
-
Hand, foot, and mouth disease (HFMD) is an infectious disease caused by various enterovirus (EV) serotypes and occurs in infants and children across the world. The mild symptoms of HFMD include fever and a maculopapular rash on the hands, feet, mouth, and buttocks. However, in some patients, this infection causes severe neurological syndromes and even death. EVs belong to the Enterovirus genus (family Picornaviridae, order Picornavirales), of which there are 15 species: Enterovirus A (EV-A), EV-B, EV-C, EV-D, EV-E, EV-F, EV-G, EV-H, EV-I, EV-J, EV-K, EV-L, Rhinovirus A, Rhinovirus B, and Rhinovirus C. Since the 2008 outbreaks, enterovirus 71 (EV-A71) and coxsackievirus A16 (CVA16) have been considered to be the two major HFMD pathogens in China [1]. Nevertheless, other prevalent EV serotypes have caused large-scale outbreaks (e.g., CVA4, CVA6, CVA10, and Echovirus 11). Therefore, increasing attention has recently shifted to other EV-A and EV-B pathogens to determine the spectrum of HFMD-causing pathogens besides the major causative ones.
Coxsackievirus B4 (CVB4) is an EV-B type virus. Other than its association with HFMD, CVB4 is associated with a wide range of conditions and diseases such as angina, myocarditis, pancreatitis, viral encephalitis, acute flaccid paralysis, and type 1 diabetes [2]. Six CVB4 strains were isolated in Inner Mongolia in 2010 [3] during the HFMD outbreaks. They were also reported to be isolated from HFMD patients in Shandong (2010–2011) and Guangxi (2010) [4, 5] and from aseptic encephalitis patients in Yunnan, 2009 [6]. Otherwise, there have been few reports of CVB4 outbreaks in China.
CVB4 comprises five genotypes: genotypes A–E [4]. Most European and American strains are genotypes C and D, but all the strains isolated so far from China are genotype E [3, 4]. CVB4 might have undergone intertypic recombination with other EV-B strains [3, 6]. In the present study, next-generation sequencing (NGS) by Illumina MiniSeq was used to identify an imported recombinant CVB4 strain from Tianjin, China. Based on its genetic and recombination characteristics, the strain was assigned to genotype D within a phylogenetic cluster mainly containing strains from Europe and America.
A 0316/TJ/CHN/2019 stool sample was collected from a five-year-old local male patient in Tianjin (2019), whose clinical signs and symptoms included fever (39 ℃), abdominal pain, rash, and herpes in the oral mucosa but no respiratory or nervous syndrome. The patient indicated no overseas travel history within the previous six days. The virus was isolated using rhabdomyosarcoma (RD) cells. Thereafter, two pairs of overlapping EV-B primers were used to amplify the complete genome sequence from CVB4 by RT-PCR, as described by Joffret et al. [7]. The amplification products were subjected to Illumina MiniSeq NGS amplicon sequencing.
A 7,503-nt genome sequence was assembled by de novo assembly (CLC Genomics Workbench 20.0.3, QIAGEN), with an average coverage of 14,386.94. After deleting the 5′- and 3′-terminal low-quality nucleotides, a 7,396-nt sequence was assembled as the ultimate complete 0316/TJ/CHN/2019 genome sequence, which has been deposited in GenBank (Accession No. MZ161144).
All the complete genome or coding sequences from the CVB4 isolates and 31 genomes from EV-B serotype prototypes were collected from NCBI GenBank, and all the low-quality sequences were removed. The nucleotide and aa genome similarity levels between 0316/TJ/CHN/2019 and CVB4 prototype J.V.B. Benschoten (JVB: GenBank Accession No. X05690) were 77.3% and 96.5%, respectively (Supplementary Table S1 available in www.besjournal.com). The nucleotide and aa genome similarity levels between 0316/TJ/CHN/2019 and the strains from the mainland of China were 76.7%–78.9% and 96.0%–97.4%, respectively (with the highest similarity level shared with the Guangxi JX308222 strain isolated from a fatal disease case in 2010). In addition, the nucleotide and aa genome similarity levels between 0316/TJ/CHN/2019 and the strains outside the mainland of China were 75.7%–89.6% and 96.4%–98.5%, respectively (with the highest similarity level shared with the French 2019/FR/3996 MN590273 strain isolated in 2019).
Region CVB4 prototype JVB Strains from the mainland of China Nucleotide (%) Amino acid (%) Nucleotide (%) Amino acid (%) 5'-UTR 82.1 – 81.4−82.7 – VP4 82.3 100.0 73.9–77.5 97.1–98.5 VP2 77.5 97.3 78.0–79.4 96.9–98.1 VP3 79.9 98.3 75.8–77.7 97.9–98.7 VP1 79.5 97.1 76.2–77.7 96.0–97.1 2A 70.4 90.0 68.8–70.2 88.5–89.2 2B 75.2 96.9 70.6–73.8 96.9–98.0 2C 76.1 98.2 77.2–78.2 97.2–99.1 3A 69.9 89.3 74.3–77.1 91.8–97.7 3B 68.0 95.3 70.5–90.1 85.3–95.3 3C 74.9 94.4 71.8–80.4 95.5–98.9 3D 76.7 96.7 76.5–83.5 95.3–98.9 3'-UTR 85.3 – 83.7–91.7 – CDS 76.7 96.5 76.0–78.5 96.0–97.4 Table S1. Nucleotide sequences similarities and their deduced amino-acid sequences between 0316/TJ/CHN/2019 and other CVB4 strains
Compared with the JVB CVB4 prototype, 19 aa substitutions were identified in the capsid protein (Supplementary Table S2 available in www.besjournal.com), and 57 such substitutions in nonstructural proteins (23 in P2 and 34 in P3) were detected in 0316/TJ/CHN/2019. Of the 19 aa substitutions found in the capsid protein from 0316/TJ/CHN/2019, only 9 were also found in other strains isolated within the mainland of China, whereas the remaining 10 substitutions were unique to the Chinese strains (Supplementary Table S2).
Proteins Size (aa) Substitutions VP4 69 None VP2 261 A137V, A142Ta, S154Qa, A160Ta, K163E, Y187F, S249N VP3 238 I55L, M63Ka, F213La, T234Sa VP1 284 K5D, Y76Fa, S263N, V271I, A273T, S277Na, I279Va, Y284Ca Note. aSubstitutions first detected in strains from the mainland of China. Table S2. Amino acid substitutions in the 0316/TJ/CHN/2019 capsid protein versus the CVB4 prototype JVB (X05690)
The point aa site mutation in the capsid protein from CVB4 has been reported in a few studies. One study suggested that the 16–24 aa region in the VP4 capsid protein is the target of infection-enhancing antibodies in CVB4 [8]. Ser-16 in VP4 and Met-129 in VP1 have been identified as virulence-related determinants [9, 10]. The 0316/TJ/CHN/2019 strain possesses both Ser-16 in VP4 and Met-129 in VP1 of the capsid protein, indicating that it is a virulent strain.
In EVs, VP1 is considered useful for standard molecular genotyping because it is serotype-specific. However, because intertypic recombination frequently occurs among EVs serotypes, a new genotyping method based on whole-genome sequences has been developed to enable the accurate determination of genetic relationships in these viruses. Phylogenetic analyses based on the 852-nt VP1 and full-length genome sequences were performed, and neighbor-joining trees were constructed using the Kimura 2-parameter model in MEGA 5.05. The results were tested by the bootstrap method with a bootstrap value of 1,000.
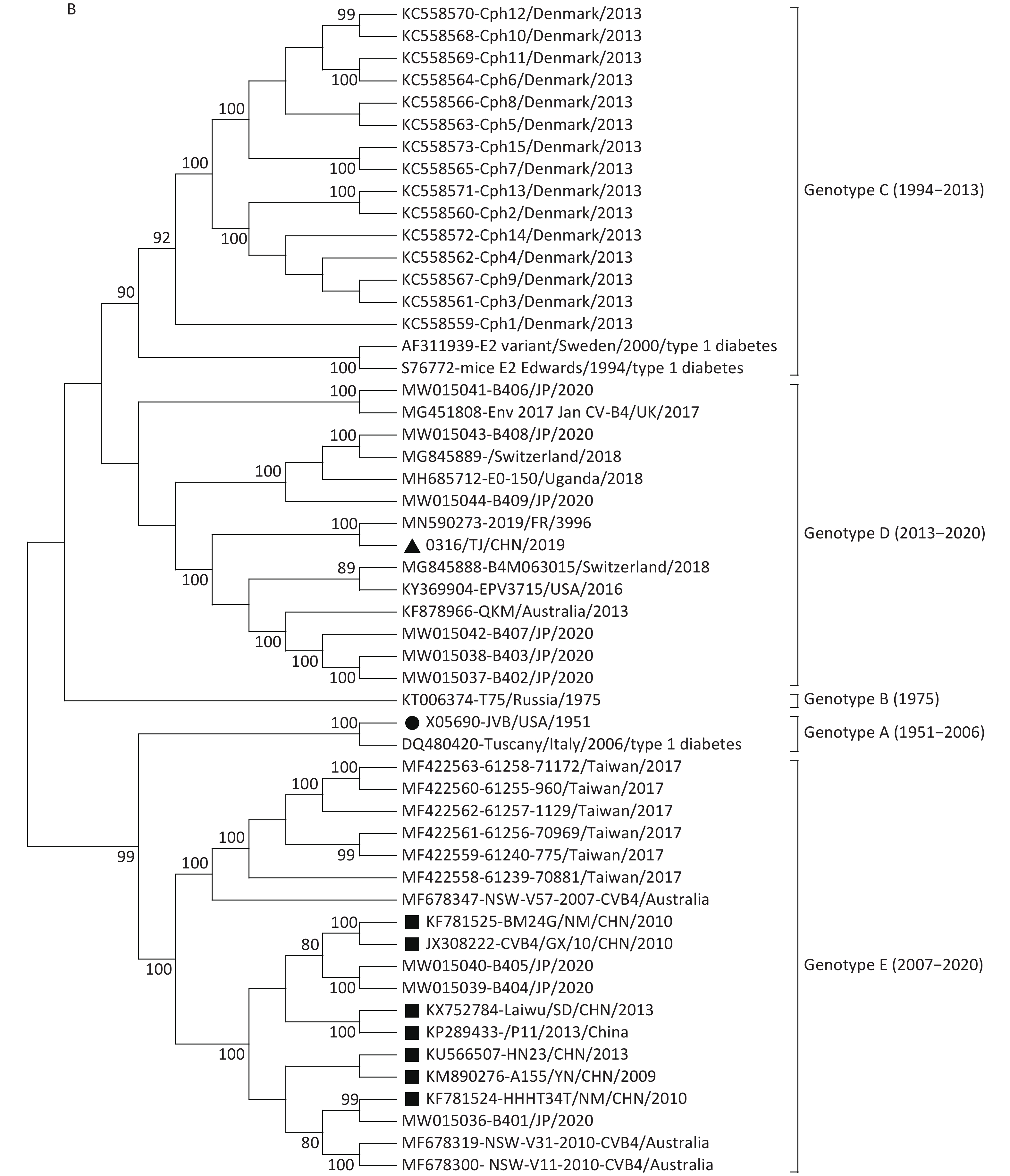
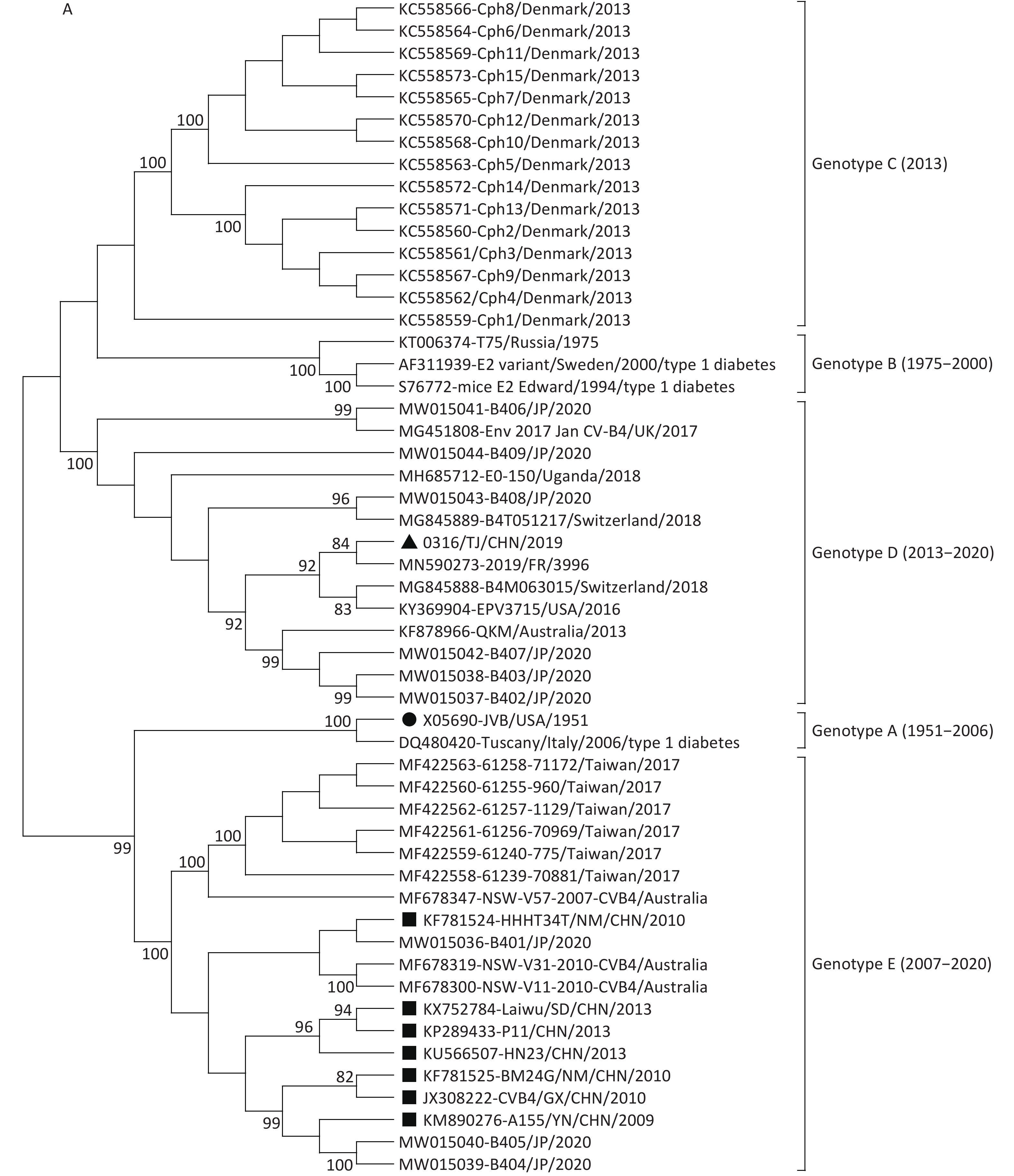
In this study, the VP1 and whole-genome sequence-based phylogenetic analyses supported the division of the CVB4 strains into five genotypes, although with a minimal difference in genotypes B and C typing (Figure 1). The genotyping difference between VP1 and the whole-genome sequences showed that substantial recombination had occurred in the CVB4 strains, highlighting the need for phylogenetic analyses to be based on the complete genome sequences.

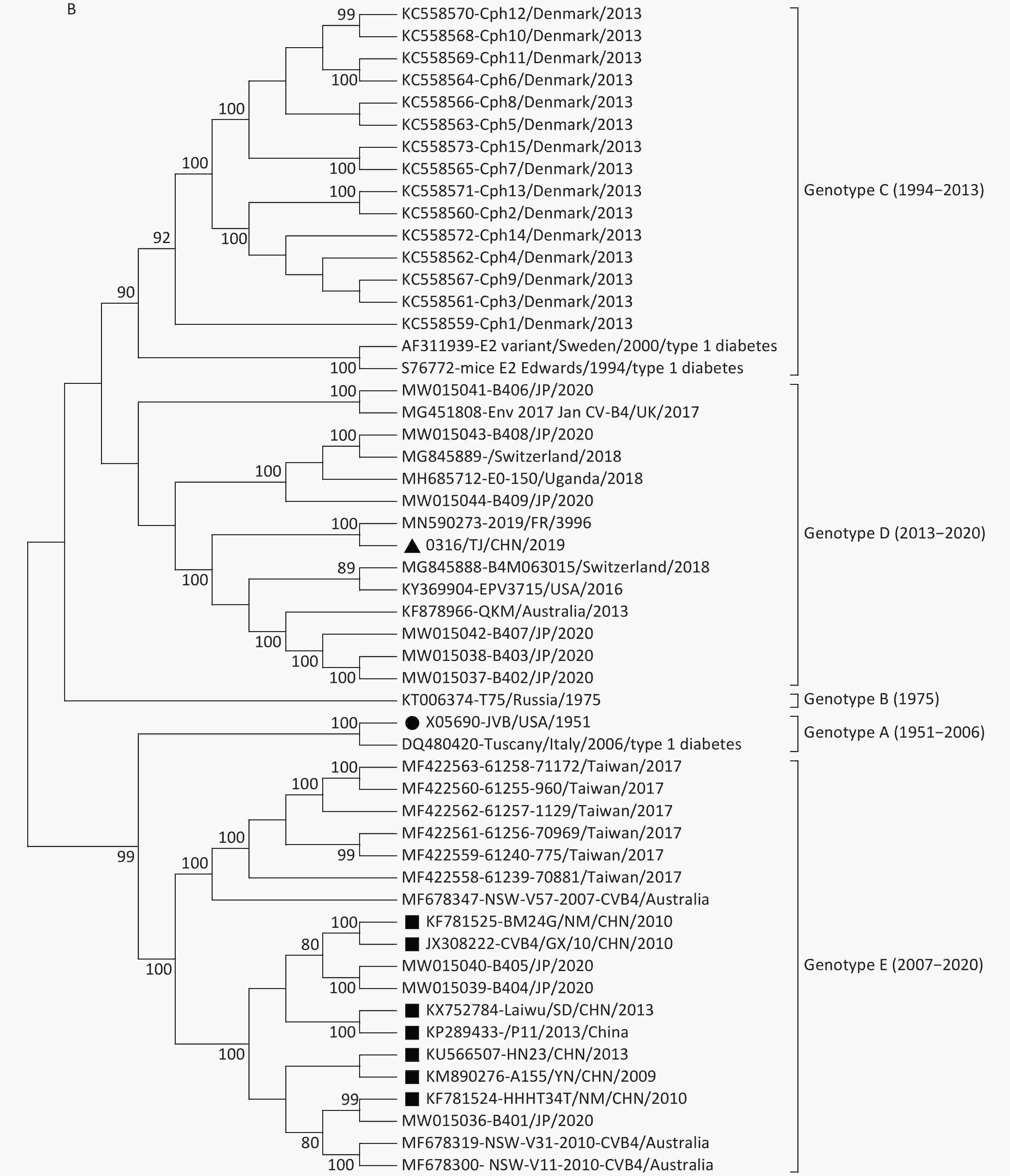
Figure 1. Neighbor-joining trees for CVB4 strains based on complete VP1 sequences (A) and full-length genome sequences (B). The strain indicated by the circle is the prototype JVB. The strain indicated by the triangle is 0316/TJ/CHN/2019. The strains indicated by squares are the isolates from the mainland of China.
In the VP1 tree, all the CVB4 strains clustered into five genotypes (genotypes A–E). The prototype JVB and a type 1 diabetes-causing strain from Italy clustered as genotype A with a similarity value of 99.8%. A Russian strain and two type 1 diabetes-causing strains clustered in genotype B (similarity values, 88.5%–98.6%). Genotype C contained 15 Denmark strains (with similarity values of 86.1%–100.0%). The 0316/TJ/CHN/2019 strain clustered into genotype D (with similarity values of 83.9%–98.1%) with strains isolated from the USA, Uganda, Australia, Japan, and European countries. Other strains from the mainland of China clustered into genotype E (similarity values, 87.6%–100.0%), with strains from Taiwan, Australia, and Japan. The mean similarity values among the different genotypes ranged from 85.6% (genotypes B and C) to 76.9% (genotypes D and E).
Both VP1 and the whole-genome sequence trees revealed that, unlike the genotype E strains from the mainland of China, the Tianjin 0316/TJ/CHN/2019 strain clustered into genotype D with the strains mainly isolated from the USA, Japan, Australia, and European countries (Figure 1). This conclusion was further supported by homology analysis, which showed that, based on the complete genome sequences, 0316/TJ/CHN/2019 shares the highest similarity (89.6%) with a French strain and, based on the complete VP1 sequence, shares the highest similarity (92.7%) with an American strain. Because the local Chinese patient did not have any overseas contact, it seems that the CVB4 genotype D strain has been imported into Tianjin.
Previous studies [3, 4] indicate that genotype E strains are the only strains derived from those circulating in the mainland of China and Australia since 2007. However, in this study, we added strains from Taiwan (2017) and Japan (2020) to the genotype E cluster (Figure 1). The similarities based on the VP1 sequences among genotype E strains ranged from 87.6% to 100.0%, conforming to the criterion normally used for genotyping EVs (i.e., no greater than 15% nucleotide sequence differences in the VP1 sequences). Our study has illustrated that the genotype E strains did not only occur in the mainland of China and Australia but also occurred in a vast region around the western Pacific Ocean, including Taiwan, China and Japan.
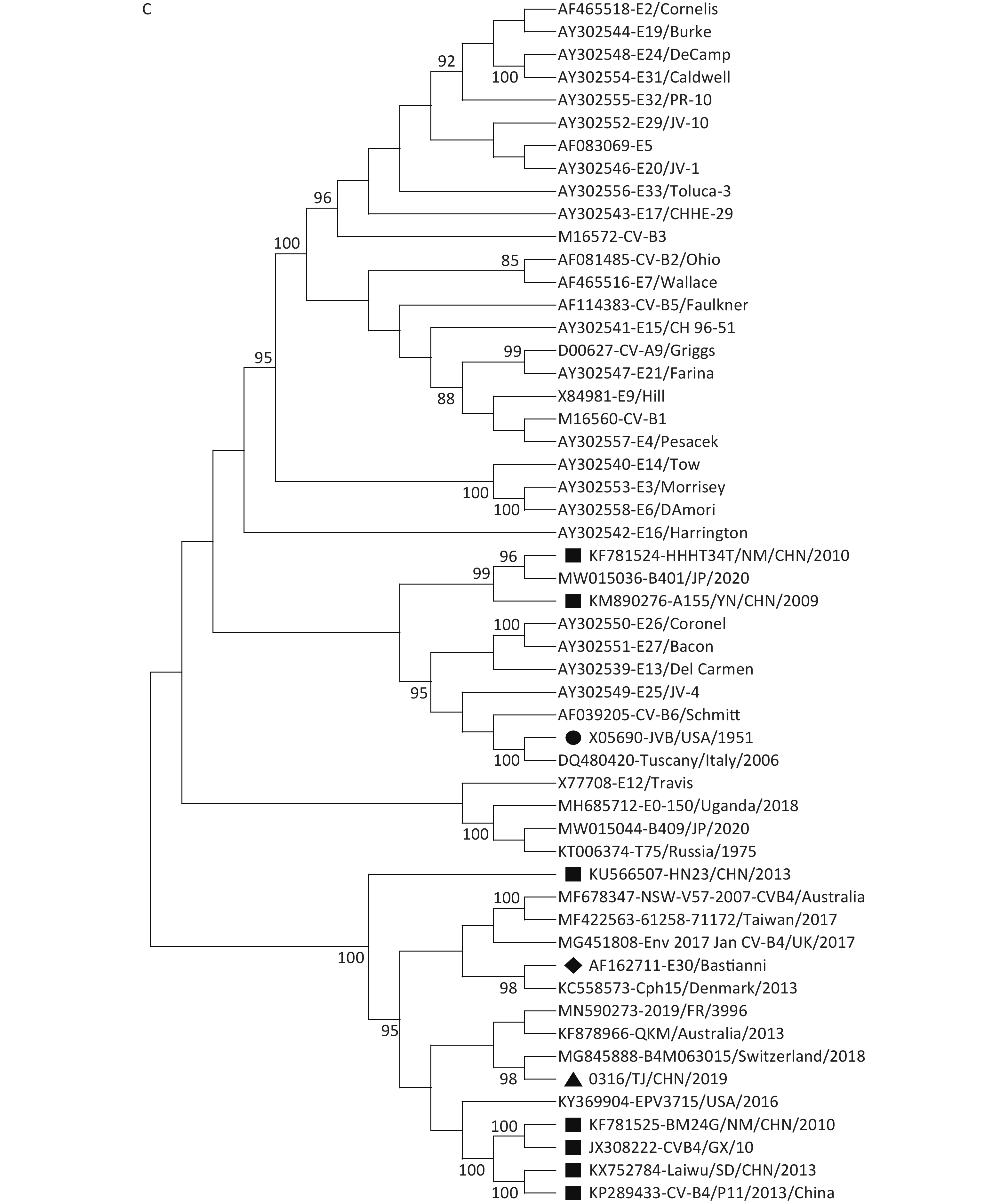
Our phylogenetic analyses based on the P1, P2, and P3 genome regions among the CVB4 strains and other EV-B strains suggested that although all the CVB4 strains shared the same cluster with the CVB4 JVB prototype in the P1 region (Figure 2A), different genetic relationships exist among the CVB4 strains in the P2 and P3 regions. In the P2 genome region, all the CVB4 genotypes B–E strains, including 0316/TJ/CHN/2019, clustered together with Echovirus 27 prototype Bacon (AY302551) and Echovirus 30 prototype Bastianni (AF162711, Figure 2B). In the P3 genome region, JVB and two Chinese CVB4 strains both clustered with Echoviruses 13, 25, 26, and 27 and CVB6. CVB4 genotype B (a Russian T75 KT006374 isolate) and two genotype D strains both clustered with Echovirus 12. Furthermore, other CV-B4 strains, including five Chinese strains and 0316/TJ/CHN/2019, shared the closest relationship with Echovirus 30 (Figure 2C). The similarity values for 0316/TJ/CHN/2019 and Echoviruses 27 and 30 in the P2 region were 76.1% and 76.9%, respectively. These values are somewhat lower than the 80.2% for the P3 region and Echovirus 30. However, 0316/TJ/CHN/2019 shared only 74.5% and 75.2% similarity with CVB4 prototype JVB in the P2 and P3 region, respectively.
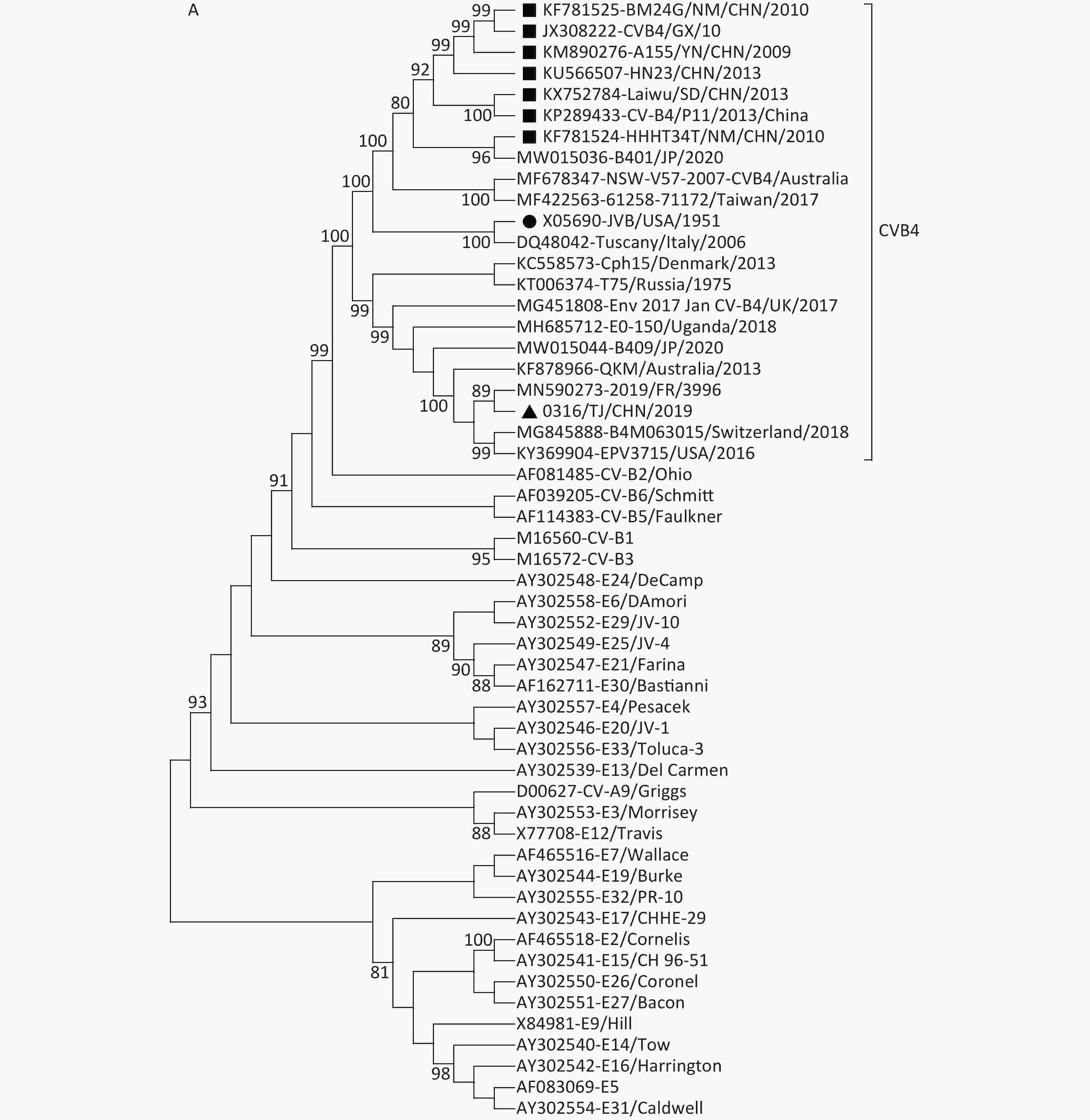

Figure 2. Neighbor-joining trees based on the P1 (A), P2 (B), and P3 (C) genome regions among the CVB4 strains and other EV-B strains. The strain indicated by a circle in each panel is prototype JVB. The strain indicated by a triangle in each panel is 0316/TJ/CHN/2019. The strains indicated by squares are isolates from the mainland of China. The strains indicated by rhomboids are Echovirus 27 and Echovirus 30.
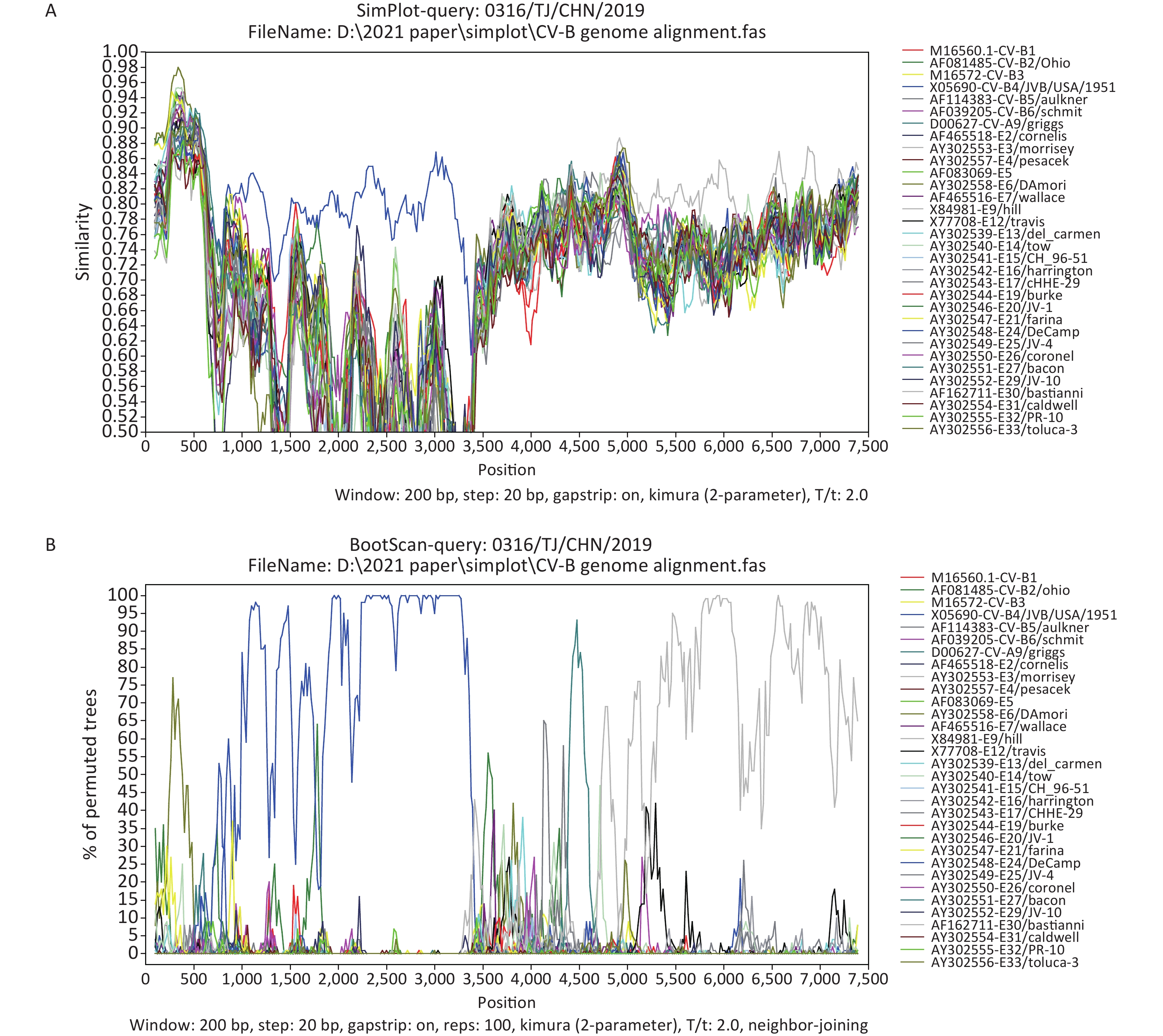
The Simplot and bootscanning analyses were constructed using Simplot 3.5.1 (Stuart Ray, Johns Hopkins University, Baltimore, MD, USA), with a sliding window of 200-bp and steps of 20-bp. The Simplot (Figure 3A) and bootscanning (Figure 3B) analyses revealed that 0316/TJ/CHN/2019 was most similar to JVB in the P1 region (average bootscanning value of 80.5%), whereas 0316/TJ/CHN/2019 and Echovirus 30 prototype Bastianni were most similar in the P3 region, with an average bootscanning value of 77.1% (Figure 3B). Both phylogenetic and bootscanning analyses suggested that recombination events have occurred between 0316/TJ/CHN/2019 and the other EV-B strains.
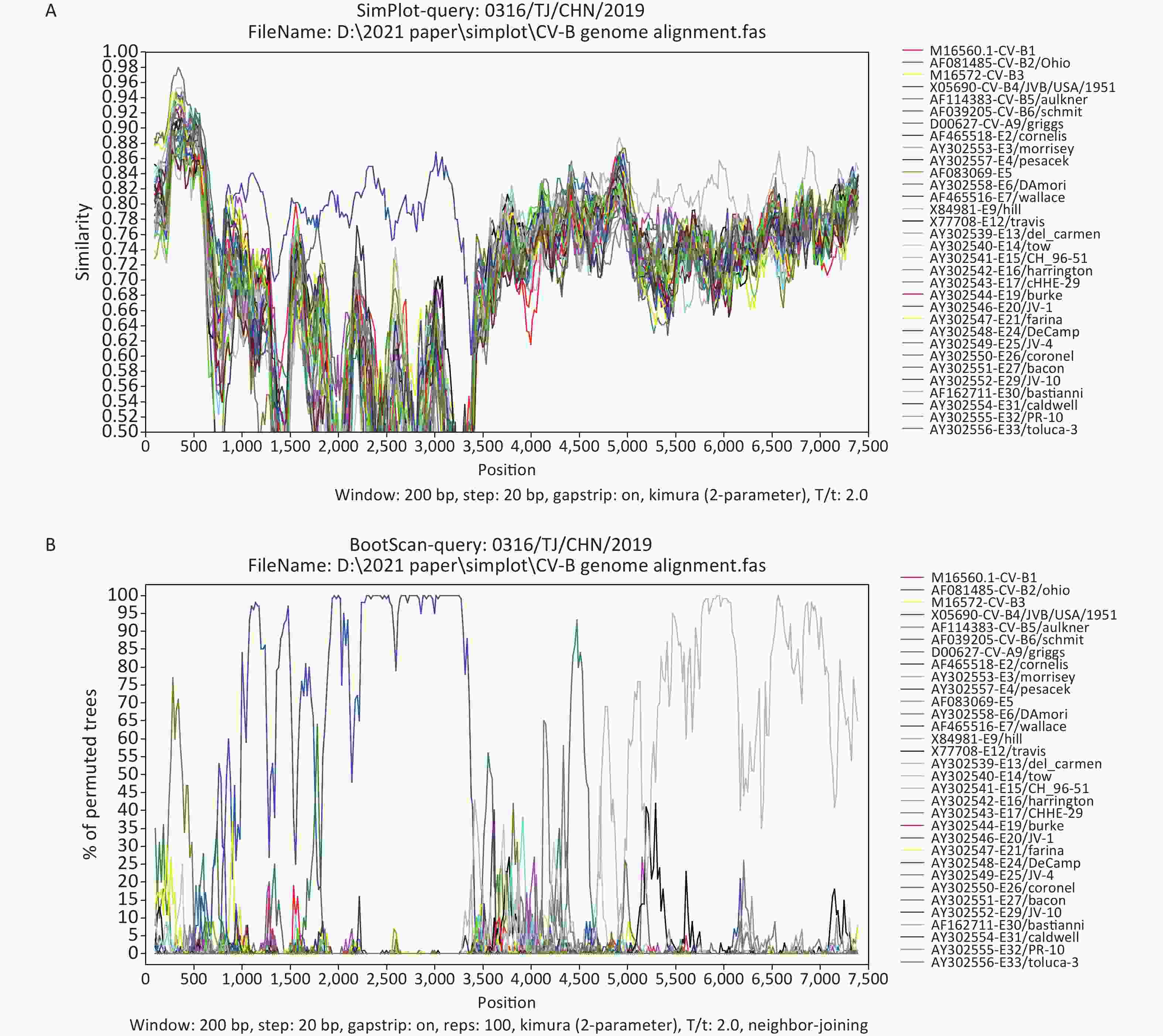
Figure 3. Similarity plot (A) and bootscanning (B) analyses based on the full-length genome sequences from 0316/TJ/CHN/2019 and EV-B strains.
Frequent recombination within the 5′-UTR, P2, and P3 regions is a common mechanism seen in the evolution of EV-B species [3, 6, 11]. Whereas the 5′-UTR sequences in EV-Bs cluster with EV-A members, the P2 and P3 genome regions have only recombined within the EV-B species, resulting in complex mosaic sequence patterning of the nonstructural protein’s coding sequences [11]. The P1 region-based phylogenetic analysis supports the monophyletic evolution of CVB4 (Figure 2A). However, P2 and P3 region-based phylogenetic trees and the bootscanning analysis both revealed that 0316/TJ/CHN/2019 is also a recombinant strain (Figure 3). Therefore, considering the high evolution rate and frequent recombination events within EVs, surveillance measures and monitoring, especially including the NGS method, should be enhanced to prevent and control the diseases caused by CVB4.
In conclusion, an imported CVB4 strain isolated in Tianjin, China, in 2019 was found to cluster into genotype D. Recombination analysis suggests that the Tianjin strain has recombined with other EV-B species, including Echovirus 30, in the P2 and P3 regions. It has also been shown that the CVB4 whole-genome sequence has advantages over the traditional methods, especially for traceability, and NGS is an effective method to trace the source of CVB4, as well as other EVs.
Acknowledgment We thank Sandra Cheesman, Ph.D., from Liwen Bianji (Edanz) (www.liwenbianji.cn) for editing the language of a draft of this manuscript.
Authors’ Contributions Designed the experiments: LI Xiao Yan. Performed the experiments: TAN Zhao Lin, LYU Li Kun. Analyzed the data: Tan Zhao Lin, XIE Tong. Collected the specimens and epidemiological information: LI Li. Wrote the paper: TAN Zhao Lin.
HTML
 21318Supplementary Materials.pdf
21318Supplementary Materials.pdf
|

|

 Quick Links
Quick Links
 DownLoad:
DownLoad: