-
Mitochondria are important intracellular organelles that carry out oxidative phosphorylation, synthesize ATP, and provide energy for cell activities. Mitochondrial dysfunction can affect the normal function of the entire cell and is thought to be a major risk factor in the onset and progression of some diseases associated with chronic inflammation and insulin resistance[1]. Reactive oxygen species (ROS) produced mainly in mitochondria have two functions, redox biology and oxidative stress, and their contribution to both physiological and pathological conditions. Redox biology involves a small increase in ROS levels that activates signaling pathways to initiate biological processes, while oxidative stress denotes high levels of ROS that result in damage to DNA, protein or lipids[2]. Various extracellular stimuli have been shown to increase the production of ROS by the mitochondrial respiratory chain, thereby causing a shift in overall redox balance and subsequent initiation of downstream signaling cascades. Mitochondrial DNA (mtDNA) is more prone to damage caused by ROS than nuclear DNA, which is likely to have an impact on the activity of those proteins containing mtDNA-encoded subunits. It is well known that ROS are involved in a wide spectrum of diseases, such as chronic inflammation. ROS generation also increases the release of pro-inflammatory cytokines, such as tumor necrosis factor α (TNFα); vice versa, TNFα and interleukin-6 (IL-6) are potent stimulators of ROS production, thus inducing oxidative stress. Oxidative stress and chronic inflammation are critical for the development of insulin resistance. As a main metabolic organ, liver oxidative stress and inflammation are closely related to metabolic disorders such as type 2 diabetes mellitus (T2DM), nonalcoholic fatty liver disease (NAFLD), hyperlipidemia, and insulin resistance[3].
Sestrins (SESN) including SESN1, SESN2, and SESN3 are highly evolutionary conserved proteins that accumulate in cells exposed to stress and have poorly defined physiological functions. In vitro, SESNs exhibit oxidoreductase activity and may function as antioxidants involved in age-associated pathological alterations. Independent of their redox activity, SESNs lead to AMP-activated protein kinase-dependent inhibition of mammalian target of rapamycin (mTOR) signaling and link genotoxic stress to mTOR regulation. Drosophila SESN is also a negative feedback regulator of mTOR whose loss results in various mTOR-dependent, age-related pathologic changes[4]. A previous study demonstrates that SESN2 ablation exacerbates glucose intolerance, insulin resistance and hepatosteatosis, which may indicate that the stress-inducible SESN protein family has an important homeostatic function in the control of glucose and lipid metabolism[5]. However, the effect of SESN1 on hepatic mitochondrial function and chronic inflammation response remains elusive.
Sirtuin 1 (SIRT1), an ortholog of the yeast Sir2 protein and a nuclear NAD+-dependent protein deacetylase, can directly couple the cellular metabolic status (via NAD+) to the chromatin structure and the regulation of gene expression. SIRT1 regulates glucose or lipid metabolism through its deacetylase activity in over two dozen known substrates, and has a positive role in the metabolic pathway through its direct or indirect involvement in insulin signaling. Growing evidence suggests that SIRT1 has been identified as an important repressor of oxidative stress and inflammation in multiple tissues, such as the liver, skeletal muscle and adipose tissues by reducing the ROS levels and improving cell survival via this antioxidant effect, thus providing beneficial effects in some disorders, such as obesity, NAFLD, T2DM and aging[6]. This study aimed to determine whether SIRT1 mediates the effect of SESN1 on improving hepatic oxidative stress and chronic inflammation.
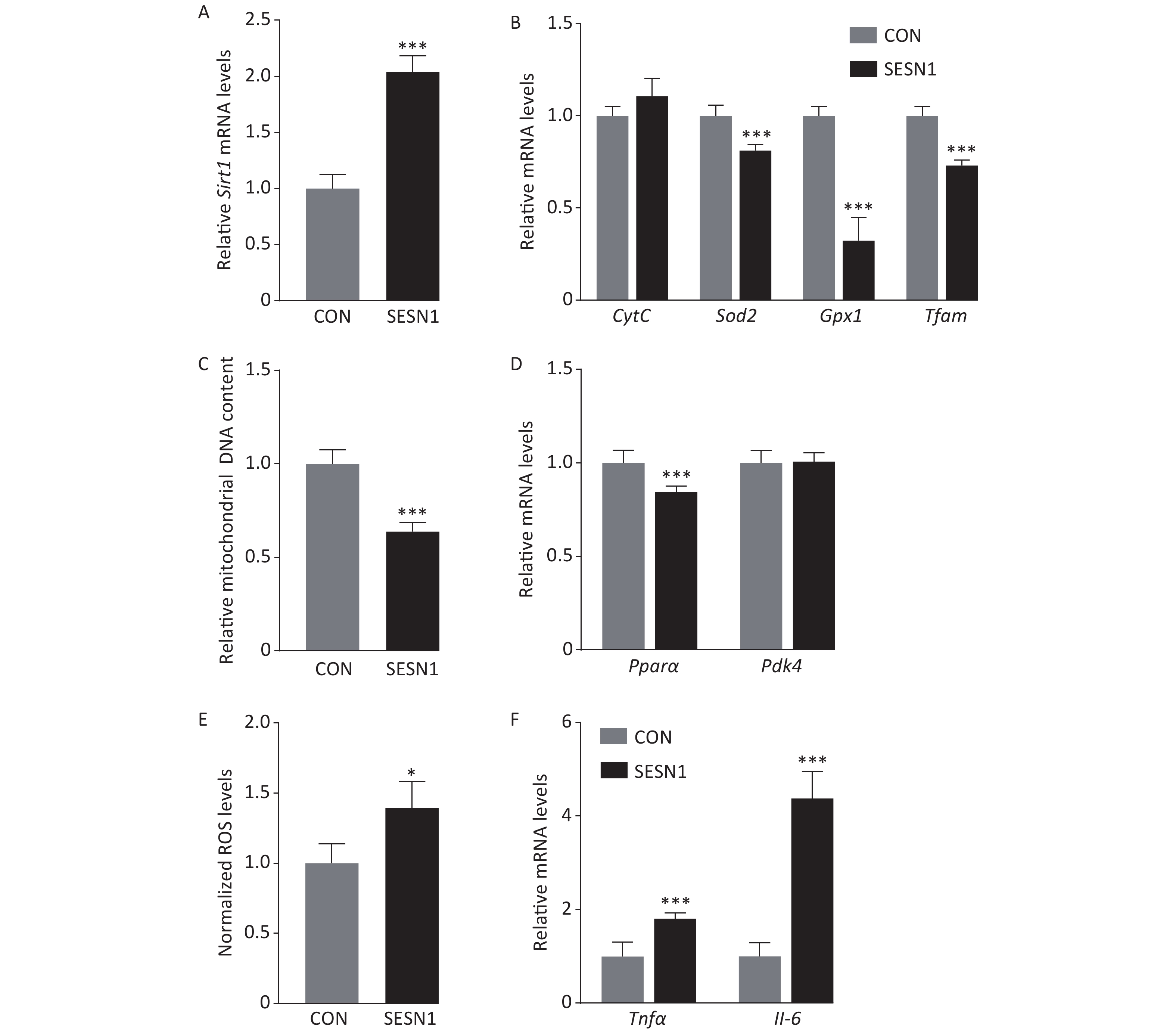
A previous study indicated that the loss of SESNs results in mitochondrial dysfunction in Drosophila[4]. Thus, we investigated the effect of SESN1 on mitochondrial functions in wild type (WT) mouse hepatocytes. Using qRT-PCR, we first confirmed that the mRNA level of Sesn1 was obviously increased when WT primary hepatocytes were infected with Sesn1 adenovirus (Figure 1A). Furthermore, the results showed that SESN1 overexpression increased the expression of mitochondrial biogenesis genes, such as cytochrome c, superoxide dismutase, and glutathione peroxidase, in WT primary hepatocytes (Figure 1B). SESN1 overexpression in WT primary hepatocytes also promoted mtDNA content using genomic DNA extraction and qRT-PCR (Figure 1C). SESN1 overexpression also suppressed ROS production measured using MitoSOX Red (Molecular Probes) (Figure 1D). Mitochondria can also contribute to the influx of fatty acids. A decrease in mitochondrial fatty acid oxidation, which is caused by mitochondrial dysfunction or reduced mitochondrial biogenesis and density, results in increased levels of fatty acyl CoA[7]. In this study, SESN1 overexpression induced the expression of fatty acid oxidation related genes, such as pyruvate dehydrogenase kinase 4
(Pdk4) and peroxisome proliferator-activated receptor (Pparα), in WT primary hepatocytes (Figure 1E). Pro-inflammatory factors TNFα and IL-6, can be transcriptionally activated by nuclear factor kappa-light-chain-enhancer of activated B cells and cause oxidation associated with diseases such as T2DM, NAFLD, and atherosclerosis[8]. As excess ROS generation can promote chronic inflammation, we detected the expression of Tnfα and Il-6. The results showed that SESN1 overexpression inhibited the expression of Il-6 and Tnfα in WT primary hepatocytes (Figure 1F). 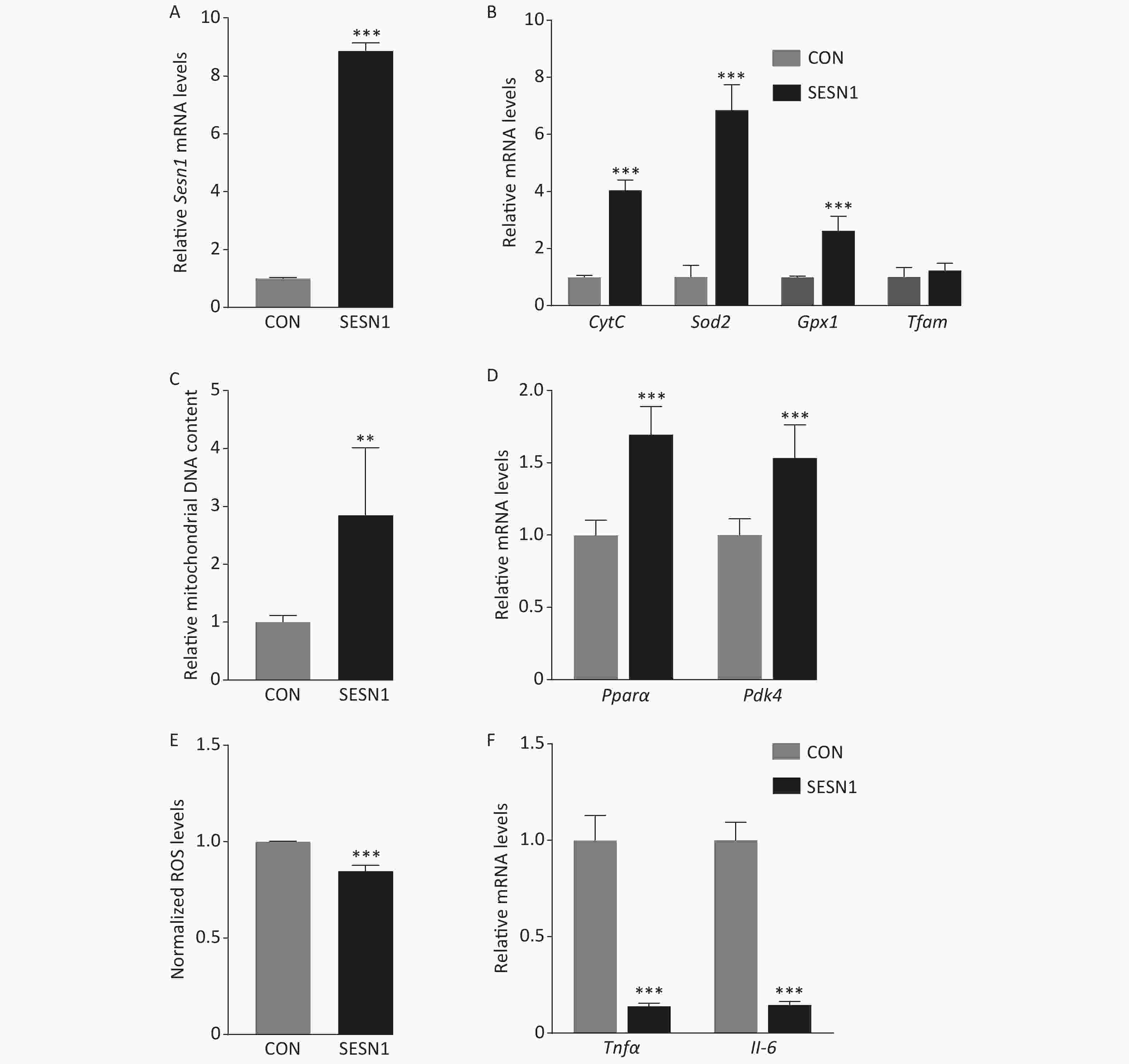
Figure 1. Sestrin1 alleviates hepatic oxidative stress and chronic inflammation. (A) Sesn1 was overexpressed in primary mouse hepatocytes. (B) SESN1 increased the mRNA levels of mitochondrial biogenesis genes in WT primary hepatocytes. (C) SESN1 stimulated mtDNA content in WT primary hepatocytes. (D) SESN1 decreased mitochondrial ROS production in WT primary hepatocytes. (E) SESN1 increased the mRNA levels of fatty acid oxidation genes in WT primary hepatocytes. (F) SESN1 decreased the mRNA levels of Tnfα and Il-6 in WT primary hepatocytes. The statistical analysis was performed using unpaired t test, all data are presented as the mean ± SEM: **P < 0.01, ***P < 0.001.
Modest overexpression of SIRT1 and its activators can reduce oxidative stress and suppress the inflammatory response[9]. Under a high-fat diet (HFD), deletion of SIRT1 in hepatocytes and whole-body insufficiency of SIRT1 induce local and systemic inflammation. In addition, an increase in the expression and activity of SIRT1 can decrease the levels of ROS and inhibit the upregulation of inflammatory markers. We asked whether SIRT1 mediates the effect of SESN1 on improving hepatic oxidative stress and chronic inflammation. Here, we estimated the effect of SESN1 on Sirt1 in WT primary mouse hepatocytes and found that SESN1 overexpression increased the mRNA expression level of Sirt1 (Figure 2A). Furthermore, we tested the role of SESN1 in primary hepatocytes from liver specific Sirt1-deficient (Sirt1-LKO) mice, which expresses a Sirt1 mutant protein in liver. Sirt1 -LKO mice bear a conditional deletion of Sirt1 exon 4, which encodes 51 amino acids of the conserved Sirt1 catalytic domain. In contrast to the role of SESN1 in WT hepatocytes, SESN1 overexpression decreased the hepatic mRNA levels of Sod2, Gpx1, and mitochondrial transcription factor A in the hepatocytes of Sirt1-LKO mice (Figure 2B). In addition, SESN1 overexpression reduced mtDNA content and increased mitochondrial ROS production in the hepatocytes of Sirt1-LKO mice (Figure 2C and 2D). A previous study demonstrated that SIRT1 is involved in mitochondrial fatty acid oxidation[10]. Different to the role in WT hepatocytes, SESN1 overexpression reduced the hepatic mRNA levels of Ppar-α, not Pdk4, in the hepatocytes of Sirt1-LKO mice (Figure 2E). Furthermore, the expression of Il-6 and Tnfα was increased in the hepatocytes of Sirt1-LKO mice (Figure 2F). These results indicated the important role of SIRT1 in mediating the protective effect of SESN1 on oxidative stress and chronic inflammation.
Figure 2. Sestrin1 alleviates hepatic oxidative stress and chronic inflammation mediated by SIRT1. (A) SESN1 induced the mRNA expression of Sirt1 in wild type primary hepatocytes. (B) SESN1 decreased the mRNA levels of mitochondrial biogenesis genes in Sirt1-LKO primary hepatocytes. (C) SESN1 decreased mtDNA content in Sirt1-LKO primary hepatocytes. (D) SESN1 increased mitochondrial ROS production in Sirt1-LKO primary hepatocytes. (E) SESN1 decreased the mRNA levels of fatty acid oxidation genes in Sirt1-LKO primary hepatocytes. (F) SESN1 increased the mRNA levels of Tnfα and Il-6 in Sirt1-LKO primary hepatocytes. The statistical analysis was performed using unpaired t test, all data are presented as the mean ± SEM: *P < 0.05, ***P < 0.001.
It is well known that oxidative stress and chronic inflammation are critical for the development of insulin resistance[3]. Here, we tested whether hepatic SESN1 plays a role in systemic insulin resistance. The insulin tolerance test (ITT) results showed that WT mice fed a normal standard chow diet (9% fat; NCD) improved insulin sensitivity when the livers of these mice were infected with SESN1 adenoviruses (1 × 109 plaque-forming units) administered by tail vein injection (Figure 3A). We also analyzed the effect of SESN1 on WT mice fed a HFD (45% fat). The results showed that SESN1 overexpression obviously protected against HFD-induced insulin resistance in WT mice fed a HFD (Figure 3B). As SIRT1 contributes to the development of insulin resistance and plays a major role in the metabolic adaptation to energy metabolism[6], we also assessed the effects of SESN1 on insulin sensitivity in Sirt1-LKO mice. Regardless of whether an NCD or an HFD was provided, SESN1 overexpression had no effect on insulin sensitivity in Sirt1-LKO mice (Figure 3C and 3D). These results indicated that SIRT1 mediated the effects of SESN1 on improving insulin sensitivity in mice.
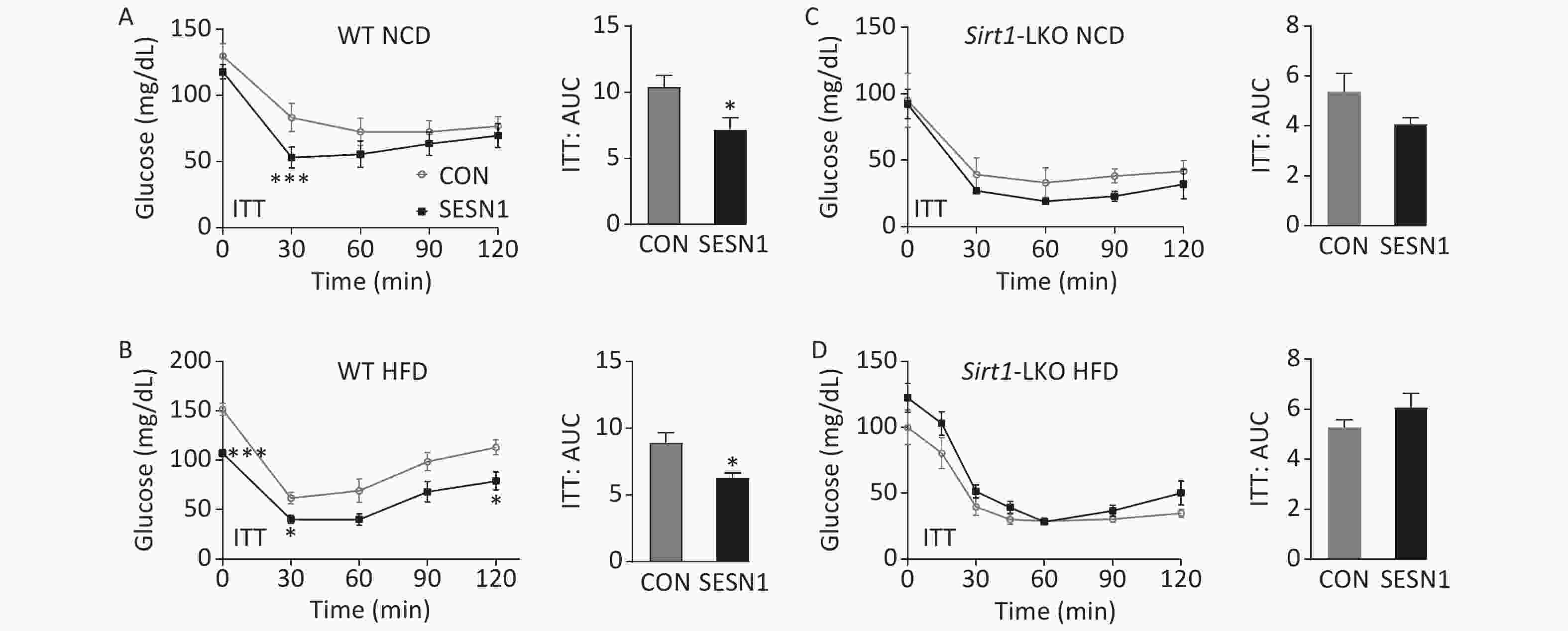
Figure 3. Sestrin1 improves insulin resistance mediated by SIRT1. The insulin tolerance test (ITT) was performed in wild type mice (n = 6 per group) and Sirt1-LKO (n = 5 per group) mice fed a normal standard chow diet (NCD) (A, C) and a high-fat diet (HFD) (B, D) after injection of the indicated adenovirus. The statistical analysis was performed using unpaired t test, all data are presented as the mean ± SEM: *P < 0.05, ***P < 0.001.
In general, SESNs have poorly defined physiological functions, especially in metabolic regulation, and require further investigation. In this study, we demonstrated that SESN1 improved insulin sensitivity by alleviating mitochondrial dysfunction and chronic inflammatory response, which were mediated by SIRT1. In summary, our work demonstrates that the stress-inducible SESN protein family has an important homeostatic function in the control of mammalian energy metabolism by improving hepatic stress and inflammation status.
Conflicts of Interest The authors declare that they have no conflicts of interest.
Acknowledgment We thank Professor LIU De Pei for providing the mice with floxed alleles of Sirt1 and the mice expressing Cre recombinase in liver.
Author contributions GENG Chao, XUE Yuan, and YANG Jia Hui performed the experiments, data analysis and writing. GAO Ming Yue, ZHAO Wei, LI Chun Mei, XIE Xiang Hong, and ZHANG Wei Hong performed the experiments and data analysis. ZHANG Hua Bing performed data analysis. FANG Fu De performed conceptualization and project administration. YAO Hong and LIU Xiao Jun designed the project and performed data analysis and writing.
HTML
Reference

 Quick Links
Quick Links
 DownLoad:
DownLoad: