





-
Infectious diseases continue to cause a substantial public health care burden in the 21st century. The constant emergence of novel pathogenic microorganisms, drug resistance pathogens, and the reemergence of classical pathogen epidemics present great challenges to the diagnostic technology of infectious diseases. In recent years, pathogens that have high mortality and transmission potential have emerged, including severe acute respiratory syndrome (SARS) in 2002–2003[1, 2], Middle East respiratory syndrome coronavirus (MERS-CoV) in 2012[3, 4], Ebola virus in 2014–2016[5-7], Zika virus in 2015–2016[8, 9], and the recent severe acute respiratory syndrome coronavirus 2 (SARS-COV-2)[10-13]. Furthermore, pathogens with resistant traits pose major threats to public health and have received considerable attention. In comparison to the pace of antibiotic development, the persistent and fast-evolving drug resistance to almost all clinically-used drugs, including last-resort drugs (e.g., colistin and polymyxin B), have become a critical issue[14]. In addition, the recently published Global Burden of Disease 2019 Study[15] provided a comprehensive analysis of the incidence, prevalence, and mortality of 369 diseases and injuries in 204 countries and territories from 1990–2019. The data revealed that infectious diseases are the main cause of disability-adjusted life-years in children, especially lower respiratory infections, diarrheal diseases, malaria, meningitis, whooping cough, and sexually transmitted infections. And HIV/AIDS is the second leading cause of disability-adjusted life-years in the 25–49 years age group. Therefore, alternative diagnostic technologies that allow more accurate and efficient diagnoses in clinical practice are imperative.
Historically, traditional culture-based methods or antigen-based tests are time-consuming, labor-intensive, costly, and only focus on individual pathogens rather than whole populations[16]. Fortunately, with the continuous advances in molecular biology, molecular-based detection technologies have become relatively common in laboratory and well-resourced clinical settings. Emerging advanced molecular diagnostic technologies, such as isothermal amplification technology[17-20], RT-qPCR[21], MALDI-TOF mass spectrometry[22-24], multiplex PCR detection[25], and high-resolution melting[26, 27], are rapidly transforming the ability to diagnose pathogens and drug-resistance genes in pathogens. A disadvantage of these techniques is an inability to recognize unknown agents and high expenses, despite the favorable sensitivity, efficiency, and specificity.
Rapid advances in sequencing technology have created new opportunities for the detection of disease-causing pathogens and drug-resistance genes. Next-generation sequencing (NGS) technology in combination with bioinformatics methods can be used to identify all microorganisms in the sample within 24 h[28, 29]. Unfortunately, the short-sequencing reads, usually < 500 bp, provided by NGS technology (e.g., Illumina and Roche 454 sequencing) make it difficult to parse the complex genome structure of microorganisms, especially the reads that contain many repeated elements. Long-read sequencing technologies, as represented by ONT[30] and Pacific Biosciences (PacBio)[31], have overcome many specific limitations, thus revolutionizing the sequencing of genomes in the clinical diagnostic arena. ONT has led to the development and commercialization of nanopore-based sequencing[32]. Compared with PacBio, nanopore sequencing is a better choice because of the low price of the instrument, portability, and potentially much longer sequence reads. In this review we provide a brief overview of the applications of nanopore sequencing technology in the clinical diagnosis of infectious diseases, as well as a discussion of current challenges and potential opportunities of this technology that can stimulate further studies on clinical pathogen diagnostic methods.
-
The basic working principle of nanopore sequencing is to observe the ionic current fluctuations caused by individual DNA or RNA molecules passing through a single nanochannel or nanopore. Subsequently, the ionic current fluctuations are decoded using base-calling algorithms to determine the molecular sequence[33, 34]. Over the past few years, a number of nanopore sequencing platforms have been developed, each with unique attributes suitable for different applications (Table 1). The pocket-sized MinION, a USB-powered sequencer produced by ONT, was launched in 2014 as the first commercially-available sequencer. Owing to portability, the MinION nanopore sequencer can be removed from the lab and used for the detection of pathogens in challenging field environments[35]. Compared with MinION, the GridION and PromethION sequencers provide higher throughput, thus enabling analysis on a larger scale and a cost-efficient way of large genome sequencing[30]. Specifically, the first laboratories to take delivery of PromethION was announced in early 2017, representing an ultra-high-throughput platform. The instrument was upgraded thereafter, and the sequencing platform currently includes 24 or 48 individual flow cells, providing a large volume of data (up to 7 or 14 Tb)[36, 37]. The low-priced Flongle flow cell sequencing device, an adapter for MinION and GridION, allows cost-efficient sequencing of smaller tests and experiments[38]. In the near future, further miniaturization and parallelization of sequencing devices will become available and provide more versatility. Smaller platforms under development at ONT include SmidgION, which is compatible with smartphones or other mobile devices[39], and the MinION Mk1D, which is designed to be an accessory keyboard with an integrated sequencer for tablet devices. In addition, Plongle, which has 96 individual, disposable flow cells, is designed for users who wish to carry out larger numbers of small, quick tests in parallel, enabling users to achieve low cost per sample.
Item Flongle MinION
Mk1BMinION
Mk1CGridION
Mk1PromethION 24 PromethION 48 Number of flow cells 1 1 1 5 24 48 Theoretical maximum output per device 2.8 Gb 50 Gb 50 Gb 250 Gb Up to 7 Tb Up to 14 Tb Theoretical maximum output per flow cell 2.8 Gb 50 Gb 50 Gb 50 Gb Up to 290 Gb Up to 290 Gb Number of channels per flow cell 126 512 512 512 2,675 2,675 Targeted sequencing Low to medium plex Medium to high plex Medium to high plex Medium to high plex Highly multiplexed Highly multiplexed Metagenomics Species ID Quantitative species ID Quantitative species ID Quantitative species ID Quantitative species ID Quantitative species ID RNA Sequencing Isoform Isoform & expression Isoform & expression Isoform & expression Isoform & expression Isoform & expression Weight 20 g 87 g 450 g 11 kg Sequencer: 28 kg
Data Acquisition unit: 25 kgSequencer: 28 kg
Data Acquisition unit: 25 kgSystem access $1,460 $1,000 $4,900 $49,995 $195,455 $285,455 Table 1. ONT sequencing platforms comparison
Compared with traditional short-read technologies, nanopore sequencing technology has the following advantages: (i) The real-time data streaming allows immediate access to actionable results and stops sequencing at any time as long as the sequencing data are sufficient. (ii) The scale-up feature with modular MinION, GridION, and PromethION is suitable for low- to ultra-high-throughput sequencing of pathogens. (iii) The portability and flexibility features[40] permits sequencing “what you want, when you want, and where you want” with portable, low-cost MinION devices. Both sequencing time and the number of sequenced samples can be adjusted. In addition, multiple samples can be sequenced simultaneously using up to 96 barcodes. (iv) The read length is unrestricted[41, 42]. Because nanopore sequencing technology does not restrict read length, successful performance in complete genomes, plasmids, and long repeat regions can be achieved. (v) The workflow is streamlined and automated. Nanopore-based tools with streamlined and automatable workflows reduce the hands-on time, thereby making nanopore-based tools more suitable for large-scale genetic investigations[43].
In addition, ONT has recently released a new Kit 12 Chemistry containing an updated sequencing enzyme enabling accuracies of > 99%. Combined with the latest Q20+ chemistries, the assembly accuracy of the Oxford Nanopore R10.4 flow cell has undergone significant improvement. Owing to the relatively high error rate of the R9.4.1 flow cell to call homopolymers, short-read polishing is essential to correct the assembly results for the generation of high-quality genomes. These additional requirements hindered the widespread use of nanopore sequencing technology. The latest Oxford Nanopore R10.4 flow cell has shown major improvement in the sequence accuracy (approximately 99%). Near perfect microbial genomes can be obtained from R10.4 data alone at a coverage of 40 x from pure cultures or metagenomes without short-read or reference polishing[44].
-
Several new and powerful strategies based on nanopore sequencing platforms have been applied to meet the needs of different experimental purposes (Figure 1). Whole-genome sequencing with nanopore sequencing platforms can improve assembly contiguity, thus allowing the investigation of the role of repeat elements in microbial function and adaptation[45]. Targeted sequencing is commonly used to aid the rapid identification of microorganisms and the detection of drug-resistance genes in pathogens. In metagenomic analysis, nanopore sequencing technology enables the assembly of complete closed-loop bacterial genomes and plasmids from complex and diverse metagenomic samples to provide unbiased and PCR-free genome sequences[46, 47].
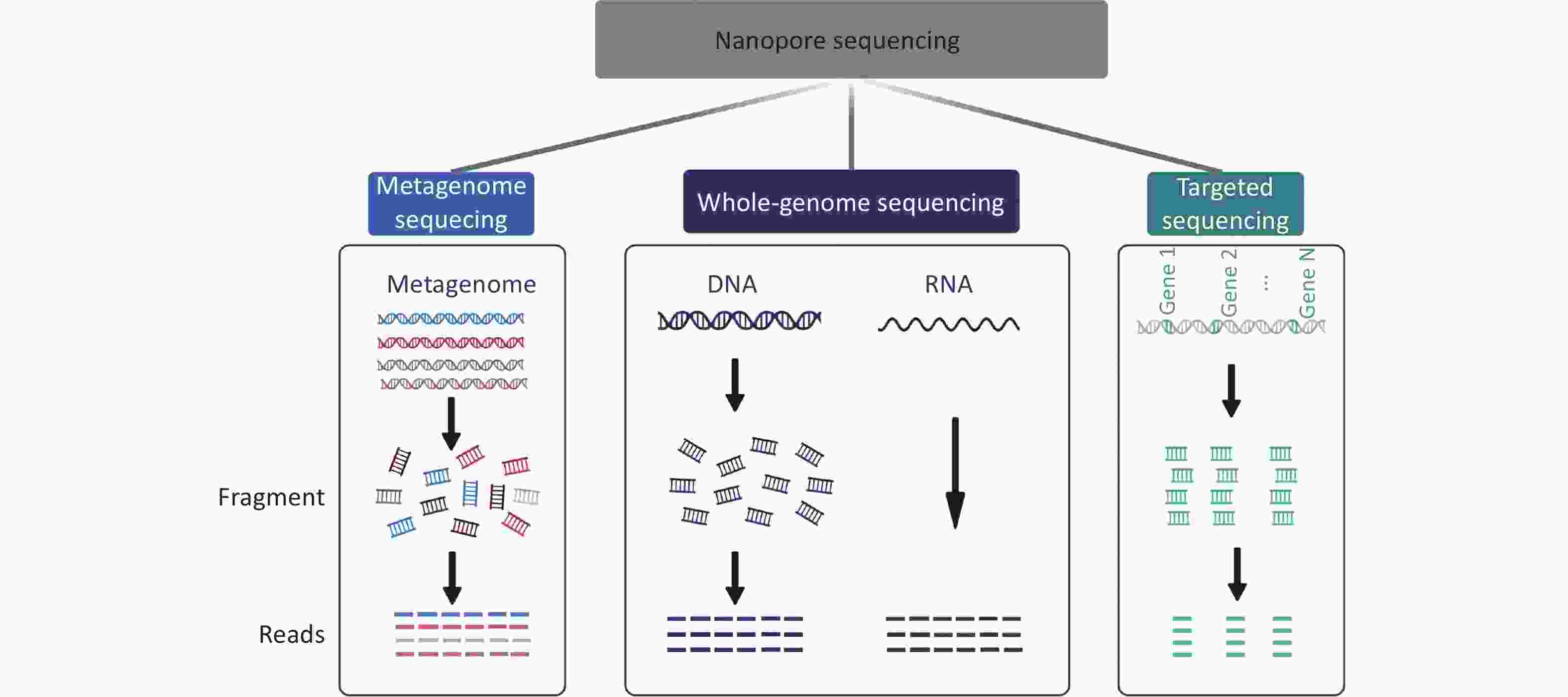
Figure 1. Strategies for nanopore-based sequencing technologies.
-
Microbial whole-genome sequencing is becoming a standard tool for diagnosing infectious diseases and providing improved resolution to monitor the spread and evolution of major pathogens within and outside hospitals in real time. Correct and complete genome assembly is crucial for understanding the effect of genome structure on genome function. Owing to their inherent length limitations, traditional short-read-based assemblies are highly fragmented and often fail to generate complete genome sequences, especially those with satellites, tandem repeats, and multigene family regions[48]. To address this challenge and enable more accurate comparative genomic studies, researchers are now utilizing nanopore devices that can sequence short to ultra-long DNA or RNA fragments to generate a more contiguous genome assembly. Compared with traditional Illumina short reads genome assembly, the hybrid assembly that combines Illumina short reads with nanopore long reads has reduced the scaffold number 51-fold. In addition, the hybrid assembly increases the number of protein-coding genes, non-coding genes, and transposable elements, thus supporting the facility to perform functional and comparative studies[49]. Moss et al. [50] proposed a sequencing process (Lathe) that combines long read long sequence assembly with short read long sequence error correction to assemble complete bacterial genomes and achieve strain identification in complex microbial communities. In a study in which 12 bacterial samples were manually mixed, 30.3 GB of data were obtained. The assembly of seven complete genomes was completed at a later stage; three genomes were assembled into four or fewer fragments, and the poorest assembled bacterial genome comprised 83% of the genomes in a single fragment.
Sequencing the RNA in a clinical sample can unlock a wealth of information, including the identity of a pathogen; however, sequences generated by methods that detect the products of a synthesis reaction (e.g., sequencing cDNA generated by reverse transcription of RNA) rather than directly detecting the RNA molecule are subject to the processing and error-rate limitations of reverse transcription and cannot detect base modifications or distinguish homopolymers[51]. The first nanopore-based direct RNA-seq, highly parallel, real-time, single-molecule method was proposed in 2018[52]. This method circumvents reverse transcription or amplification steps, yields full-length, strand-specific RNA sequences, and enables the direct detection of RNA modifications. Nanopore sequencing technology allows sequencing of the entire viral RNA sequence in one read, thus eliminating the need for assembly. Compared with other RNA-seq strategies, the direct RNA-seq method has many potential advantages, as follows: the direct RNA-seq method does not require amplification and does not have PCR or reverse transcription bias; the direct RNA-seq method produces very long sequences, which are particularly useful for the study of splice variants; and the direct RNA-seq method uses direct determination of RNA and can thus detect nucleoside analogs, which are chain specific[53].
-
The long sequencing reads delivered by nanopore technology expand the capabilities of targeted sequencing approaches beyond the analysis of single nucleotide variants, including cost-effective, high-coverage characterization and phasing of structural variants, repetitive regions, and base modifications. Targeted nanopore sequencing concentrates the data collection over a specific genomic region, and obtains more accurate identification and characterization of biological agents in complex samples[54]. It is worth noting, however, that although this strategy improves the detection sensitivity of target microorganisms, the breadth of the potential pathogens being detected is limited.
Nanopore-targeted amplicon sequencing uses highly conserved primers to amplify specific genes of microorganisms in clinical samples[55], which provides unprecedented insight into the microbial communities[56] and drug-resistance determinants[54]. Sequencing taxonomic-related genes in the entirety, such as 16S and 23S for bacteria and 18S for fungi[57], improves the resolution of identification. The method relies on multiplex PCR for targeted enrichment of viral genomes, thus allowing viral consensus sequences 1–2 days from clinical samples[58]. Another advantage of nanopore-targeted sequencing is that nanopore-targeted sequencing combines targeted amplification and long-read nanopore sequencing, thus allowing the detection of SARS-CoV-2 and other respiratory viruses simultaneously within 6–10 h. The limit of detection was 10 standard plasmid copies per reaction, and the detection specificity for SARS-CoV-2 was 100%. Therefore, this strategy is suitable for COVID-19 diagnosis and can be further extended to diagnose other pathogens[59]. A method for SARS-CoV-2 genome sequencing in clinical samples [rapid sequencing long amplicons (RSLAs)] uses random primers to generate cDNA from RNA purified from clinical samples, followed by single or multiplex PCRs to generate longer amplicons of the SARS-CoV-2 genome. This protocol can identify SARS-CoV-2 and provide improved sensitivity in identifying viruses in clinical samples, which may be false-negatives when other nucleic acid-based methods are used[60]. Sequencing, which combines multiplex amplicon PCR and nanopore technology to sequence drug-resistance genes directly from clinical samples, is a versatile and convenient culture-free diagnostic method and has the advantages of high sensitivity and accuracy[54]. Furthermore, mutations with no known association with phenotypic drug resistance and novel mutations can also be detected[61].
-
Recently, metagenomics analysis was reported to address the composition of a microbial community[62, 63]; there are several strategies for metagenomic analysis.
-
Long-read nanopore sequencing technology improves the traditional gene-level shotgun metagenomic analysis provided by short-read sequencing approaches to enable unbiased assembly of complete, closed genomes and plasmids from clinical research and microbiome samples[64]. In addition, whole-genome metagenomic analysis based on nanopore sequencing technology helps discriminate closely related species, resolve challenging repeat regions and structural variants, and delineate plasmid and genomic drug-resistance genes[65].
-
Targeted metagenomics with a long sequencing sequence improves the precision of targeted metagenomic species identification[66]. By enabling the sequencing of complete genes or operons, such as repetitive regions, long nanopore sequencing reads have been shown to offer a more comprehensive identification of species in mixed microbial communities[67, 68].
-
The long reads provided by nanopore technology enable sequencing of full-length transcripts in single reads, precluding the requirement for complex and often inaccurate post-sequencing transcript assembly. The unambiguous identification of transcript isoforms significantly simplifies studying gene expression in mixed microbial communities and provides an alternative method for identifying pathogens from metagenomic samples. Nanopore meta-transcriptome sequencing excels in read length and deals with complex microbiome and non-bacterial transcriptome backgrounds[69].
-
Over the past decade nanopore-based sequencing methods have been widely investigated for the diagnosis of infectious diseases. The application of high-throughput sequencing has been described in detail in different clinical fields[70, 71]. Additionally, the growing application of ONT platforms has driven development towards an automation-oriented format.
-
Rapid and accurate identification of pathogenic microorganisms is key to diagnosing and treating infectious diseases as the outbreak progresses. Currently, culture-based techniques are still the main method for clinical microbial detection; however, culture-based techniques are time-consuming and laborious, which leads to a diagnostic lag. Nucleic acid amplification tests (NAATs) have been applied in recent years for the rapid detection of pathogenic microorganisms, such as real-time PCR assays; however, these techniques have limitations. For example, NAAT assays are designed based on known sequences, thus there is a risk for false-negative results when detecting organisms with high mutation rates. It is impossible to use existing assays to detect unknown or emerging targets. Moreover, most NAATs, like real-time PCR, cannot obtain targeted genome sequences. Therefore, in-depth analysis of pathogen transmission and evolution may be not possible. Indeed, nanopore sequencing technology overcomes these drawbacks. Novel infectious diseases are potentially serious threats to public health because novel infectious diseases may cause a large number of human infections, morbidity, and heavy economic burden, as illustrated by the ongoing coronavirus disease 2019 (COVID-19) pandemic.
During the Ebola outbreak in Guinea, Quick et al.[72] designed an entire sequencing system based on the ONT MinION platform, which could be accommodated in a single ordinary test bed for real-time genome monitoring of the epidemic. During the Zika outbreak in Brazil[73], the research team performed PCR and sequencing using the Oxford Nanopore MinION platform and undertook sequencing of a set of samples. The identification and characterization of ZIKV complete genomic DNA in positive samples provide a framework for reconstructing the epidemic trajectories of ZIKV and tracking its spread into other regions. More recently, the outbreak of COVID-19 caused by SARS-CoV-2 has had an enormous impact worldwide. Accurate detection of SARS-CoV-2 is critical for effective therapy and control of the pandemic[74-77]. Although the SARS-CoV-2 (WHHuman_1) genome sequencing was completed in China in January 2020 and the results have been shared with other countries[78], mutations and recombination have occurred during the replication of SARS-CoV-2[79, 80]. Sequencing of the viral genome using nanopore technology revealed that a number of patients had a proportion of viral genomes with deletions, which is likely to result in false-positive results when tested using other nucleic acid-based assays[60]. Currently, reverse real-time reverse transcription-polymerase chain reaction (RT-PCR) assays, the most common clinical diagnostic tool, for SARS-CoV-2 detection targets the nucleocapsid (N), envelope (E), and open reading frame 1a or 1b genes. When the binding sites of primers and probes are mutated in these genes, the sensitivity and accuracy of the assay will be greatly compromised. In a study of whole genome sequencing of SARS-CoV-2 based on the Oxford Nanopore platforms, researchers found that the nonstructural protein 1 (nsp1) gene, located at the 5' end of the SARS-CoV-2 genome, is highly expressed in the nasopharyngeal or saliva specimens of COVID-19 patients with different degrees of clinical severity. Therefore, a novel nsp1 real-time RT-PCR assay was added to the multitarget detection of SARS-CoV-2, which can avoid false-negative results due to mutations at the primer or probe binding sites of currently available RT-PCR assays[81]. In addition, co-infection with SARS-CoV-2 and other respiratory viruses pose a major challenge in the detection of SARS-CoV-2. Wang et al.[59] developed an innovative nanopore targeted sequencing detection method, which can detect SARS-CoV-2 and other respiratory viruses simultaneously in 6–10 h with a limit of detection of 10 standard plasmid copies per reaction. Nanopore sequencing methods can be applied, not only for pathogen identification and genome sequencing, but also for pathogen typing. The nanopore sequencing system can identify various types of viral targets simultaneously based on the established SARS-CoV-2 GenBank, thus allowing researchers to track disease transmission and pathogen evolution[82].
A metagenomic study can provide comprehensive pathogen detection, not just the causative pathogens, thus revealing additional taxonomic and functional diversity. Metagenomic sequencing with a nanopore sequencer for direct and rapid pathogen detection has become increasingly popular in clinical diagnosis, especially in the detection of infected body fluid samples. A metagenomic intra-species typing tool (MIST;
https://github.com/pandafengye/MIST ) was established to estimate strain-level compositional abundance. MIST can accurately predict the strain composition at a 99.9% average nucleotide identity resolution. Notably, this is the first time that infected body fluids have been systematically characterized at the strain level[83]. Another study developed a metagenomic sequencing test using cell-free DNA from body fluids to identify pathogens with Illumina and nanopore sequencing platforms. The sensitivity and specificity of the test for bacteria and fungi detection using Illumina and nanopore sequencing platforms were comparable; however, nanopore sequencing has a shorter turnaround time (< 6 h), which is essential for infections that demand a rapid response and timely diagnosis[84]. In brief, clinical metagenomic sequencing with a nanopore sequencer can greatly benefit the rapid identification of multi-strain infection from body fluids, and guide infection prevention and control practices. -
The inappropriate use of drugs has led to an alarming trend in the increasing frequency of drug resistance among pathogens that cause nosocomial and community-acquired infections. Infectious diseases caused by drug-resistant pathogens lead to severe disease and death. Thus, there is an urgent need for rapid and comprehensive methods that can accurately describe specific drug-resistance gene profiles. Early clarification of bacteria-associated antimicrobial resistance (AMR) is considered to be a prerequisite for effectively treating diseases and limiting the spread of AMR[85, 86]; however, microbiological culture usually requires > 48 h, which leads to delayed diagnosis and a wide distribution of drug-resistant bacteria. Although a variety of molecular biological detection methods have been used to resolve this problem, it is impossible to cover all AMR determinants and identify novel mutations. Nanopore-based analysis is currently an area of great interest in many disciplines with the potential for AMR gene detection.
Gonorrhea is the second most common sexually-transmitted bacterial infection worldwide[87]. AMR of Neisseria gonorrhoeae (NG), which is the causative agent of gonorrhea, hinders the prevention and control of this disease and has aroused global health concern[88, 89]. The complete genome sequence of N. gonorrhoeae was obtained for the first time by our group using the MinION sequencer. By comparison, most AMR determinants identified by a nanopore-based assembly alone were the same as the AMR determinants identified by hybrid assemblies containing Illumina and MinION reads. Sequentially, AMR profiles related to seven classes of antibiotics can be acquired using Pathogenwatch and a BLAST-based workflow. Moreover, a potentially novel antimicrobial-related mutation located in mtrR was found, indicating that this assay is a potential approach for discovering new AMR determinants[90]. In addition, multiplex PCR amplicon nanopore sequencing coupled with a bioinformatics analytical pipeline enables more efficient enrichment of AMR genes from clinical samples[91]. Based on this strategy, our group established another method that could simultaneously sequence 13 genes associated with AMR in N. gonorrhoeae by starting from clinical samples within a 7 h 40 min to 10 h 40 min timeframe. Compared with the Sanger sequencing results, this method had a base accuracy of > 99.5%, and the AMR sites were correctly identified[54].
At the same time, this method also provides an avenue for the detection of AMR genes in poorly cultivated pathogens, such as Mycobacterium tuberculosis (MTB). In the 2020 Global Tuberculosis Report published by the World Health Organization (WHO), data showed that approximately 10.0 million people fell ill with tuberculosis (TB) in 2019[92]. Drug-resistant MTB, the causative agent of TB, is a great challenge in the treatment of TB. Most patients with TB are multidrug-resistant[93, 94]. In one study, an existing TB targeted next-generation sequencing assay kit and drug susceptibility testing analytics solution (Deeplex Myc-TB), which was optimized primarily for Illumina sequencing platforms, was evaluated for use on the ONT MinION sequencer. It was demonstrated that Deeplex Myc-TB can be successfully implemented on a portable MinION sequencing device. The method of applying the Deeplex Myc-TB to MinION is a promising solution for low capital costs and rapidly provides clinically relevant data, despite the higher raw error rates on MinION[95]. More broadly, this platform is suitable for use to a wider extent in clinical drug-resistant bacterial pathogens.
Bloodstream infections and sepsis are major causes of morbidity and mortality worldwide. Phenotypic determination of antibiotic susceptibility requires 2−3 days after 1−2 days of blood culture-based diagnostics for identification of bacterial agents. Whole-genome sequencing based on nanopore platforms represents a genotypic diagnostic approach with the ability to rapidly identify pathogens and detect plasmids and AMR-encoding genes within 1−2 h[96, 97], which shows promise for use in clinical diagnosis. Nanopore sequencing was used to monitor the transfer and rapid evolution of AMR plasmids within and across multiple species. The transfer of mobile genetic elements between different bacterial species is an important method of AMR transmission; however, surveillance is difficult until recent advances in sequencing technologies. A recent study[98] on nanopore sequencing technology was used to detect a long-term outbreak of multidrug-resistant Pseudomonas aeruginosa, Citrobacter freundii, and Citrobacter cronae in a German hospital over 6 years. A novel computational pipeline was developed, which enabled the assembly of genomes and plasmids, AMR gene annotation and visualization, and comparative analysis. A 40-kb plasmid carrying blaIMP-8, first detected in P. aeruginosa, was identified as C. freundii, underwent further evolution and plasmid fusion, which resulted in a 164-kb megaplasmid that was transferred to C. cronae. In summary, nanopore-based sequencing methods can detect plasmid transfer between different bacterial species, plasmid fusion, and rearrangements of the AMR gene cassette, which mediate the rapid evolution of opportunistic pathogens, enabling successful countermeasures to contain plasmid-mediated outbreaks.
In fact, there is an urgent need to develop software that can efficiently analyze all known gene targets and identify mutations associated with AMR to predict antimicrobial susceptibility of pathogenic microorganisms in clinical practice. BacWGSTdb 2.0 (http://bacdb.org/BacWGSTdb), a free bacterial whole-genome sequence typing and source tracking database, provides essential insight for surveillance of AMR bacteria evolution[99]. High quality complete genomes can be obtained by hybrid assembly of short and long reads generated using Illumina and nanopore sequencing technology, respectively[100]. Using the AMR prediction tool, these whole-genome sequences may help to better understand the AMR mechanisms, genomic features, and transmission dynamics of antibiotic-resistant organisms in clinical settings[101-103]. Therefore, the AMR prediction tool can help develop effective control strategies and prevent further dissemination of AMR.
In addition, genome sequencing of parasites in clinical samples is an important step to track the spread of drug resistance, and to monitor evolutionary changes in the parasite population. A method[104] exists that combines PCR amplification and nanopore sequencing technology for sequencing nine drug-resistance genes of the malaria parasite, Plasmodium falciparum. There was a significant increase in precision and recall, and a decrease in the error rate with the use of further improved chemistry. This examination method will be extremely beneficial in expanding the database repertoire of the parasites. Furthermore, this method will enrich our knowledge about the geographic distribution and changes in parasites over time.
-
The microbial community is important for the establishment and maintenance of human health[105]; however, the balance between the human body and the microbial community is easily upset by changes in the internal pathologic conditions and external interference. In most cases, clinical infections are complex and co-infections are not surprising, especially in respiratory and intestinal diseases[106]. Sequencing from clinical samples could be superior to current molecular methods of pathogen identification because sequencing has the potential to detect known and novel potential pathogenic species in a single application[107]. Metagenome sequencing is an important sequencing strategy for the characterization of microbial communities[108]. In a study[109] focusing on the bacterial composition of complex microbial communities, nasal microbiota results were compared at the genus level using both Illumina and nanopore 16S rRNA gene sequencing. At the genus level, the nanopore sequencing platform is comparable with the Illumina platform in detecting bacterial genera of the nasal microbiota, but the nanopore platform does have problems in detecting bacteria within the genus, Corynebacterium. Another study[110] examined the feasibility and clinical validity of metagenomics with nanopore sequencing of clinical respiratory specimens. The results showed that nanopore sequencing is in striking concordance with positive microbiologic cultures for a diagnosis of severe pneumonia.
Metagenomics based on 16S rRNA sequencing is frequently used to analyze the density of bacteria found in the human body and their relationship with disease[111]; however, the usual approach of sequencing short regions of the 16S rRNA gene fails to assign taxonomy at the genus and species levels. Therefore, long-amplicon PCR-based approaches based on nanopore sequencing have been developed to achieve increased taxonomic resolution. Two different genetic markers (the full-length 16S rRNA and 16S-ITS-23S region from the rrn operon) were assessed. In the sequencing of a clinical isolate of Staphylococcus pseudintermedius, two mock communities and two pools of low-biomass samples (dog skin), targeting the 16S-ITS-23S of the rrn operon was shown to be the better choice for increased resolution at the species level[112]. The accuracy of identification of bacterial species is hindered by the read length in short-read sequencing. Methods based on next-generation sequencing tend to amplify 1–3 hyper-variable regions within the 16S rRNA gene and identify bacterial species using existing 16S rRNA gene sequence databases[111, 113, 114]. Given the advantage of the long read length of the nanopore sequencing platforms, longer target sequences can be obtained, which not only improve the resolution of species identification, but also improve the precision of microbial composition identification in a sample, thus enabling a more realistic reduction of microbial community structure in a clinical sample.
With the application of genomics and metagenomics based on nanopore sequencing technologies to infectious disease epidemiology and human microbiome research, opportunities for specifically-targeted treatments and prevention of diseases caused by pathogenic microorganisms will increase considerably. In addition, these well-established methods can be further generalized to more comprehensive and faster specific detection of microbial species and strains in complex environments[115].
-
The advent of the nanopore long-read sequencing technique has provided a powerful tool for the diagnosis, investigation, and monitoring of infectious diseases. Long-read sequences generated using nanopore sequencing can obtain a rich collection of sequence information and increase pathogen classification accuracy. Target nanopore-based sequencing is a rapid and comprehensive method for accurately describing specific drug-resistance profiles, and whole-genome sequencing is vital for revealing the rapid temporal and spatial evolution of drug-resistance in bacterial pathogens. Nanopore-based metagenomic analyses are currently being explored for expanded use in bacterial community characterization, but have also found pronounced biases in the recovered taxon abundance[116]. Further research and time are needed to fully explore the power of this approach in the clinical identification and management of infectious diseases. There is no doubt that the nanopore sequencing methodology can also be expanded to infectious diseases and serve as a technical reserve for rapid pathogen identification during future pandemics. Recent developments in ONT sequencing platforms that combine miniaturization technologies with long-read sequencing, such as SmidgION, have led to more portable, low-cost, and efficient sequencing analyses with ease of use in the field.
Nanopore sequencing technology can serve as a versatile and convenient culture-free diagnostic method with the advantages of high sensitivity and accuracy, and holds great promise for future applications in clinical infectious diseases and for healthcare surveillance purposes; the data obtained can also serve as a reservoir for the detection of drug-resistance genes[117]. Every technique or diagnostic method has advantages and limitations, thus it is not possible to always replace existing molecular methods with novel methods. An increasing number of studies have found that a hybrid approach combining the strengths of NGS and nanopore sequencing yields less erroneous outcomes at lower costs. Appropriate sequencing protocols should be constructed according to specific research purposes and clinical needs.
-
The authors report no conflicts of interest in this work.
-
PENG Jun Ping participated in the coordination of the study and reviewed the manuscript. ZHANG Lu Lu performed the literature search and completed the first draft. ZHANG Chi was responsible for the final revision of the draft. All the authors reviewed the manuscript and provided critical input for the revision.
Funds:
This work was supported by CAMS Innovation Fund for Medical Sciences (CIFMS) [2021-I2M-1-038]

 Quick Links
Quick Links
 DownLoad:
DownLoad: