the Differentially Expressed Proteins in 16HBE Cells Exposed to Beryllium Sulfate[J]. Biomedical and Environmental Sciences, 2021, 34(11): 926-930. doi: 10.3967/bes2021.128

|

 Quick Links
Quick Links
 DownLoad:
DownLoad: