-
Enteroviruses (EVs) belong to the family Picornaviridae and are divided into 15 species: enterovirus A–L and rhinovirus A–C (
http://www.picornaviridae.com ). EV-C consists of 23 serotypes, including poliovirus 1-3, CVA1, CVA11, CVA13, CVA17, CVA19, CVA20, CVA21, CVA22, CVA24, EV-C95, EV-C96, EV-C99, EV-C102, EV-C104, EV-C105, EV-C109, EV-C113, EV-C116, EV-C117, and EV-C118, which can cause diseases as herpangina, hand, foot and mouth disease, acute hemorrhagic conjunctivitis (AHC), aseptic meningitis, and acute flaccid paralysis (AFP)[1-3]. CVA1 is only rarely detected during EVs monitoring, so the data of this virus is very limited. Up to now, only six strains of CVA1 with whole genome or polyprotein gene sequence have been published. The prototype strain Tompkins was identified from the fecal sample of a patient with paralytic disease in the Coxsackie, USA in 1947 which was pathogenic for lactating mice and hamsters but not for rhesus monkeys. It caused significant damage to the skeletal muscle of experimental animals, but not to the central nervous system[4]; HT-THLH02F/XJ/CHN/2011 strain and KS-ZPH01F/XJ/CHN/2011 strain were isolated from two healthy children’s fecal samples in the Xinjiang Uygur autonomous region of China, in 2011. they could elicit cytopathic effects (CPE) in a human rhabdomyosarcoma (RD) cell line[5]; BBD34 strain was identified from a patient with gastrointestinal disease in Bangladesh in 2009[6]; V18A strain was isolated from fecal sample in Venezuela in 2015; ETH_P8/A1_2016 strain was isolated from the excrement of children who participated in a clean water intervention experiment in Ethiopian, in 2016[7]. In order to obtain the in-depth understanding of CVA1 genetic diversity and epidemics, more information about CVA1 strains from throughout the world is needed. Here, we report the entire genome sequence of the CVA1 strain 1222/YN/CHN/2010, isolated from a patient with severe HFMD in Yunnan, China, 2010. The findings of the present study will enrich the knowledge of CVA1.During the HFMD surveillance in Yunnan Province, China in 2010, a CVA1 case was only detected by reverse transcription nested/semi-nested PCR (RT-n/snPCR) from the 1,220 stool samples using several pairs of primers (Supplementary Table S1 available in www.besjournal.com). The patient was a 3.5-year-old child presenting with a fever of 39 °C, oral ulcer, hand and foot rash, limb tremor, headache, and encephalitis. The use of the sample did not directly affect the patient and consent was not required. This study has passed the ethical review, and all the experimental work was supervised by the Institutional Ethics Boards of the Institute of Medical Biology, Chinese Academy of Medical Sciences and Peking Union Medical College. RD, human embryonic lung diploid fibroblasts (KMB17) and human lung cancer (A549) cell lines were used for viral isolation. The three cell lines were conserved by Institute of Medical Biology, Chinese Academy of Medical Sciences and Peking Union Medical College.
Primer Sequence (5’→3’) Position Species 1[1] 222
224
AN89
AN88CICCIGGIGGIAYRWACAT
GCIATGYTIGGIACICAYRT
CCAGCACTGACAGCAGYNGARAYNGG TACTGGACCACCTGGNGGNAYRWACAT2969−2951
1977−1996
2602−2627
2977−2951all 2[2] A-OS
A-OAS
A-IS
A-IASCCNTGGATHAGYAACCANCAYT
GGRTANCCRTCRTARAACCAYTG
TNASNATYTGGTAYCARACANAYT
GANGGRTTNGTNGKNGTYTGCCA2268−2291
3109−3086
2332−2356
3016−2993EV-A 3[2] B-OS
B-OAS
B-IS
B-IASGGYTAYATNCANTGYTGGTAYCARAC GGTGCTCACTAGGAGGTCYCTRTTRTARTCYTCCCA CTTGTGCTTTGTGTCGGCRTGYAAYGAYTTYTCWG TCYTCCCACACRCAVTTYTGCCARTC 2324−2351
3505−3469
2392−2428
3477−3451EV-B 4[3] EntAF
EntARo
EntARiTNCARGCWGCNGARACNGG
ANGGRTTNGTNGMWGTYTGCCA
GGNGGNACRWACATRTAYTG2571−2589
2957−2936
2898−2879EV-A 5[3] EntBF
EntBRo
EntBRiGCNGYNGARACNGGNCACAC
CTNGGRTTNGTNGANGWYTGCC
CCNCCNGGBGGNAYRTACAT2610−2630
3006−2986
2970−2951EV-B Note. 1. Nix WA, Oberste MS, Pallansch MA. Sensitive, seminested PCR amplification of VP1 sequences for direct identification of all enterovirus serotypes from original clinical specimens. J Clin Microbiol, 2006; 44, 2698−704. 2. Leitch EC, Harvala H, Robertson I, et al. Direct identification of human enterovirus serotypes in cerebrospinal fluid by amplification and sequencing of the VP1 region. J Clin Virol, 2009; 44, 119−24. 3. Iturriza-Gomara M, Megson B, Gray J. Molecular detection and characterization of human enteroviruses directly from clinical samples using RT-PCR and DNA sequencing. J Med Virol, 2006; 78, 243−53. Table S1. Primers for amplification and sequencing of VP1 of enterovirus
We designed specific primers according to CVA1 for amplification and sequencing of the isolate. The viral RNA was extracted from the fecal suspension using AxyPrep™ Body Fluid Viral DNA & RNA Purification Miniprep Kits (Axygen, USA), then the cDNA was directly amplified by PrimeScript™ II High Fidelity RT-PCR Kit (TaKaRa, Dalian, China). The VP1 gene was amplified and sequenced for serotype identification by specific primer pairs CA1VP1f(TTACTTAAAGACTCCCCCCA)/CA1VP1r(AATTGCAGATCTTGTAGCCCG). The PCR amplification was conducted under the following conditions: 55 °C for 30 min, 94 °C for 2 min, 35 cycles of 94 °C for 30 s, 52 °C for 30 s, and 72 °C for 1 min followed by 72 °C for 5 min. The VP1 of the isolate was sequenced by Tsingke Sequencing Company (Kunming, China) using the same primers as VP1 amplification through Sanger’s sequencing.
The entire genome 1222/YN/CHN/2010 was amplified and sequenced as described previously [8]. The primers used for whole genome amplification and sequencing were listed in Supplementary Table S2 available in www.besjournal.com. All the sequenced fragments were read and edited using the SeqMan in DNAStar v7.1. The whole genome was assembled manually. Primer Sequence (5’→3’) Position Orientation CA1VP1f TTACTTAAAGACTCCCCCCA 2400−2420 Forward CA1VP1r AATTGCAGATCTTGTAGCCCG 3391−3371 Reverse CA11F TTAAAACAAGCCCTTGGGTG 1−20 Forward CA12R GCACCTCTTTTGAGTGGGTT 2552−2533 Reverse CA13F AAACCAAAACACATCCGTGC 3212−3231 Forward CA18R CCCTCCGAATTAAAAGAAAA 7396−7377 Reverse CA11R AACCCCCAATTGTTATGTTTG 1596−1576 Reverse CA11r ACCAGCAGCCACATTCTGATT 772−752 Reverse CA14F TGCAATATGGTTGGCATTCC 4037−4056 Forward CA15F ATTGCCACAAACCAGCAAAC 4884−4903 Forward CA17R TGACCAAAAGCCTGTCTCAT 6516−6497 Reverse CA18f TGAATCAATCAGGTGGACCAA 7132−7152 Forward Table S2. Primers for amplification and sequencing of CVA1 complete genome Sequence
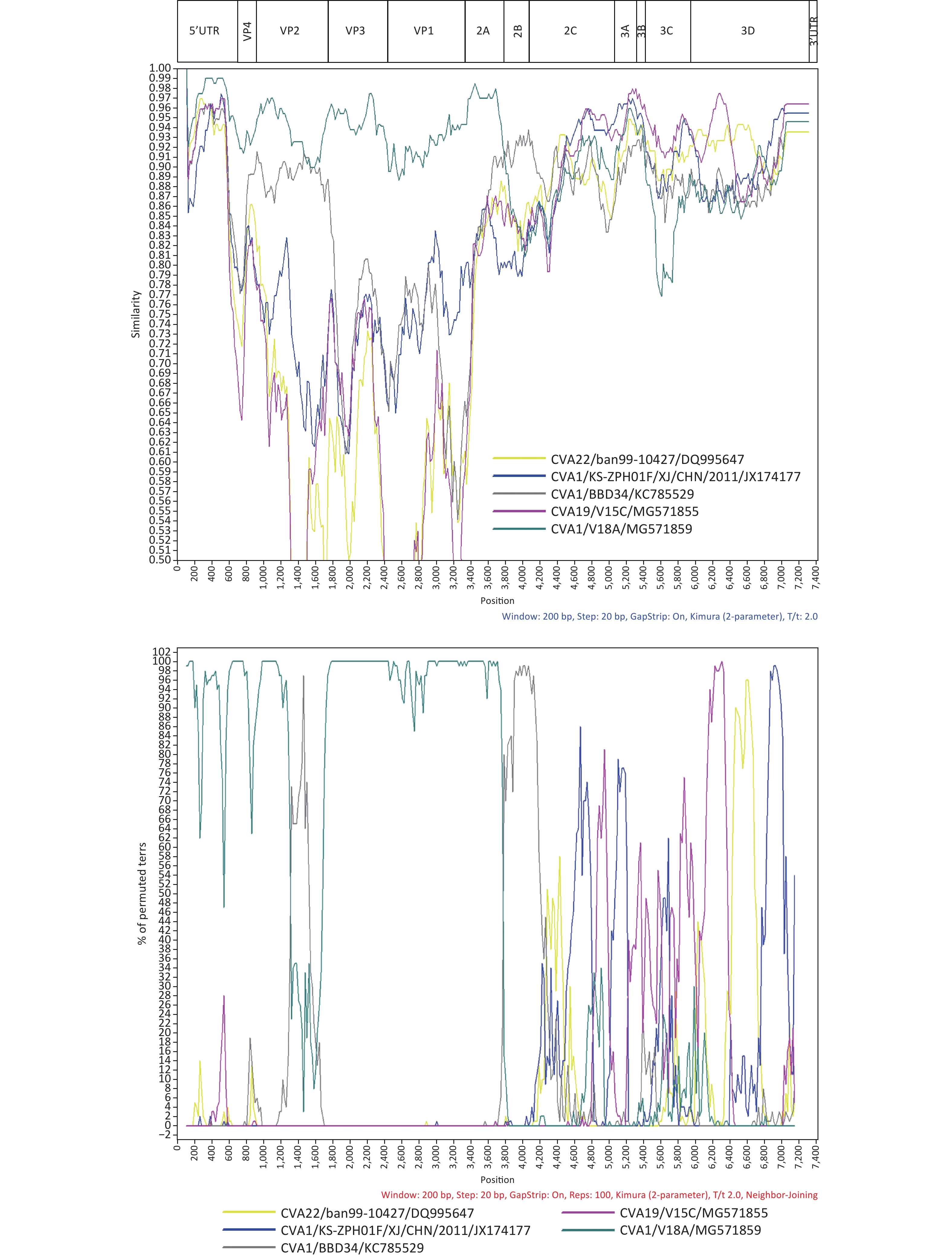
And then we performed bioinformatics analyses of 1222/YN/CHN/2010. Multiple sequence alignment of CVA1 was carried out using BioEdit v7.0.5. The nucleotide and amino acid sequences of all genomic regions of Yunnan isolate were compared with other CVA1 strains in the corresponding region using Geneious Prime 2020.1.2. Phylogenetic analyses of CVA1 based on partial VP1 fragments and the entire genome sequences were performed using the Neighbor-Joining method with the Kimura two parameter model of nucleotide substitution in MEGA 7.0, with a bootstrap value of 1,000. BLAST online was used to identify sequences with the highest similarity to the full-length sequence and each segment of the 1222/YN/CHN/2010 genome. Simplot (version 3.5.1) was used to perform similarity plots and a bootscanning analysis with a 200-nt window moving in 20-nt steps and a Jukes-Cantor correction.
Acquired experiment results showed that the CVA1 strain was unable to be isolated by using KMB17, RD, and A549 cell lines, but its whole genome sequence was successfully amplified from fecal samples by reverse-transcription PCR (RT-PCR). Complete VP1 sequence identified through BLAST (www.ncbi.nlm.nih.gov/blast) indicated that the reverse transcript product was CVA1. The complete genome of the CVA1 strain 1222/YN/CHN/2010 has been submitted to GenBank (GenBank accession numbers: MK250423). Previous reports suggested that CVA1 may cause enteric and neurological disease, but seldom detected from HFMD samples [5, 6, 9]. The Yunnan isolate 1222/YN/CHN/2010 was detected from stool sample of a patient with acute infection phase of typical severe HFMD, and by three passages, CPE was not be observed in these 3 cell lines, respectively. Meanwhile, only one partial VP1 sequence of CVA1 was amplified from this specimen with different primers (Supplementary Table S1). Thus, these results indicated that CVA1 may be associated with HFMD. However, whether there is co-infection with other EV serotypes remains to be further investigated.
Complete genome sequence analysis showed that strain 1222/YN/CHN/2010 comprised of 7,396 nucleotides, which consisted of a 5’UTR of 709 nucleotides and a 3’UTR of 72 nucleotides and an open reading frame (ORF) which encoded a polyprotein of 2,204 amino acids. It shared the highest nucleotide similarity to the Venezuela strain V18A/JX174177 (91.8%), isolated from fecal sample in Venezuela in 2015. 1222/YN/CHN/2010 strain exhibited low nucleotide similarity (< 91.8%) but high amino acid similarity (> 96.0%) to other CVA1 strains. However, in the 5’UTR and 3’UTR, nucleotide sequences of CVA1 were highly conserved, and the nucleotide similarity among them ranged from 91.7% to 100.0%. The VP1 region of the 1222/YN/CHN/2010 strain had 78.0% nucleotide identity (90.2% amino acid identity) to Tompkins strain and 77.5%–92.2% nucleotide identity to other CVA1 strains (86.1%–97.0% amino acid identity) (Table 1).
Genomic
regionTompkins Other CVA1 strains Nucleotide identity (%) Amino acid identity (%) Nucleotide identity (%) Amino acid identity (%) 5’UTR 91.7 / 92.4−95.6 / VP4 89.9 100.0 86.0−96.6 100.0 VP2 78.4 94.1 77.8−94.0 93.7−99.6 VP3 78.0 93.2 76.7−95.1 99.6 VP1 78.0 90.2 77.5−92.2 86.1−97.0 2A 85.9 95.3 85.0−97.3 96.6−100.0 2B 83.2 94.8 82.3−92.1 96.9−97.9 2C 85.7 98.5 89.6−91.5 98.9−99.1 3A 83.7 97.7 90.9−96.2 97.7−100.0 3B 84.6 100.0 95.5−97.0 95.2−100.0 3C 82.8 95.1 88.5−93.6 95.6−96.2 3D 89.1 98.9 90.0−91.2 98.3−98.9 3’UTR 98.6 / 92.8−100.0 / Genome 84.1 95.7 86.1−91.8 96.0−98.3 Table 1. Nucleotide and amino acid identity between 1222/YN/CHN/2010 strain, Tompkins and other CVA1 strains in sequenced genomic regions
Phylogenetic tree based on the partial VP1 sequence (nucleotide positions 2590–2889) of 1222/YN/CHN/2010 and 25 CVA1 strains available at GenBank database showed that the 26 CVA1 strains were categorized into two clusters, and the 1222/YN/CHN/2010 strain was not in the same branch with other Chinese strains (Figure 1A). This is consistent with the phylogenetic analysis based on the entire genome of other EV-C reference strains and all CVA1 strains available (Figure 1B). Now, genotyping of CVA1 has not yet been standardized. In order to further study the phylogeny of CVA1 and establish a reasonable and unified genotyping approach, monitoring of CVA1 should be strengthened to obtain more full-length VP1 and genome sequences of CVA1 from throughout the world.


Figure 1. The phylogenetic trees based on partial VP1 sequences of 26 CVA1 strains (A) and the entire genome sequences of all CVA1 strains available and other EV-C reference strains (B), generated by the neighbor-joining algorithm implemented in MEGA (version 7.0) using the Kimura two-parameter substitution model and 1,000 bootstrap pseudo-replicates. Only strong bootstrap values (> 70%) are shown. ▲represents strains isolated in this investigation. ●represents CVA1 strains isolated in China. ■represents the prototype strain of CVA1.
We conducted a preliminary screening with BLAST online for strains which possess a high similarity with the 1222/YN/CHN/2010 strain in different coding regions, and found that the 1222/YN/CHN/2010 strain had the highest identities with other EV-C serotypes as the CVA19 strain V15C (MG571855) (94.0%–97.0%) and CVA22 strain ban99-10427 (DQ995647) (93.0%) in the 3B–3C, and 3D respectively (Supplementary Table S3 available in www.besjournal.com). Similarity plot and Bootscaning analysis further revealed that the intertypic recombination events occurred between CVA1 and other EV-C serotypes as CVA19 and CVA22 in the 2C–3D and 3D regions, respectively (Figure 2). At present, only one study reported that CVA1 could grow and produce CPE in RD cells, and most studies, including our study have shown that CVA1 cannot be isolated from cell lines [5]. The two Chinese CVA1 strains (HT-THLH02F/XJ/CHN/2011 and KS-ZPH01F/XJ/CHN/2011), which could produce CPE in RD cells, recombined with CVA22 strains 10427/BAN/1999 (DQ995647) and 438913/HK/CHN/2010 (JN542510) in the P2 and P3 coding regions. In this study, recombination events were detected between 1222/YN/CHN/2010 and CVA19 and CVA22 in the 2C–3D and 3D regions, respectively. Previous literature on enteroviruses have shown that the nonstructural proteins regions (particularly 3D RNA polymerase) had various functions in viral replication. The potential intertypic and interspecies recombination generally occurred in the nonstructural, and recombination played an important role in the evolution of enteroviruses [10]. Therefore, further investigation is needed to research whether the difference in pathogenicity of CVA1 on RD cells is due to the difference of recombination pattern. Moreover, intra- and inter-serotypic recombination events were increasingly reported in enterovirus, which may lead to the emergence of a novel genotype that can enhance the virulence of less common virus. It may provide selective advantages to better adapt to the environment, even form a novel epidemic strain as some epidemic EV serotypes, which may cause relative disease, even an outbreak. Therefore, further analysis of the recombinant strain isolated in this study will help to grasp the dynamics variation of CVA1.
Figure 2. Similarity plot and Bootscaning analysis of the 1222/YN/CHN/2010 with closely related strains.
Genomic region 1222/YN/CHN/2010 Serotype/strain %Nucleotide identity GenBank
accession number5’UTR CVA1/V18A 95.5 MG571859 VP4 CVA1/B1205 96.6 KY079224 VP2 CVA1/ V18A 94.0 MG571859 VP3 CVA1/ V18A 95.1 MG571859 VP1 CVA1/V18A 92.4 MG571859 2A CVA1/V18A 97.3 MG571859 2B CVA1/BBD34 92.1 KC785529 2C CVA1/KS-ZPH01F/XJ/CHN/2011 91.5 JX174177 3A CVA1/KS-ZPH01F/XJ/CHN/2011 96.2 JX174177 3B CVA19/V15C 97.0 MG571855 3C CVA19/V15C 93.6 MG571855 3D CVA22/ban99-10427 93.0 DQ995647 3’UTR CVA1/KS-ZPH01F/XJ/CHN/2011 100.0 JX174177 Genome CVA1/V18A 91.8 MG571859 Table S3. The strains with the highest similarity of the nucleotide sequences in all the genomic regions of the 1222/YN/CHN/2010 strain using BLAST
Authors’ Contributions Designed the experiments: MA Shao Hui, YANG Zhao Qing. Performed the experiments: FENG Chang Zeng, ZHANG Ming. Analyzed the data: FENG Chang Zeng, XU Dan Han, Guo Wei, SUN Hao. Wrote the paper: FENG Chang Zeng.
HTML
 21370Supplementary Materials.pdf
21370Supplementary Materials.pdf
|

|

 Quick Links
Quick Links
 DownLoad:
DownLoad: