-
Triclosan (TCS) is widely used in personal-care products because of its bactericidal and antibacterial properties. However, TCS and its toxic byproducts have been detected in aquatic environments, animals, and plants worldwide. TCS is a common phenolic environmental endocrine disruptor, and TCS in the environment enters the body mainly through diet and water. Most of the absorbed TCS is metabolized by the human kidneys and is eliminated in the urine[1]. Some epidemiological studies have suggested a positive correlation between the TCS exposure level in urine and albumin (a biomarker of renal function), which could cause renal dysfunction[2]. However, the potential effects of TCS on the mammalian kidney and its mechanisms are currently unknown. Consequently, we initiated an in vitro trial to study the molecular mechanism of cytotoxicity of TCS to glomeruli.
Human renal glomerular endothelial cells (HRGECs) constitute the inner layer of the glomerular capillary wall and play an essential role in regulating blood cell permeability, but various environmental pollutants potentially cause glomerular barrier disorders. Previous studies have demonstrated that exposure to TCS is cytotoxic, including the induction of inflammatory factors, oxidative stress, and cell apoptosis in vitro. Thus, the molecular mechanisms underlying TCS-induced nephrotoxicity warrant further research. In the current study, we investigated the effects of TCS on HRGECs, such as TCS-induced reactive oxygen species (ROS) production, the collapse of the mitochondrial membrane potential (MMP), induction of autophagy, and changes in PI3K/Akt signaling. Additionally, we discussed the mechanism of TCS-related regulation of apoptosis. This study provides a theoretical basis for further clarifying the nephrotoxic characteristics of TCS.
HRGECs were cultured in extracellular matrix (ECM) supplemented with endothelial cell growth supplement (Cat #1052) and 25 mL fetal bovine serum (ScienCell, Carlsbad, CA, USA) in 5% CO2 at 37 °C. Cell IC50 and viability were measured by the CCK-8 method. According to the results, HRGECs entered the logarithmic growth phase after 24 h of culture at a density of 5 × 105 cells/mL (Supplementary Figure S1A, available in www.besjournal.com). In addition, the proliferation rates of HRGECs treated with different concentrations of TCS (5, 15, 20, 30, 40, 50, 75, and 100 µmol/L) for 24 h were 99.9%, 97.2%, 92.5%, 82.5%, 72.3%, 63.2%, 37.4%, and 3.1%, respectively compared to the control group (Supplementary Figure S1B). The results show that TCS inhibited HRGEC viability, and the 24 h IC50 of TCS in HRGECs was 56.5 µmol/L (95% IC50 53.3–59.96 µmol/L). Subsequently, the HRGECs were treated with 0, 15, 20, or 30 µmol/L TCS for 24 h in the follow-up experiments.
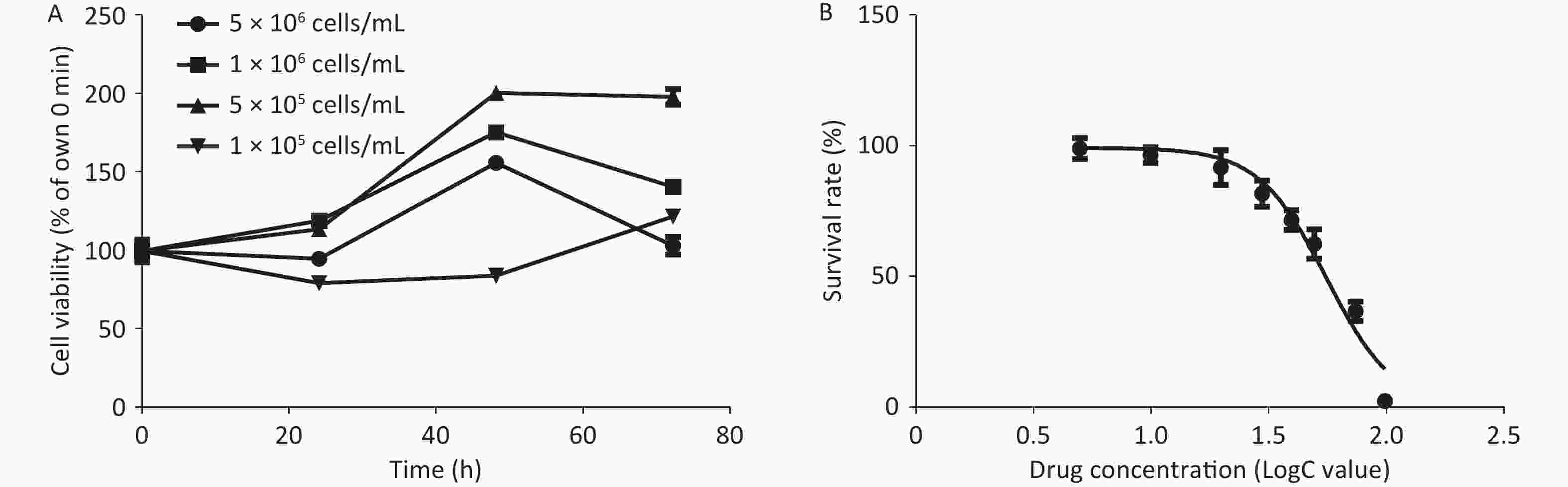
Figure S1. The growth curves of HRGEC, and the survival rate curve of HRGEC incubated with TCS of different concentrations. (A) HRGEC culture (0–72 h). Twenty-four hours after the inoculation HRGECs into 96- well plates at a density of 5 × 105 cells/mL, the cells entered the logarithmic growth phase. (B) The inhibition rate of CCK-8 on HRGECs was evaluated after treatment with 5–100 μmol/L TCS for 24 h.
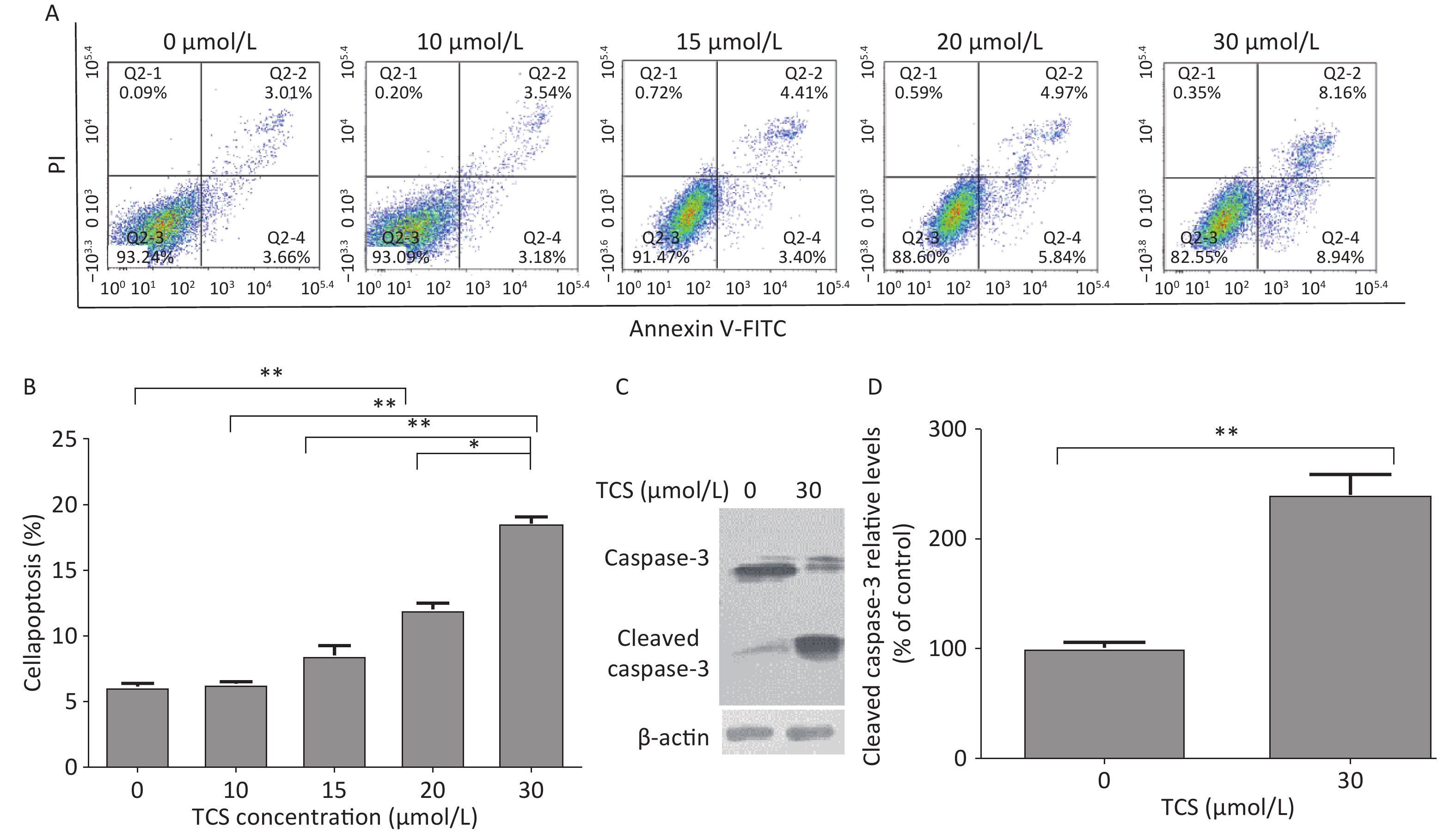
The apoptosis rates of the cells were determined by flow cytometry by measuring Annexin V-FITC/propidium iodide double staining 24 h after the TCS treatment (Figure 1A). The apoptosis rate of the 10 µmol/L TCS treatment group was not significantly different from the control group. The apoptosis rate of cells treated with 15, 20, or 30 µmol/L TCS increased in a dose-dependent manner. In addition, TCS increased cleaved caspase-3 activity in HRGECs compared to that in the control group after exposure to 30 µmol/L TCS for 24 h (Figure 1B). Caspase 3 (35 kD) was cleaved into cleaved caspase 3 (20 kD) and activated, and is a key element in apoptosis. These findings suggest that TCS may inhibit proliferation and promote apoptosis of HRGECs and the expression of cleaved caspase-3.
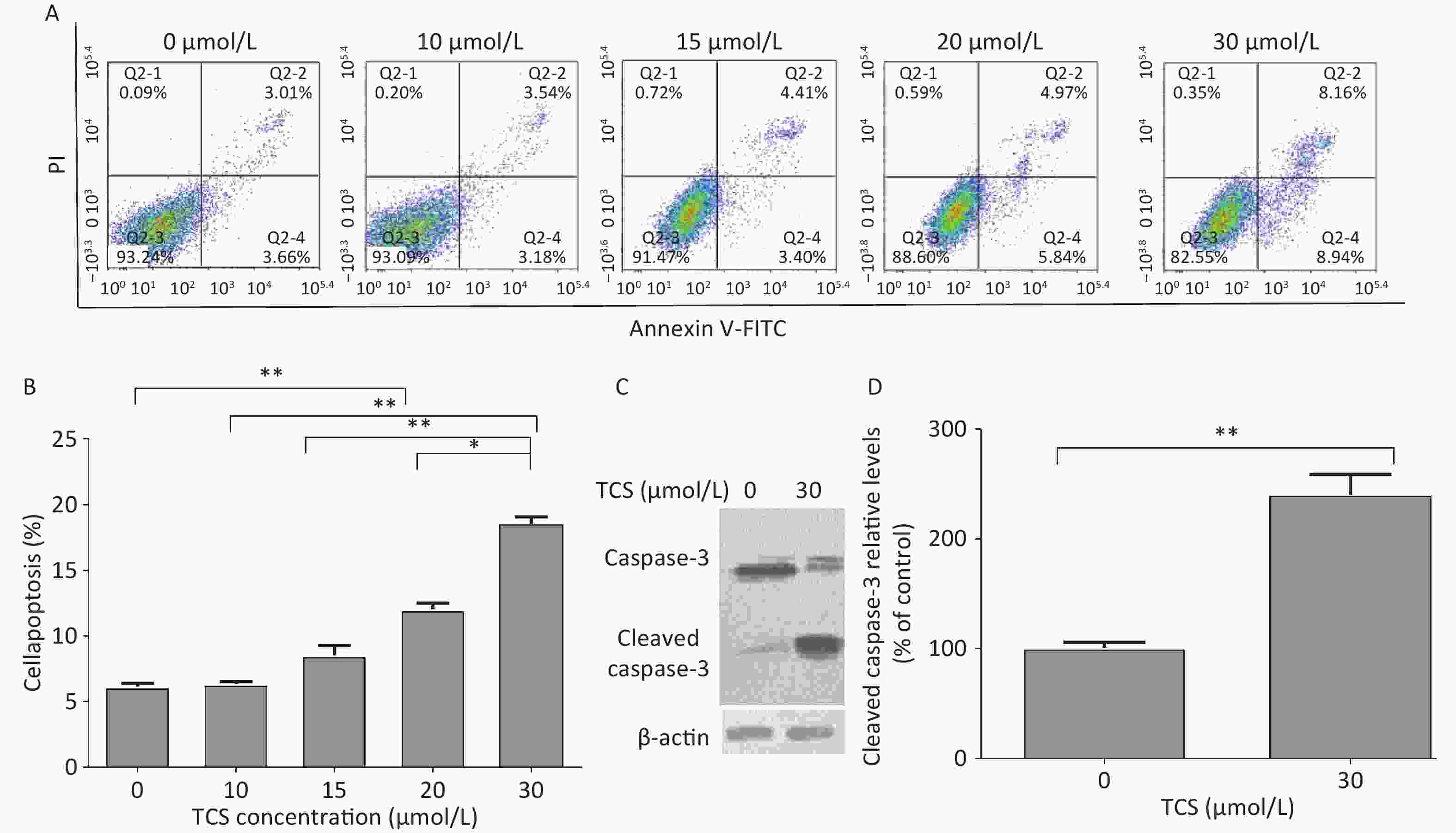
Figure 1. Effects of TCS on HRGEC apoptosis and caspase-3. The apoptotic rate of HRGECs treated with 0–30 µmol/L TCS was measured by flow cytometry, as shown in (A) and (B). TCS promoted the expression of cleaved caspase-3, which is shown in the electrophoretogram (C) and (D). Data in the bar charts are mean ± SD, n = 6. *P < 0.05 significantly different between the groups, **P < 0.01 significantly different between the groups.
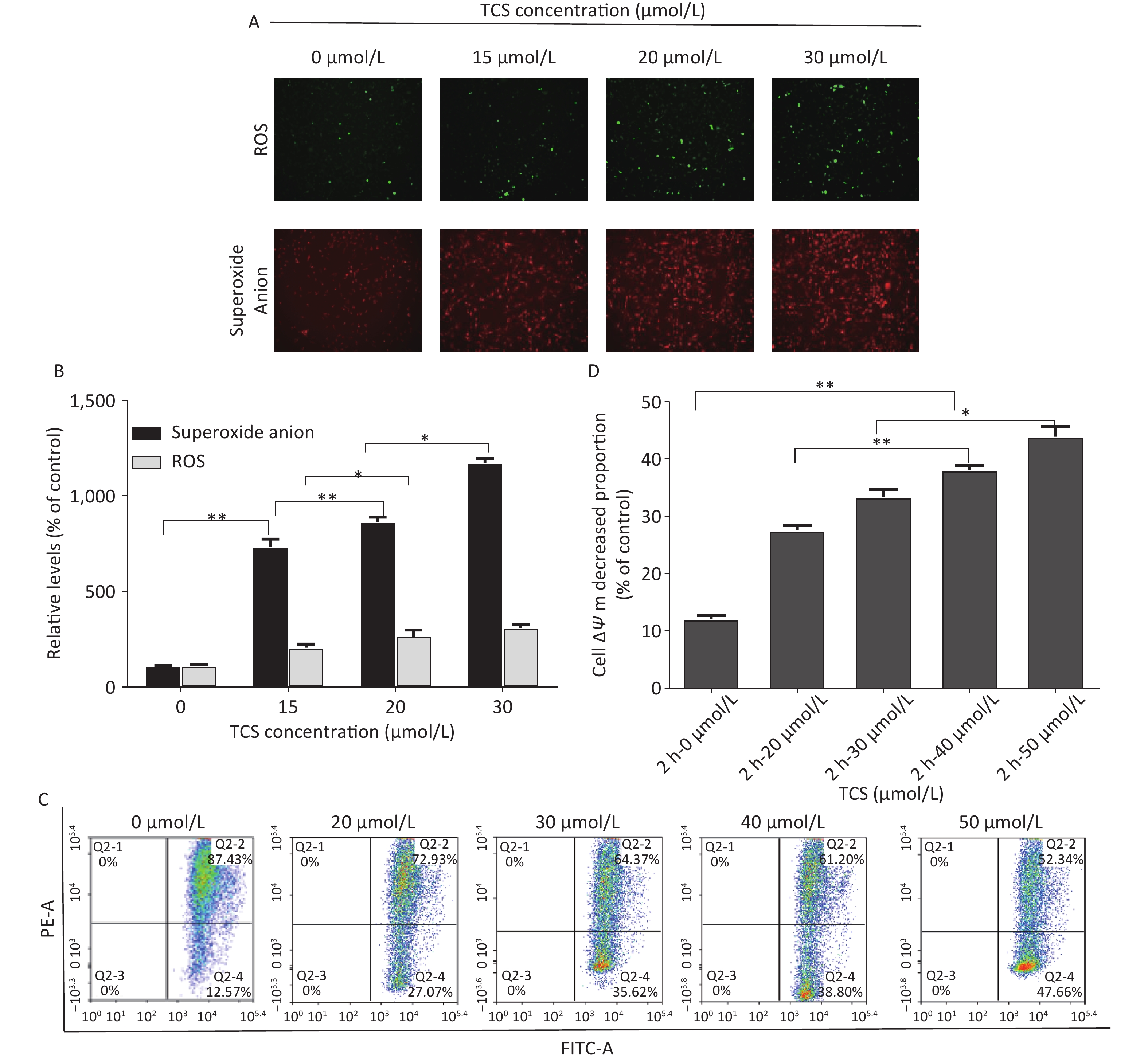
Oxidative stress in HRGECs induced by TCS was observed by fluorescence microscopy. DCFH-DA is a fluorescent stain used to measure ROS in cells, and the dihydroethidium (DHE) fluorescent probe was used to measure superoxide anion levels. Superoxide anion production is triggered by the leakage of electrons from the mitochondrial respiratory chain, and it is a type of free radical that exists longer and is more harmful than other types of ROS. In this study, after treating the HRGECs with 0, 15, 20, or 30 µmol/L TCS for 12 h, ROS levels (using DCFH-DA as an indicator) and the intracellular superoxide anion level (using DHE as an indicator) increased and were closely correlated with the TCS exposure dose. The ROS levels of 15, 20, and 30 µmol/L TCS treatment were 198.2%, 257.1%, and 310.5% compared to the control group, respectively. In particular, the levels of intracellular superoxide anions increased by 725.4%, 865.3%, and 1162.1%, respectively (Figure 2A). TCS promoted the generation of excess ROS in HRGECs in a dose-dependent manner, particularly superoxide anions. In previous studies, one of the key features of TCS-induced cell dysfunction was the accumulation of mitochondrial ROS in normal rat mesenteric arteries and aortic cells. ROS attacking mitochondrial genetic material or functional proteins can cause irreversible oxidative stress, leading to adverse events, such as destruction of the mitochondrial respiratory chain, the collapse of the MMP, or an ATP synthesis disorder[3]. The same effect also occurs in some cancer cells, such as TCS aggravated mitochondrial oxidative stress response and induced autophagy in A375 melanoma cells[4].
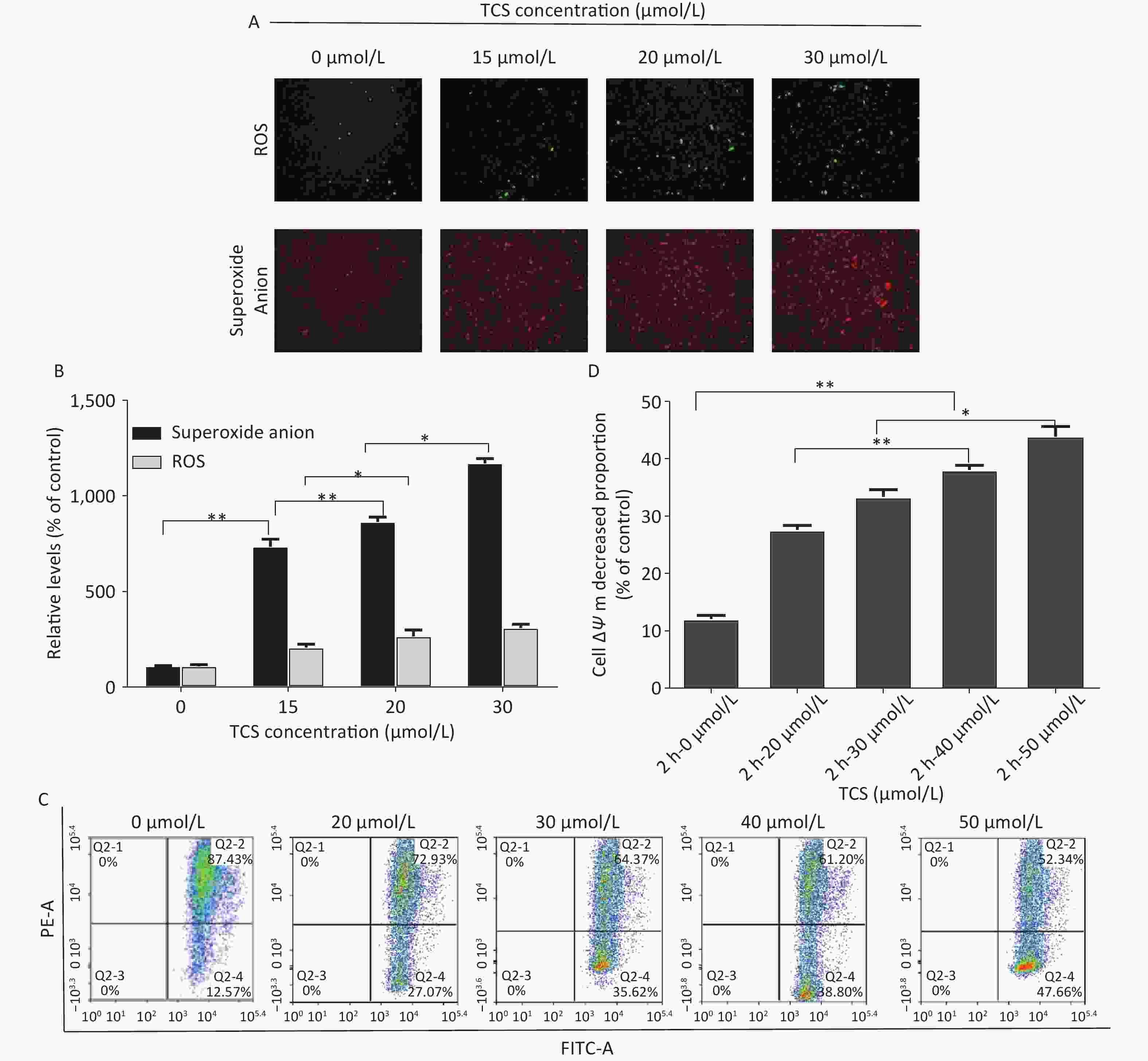
Figure 2. HRGEC mitochondrial damage induced by TCS. The levels of HRGEC ROS and superoxide anions are shown in (A) and (B). (C) The MMP (ΔΨm) of the HRGECs treated with TCS was measured by flow cytometry, as shown in (D) and (E). Data in the bar charts are mean ± SD, n = 6. *P < 0.05 significantly different between the groups, **P < 0.01 significantly different between the groups.
The excessive production of ROS and superoxide anions may be related to mitochondrial dysfunction, so we investigated the effects of TCS on HRGEC mitochondria by examining variations in the MMP (ΔΨm) and mitochondrial autophagy levels. After a 2 h TCS treatment (0–50 µmol/L), the proportions of HRGECs with a lower MMP were 12.2%, 27.8%, 33.7%, 38.4%, 44.3% (vs. control, P < 0.01). In brief, TCS induced a decrease in the MMP of HRGECs (Figure 2B). These results suggest that the cytotoxicity of TCS was complemented by the generation of ROS, loss of the MMP, and the induction of apoptosis. Confocal fluorescence microscopy revealed that the HRGEC mitochondria treated with 30 µmol/L TCS were enlarged, and autophagy was enhanced. In addition, compared to HRGECs treated with TCS alone, 1 mmol/L N-acetylcysteine (NAC)-pretreated HRGECs had a lower autophagy rate and better cell viability. The primary role of NAC as an antioxidant originates from its ability to increase the intracellular concentration of glutathione, thus, preventing or reducing oxidative stress. These results suggest that oxidative stress affects the autophagy of HRGECs. TCS-induced autophagy may be a clearing mechanism induced by oxidative stress injury.
Oxidative stress and apoptosis play fundamental roles in some toxin-induced renal injuries[5]. Hence, simple linear regression was used to analyze the relationship between ROS and the HRGEC apoptosis rate. The results showed that the level of ROS was positively correlated with the cell apoptosis rate (r = 0.864, P = 0.042, (P < 0.05), and suggest that ROS may be a risk factor for HRGEC apoptosis.
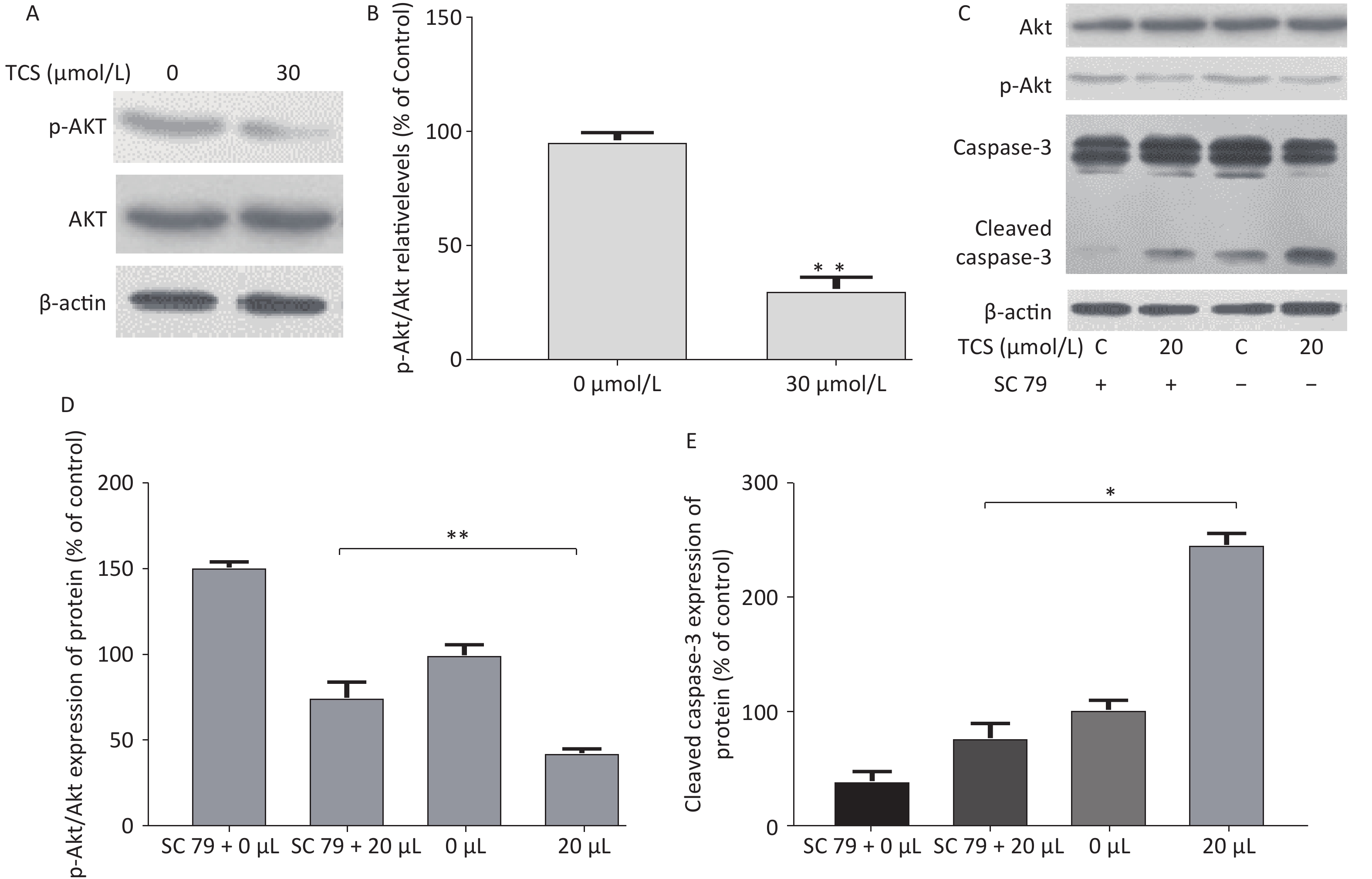
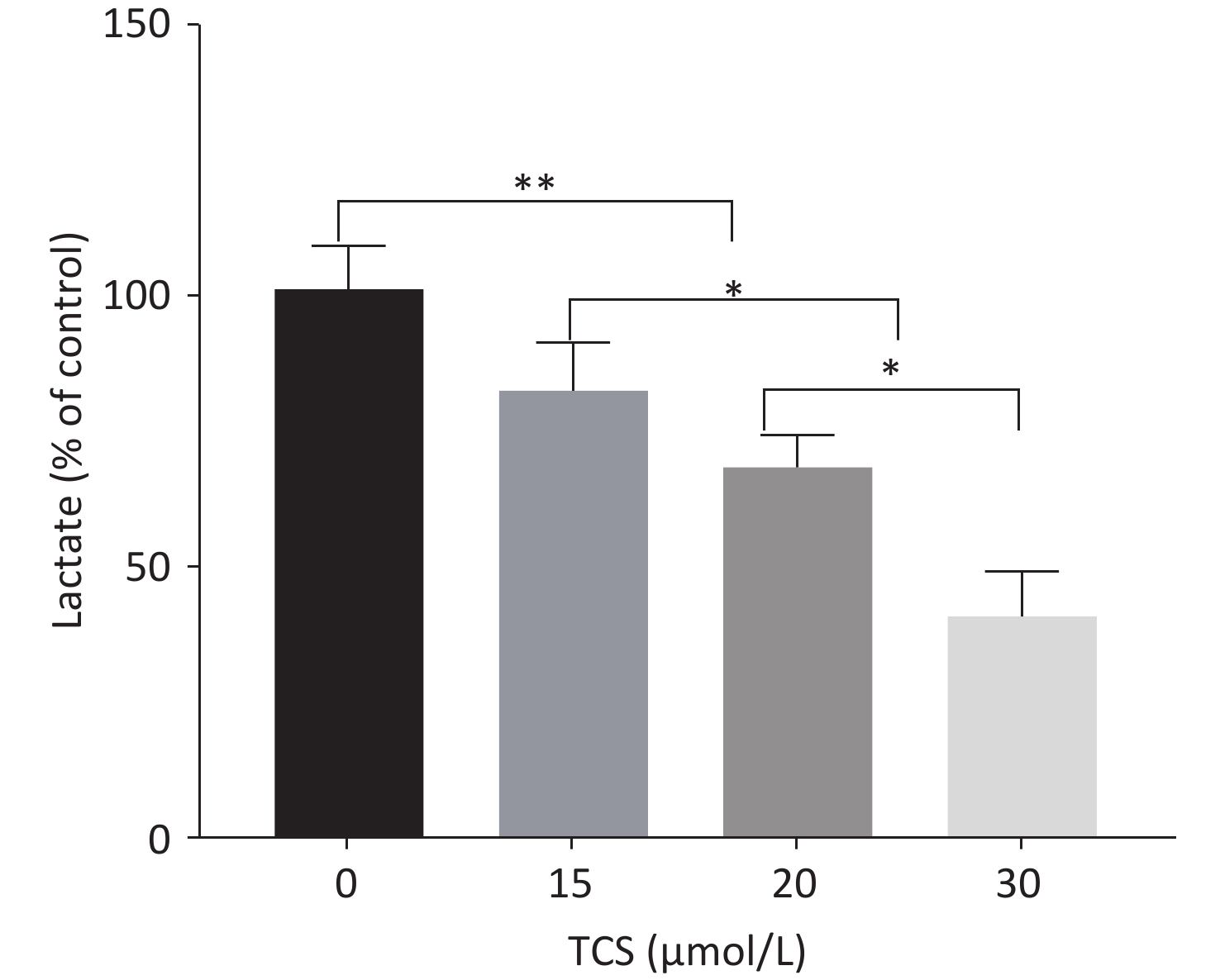
This study used a previously reported derivatization-gas chromatography-mass spectrometry method[6] to detect lactate in the supernatant (excluding lactate content in fetal bovine serum). The proportion of lactic acid in the supernatant was detected after 48 h in response to different concentrations of TCS (15, 20, and 30 µmol/L) were 81.6%, 67.7%, 40.5% (vs. control group, P < 0.01) (Supplementary Figure S2 available in www.besjournal.com). Lactate in the supernatant is often used as an indicator of cell glycolysis. The results show that compared to the control group, the 24 h 30 µmol/L TCS treatment significantly inhibited the expression of p-Akt (phospho-serine/threonine-protein kinase, p-Akt) in HRGECs, and the p-Akt level decreased to 30.2% (Figure 3A).
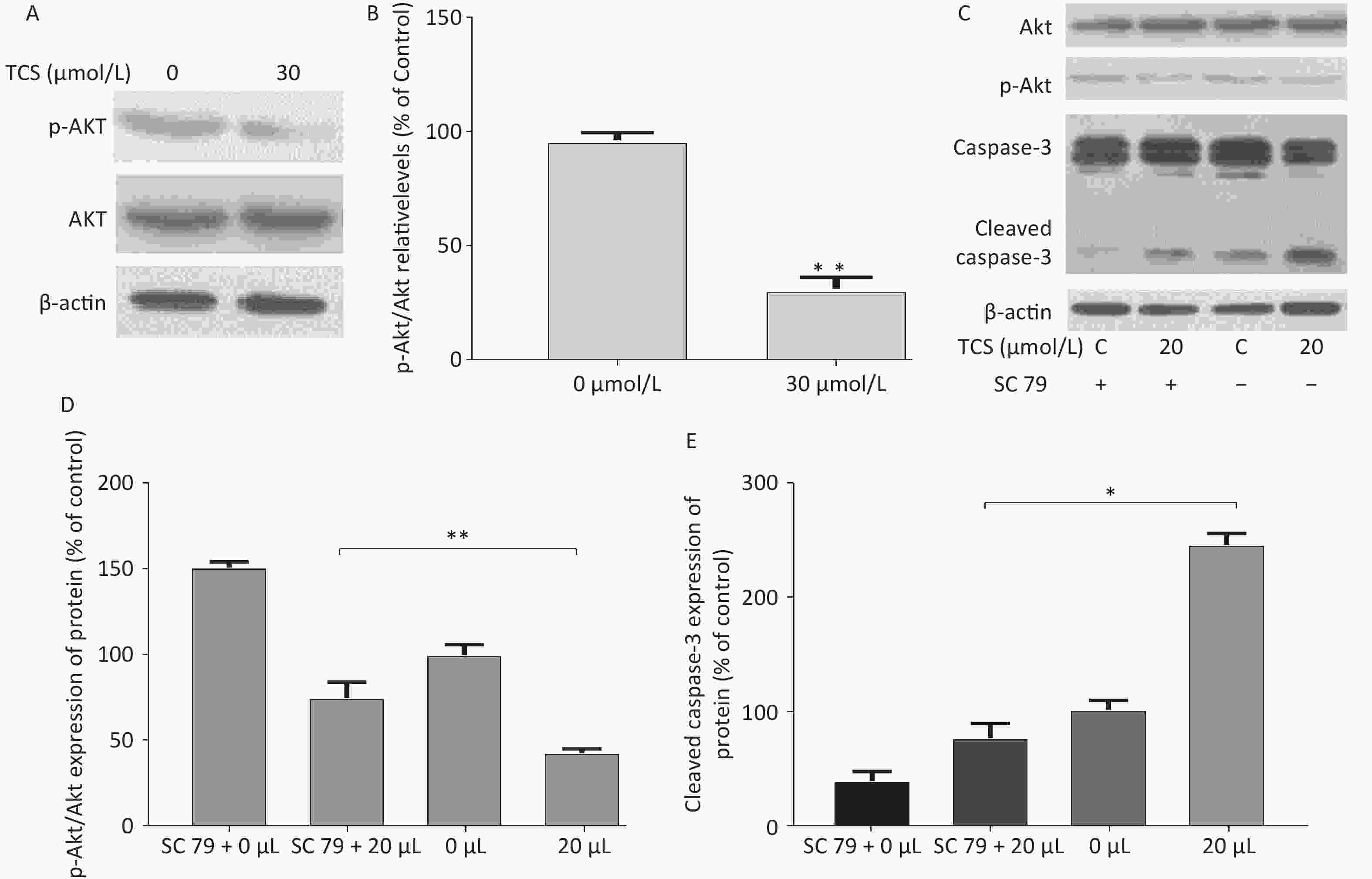
Figure 3. p-Akt/Akt and caspase-3 expression by HRGECs induced by TCS on western blot. Representative images of p-Akt/Akt expression after incubation with TCS are shown in the electrophoretogram (A) and (B). Effects of TCS on the expression of p-Akt, Akt, caspase-3, and cleaved caspase-3 after intervention with sc 79 are shown in (C) and (D, E). Data in the bar charts are mean ± SD, n = 6. *P < 0.05 significantly different between the groups, **P < 0.01 significantly different between the groups.
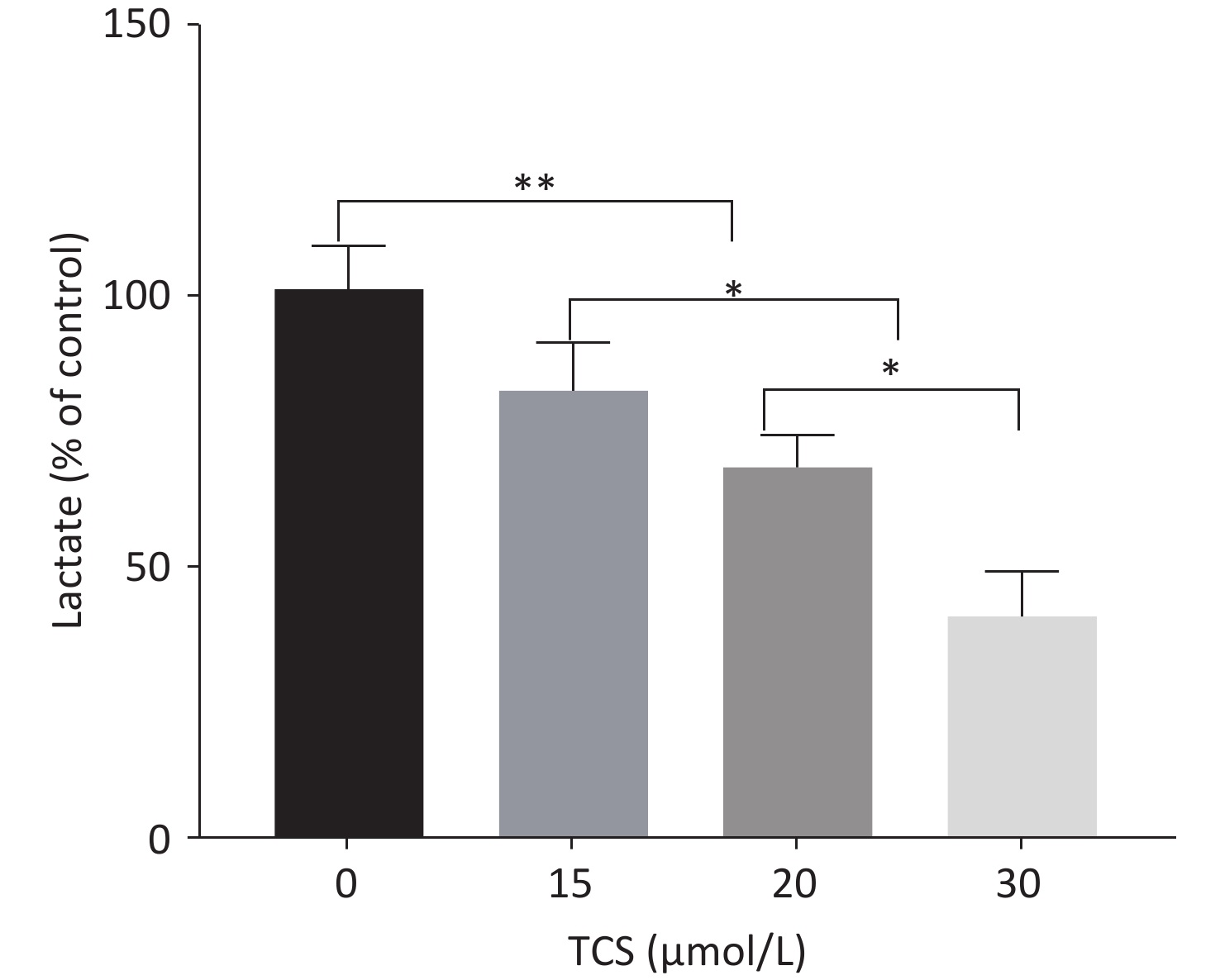
Figure S2. The ratio of lactic acid level in the medium supernatant was determined.
In addition, to further investigate the effect of the PI3K/Akt pathway on apoptosis, sc79 as a specific activator of Akt was used in subsequent experiments. The protein expression levels of cleaved caspase-3 in different groups (sc 79, 20 µmol/L + sc 79, 20 µmol/L) were 35.8%, 76.2%, and 243.3%, respectively (vs. control group, P < 0.01) (Figure 3B). These findings suggest that TCS may downregulate p-Akt expression, thereby inhibiting glycolysis and inducing activation of cleaved caspase-3. Caspase-3 apoptosis signaling may be involved in TCS-induced apoptosis of HRGECs in this study.
Notably, TCS caused significant differences in glucose metabolism between cultured normal and cancer cells. Zhang Min (2020) suggested that a high-concentration TCS (50 µmol/L for 24 h) treatment increased the expression of ROS in human umbilical vein endothelial cells in vitro and stimulated injury of human vascular endothelial cells by inhibiting the expression of p-Akt/Akt[7]. In contrast, some experiments have shown that TCS generally promotes Akt expression in cancer cells. Liu Jieyu reported that a 20 µmol/L TCS treatment for 24 h significantly increased glucose consumption and lactic acid production by mouse glioma cells (BV-2) and suggested that TCS can induce glycolysis in cancer cells through Akt-related signing pathways[8]. It is well known that Akt (serine/threonine-protein kinase, Akt) plays an important role in regulating many downstream pathways, such as the glycolysis/gluconeogenesis and apoptosis pathways. These differences in internal p-Akt expression after TCS treatment may be related to the different metabolic processes of normal and cancer cells. Different from normal cells, cancer cells treated with TCS rely more on glycolysis. Glycolysis is a poorly efficient process to obtain energy, but it can provide cancer cells with a variety of essential metabolites, such as glutamine. These findings show that TCS is deeply involved in cell proliferation, growth, metabolism, and carcinogenesis through the Akt-related signaling pathway. Moreover, pathological or acute stress conditions, such as mitochondrial oxidative stress injury, lead to mitochondrial electron transport chain leakage, excessive ROS generation, the release of cytochrome c[9], and possibly the induction of autophagy by depolarization of the mitochondria[10]. The release of cytochrome c from the mitochondria may activate caspase-3, leading to cell apoptosis.
In conclusion, we investigated the effects of TCS exposure on HRGECs by analyzing ROS production, the decrease in the MMP, apoptosis, and activation of autophagy. Exposure to TCS led to mitochondrial dysfunction in HRGECs. These findings provide the first evidence of the adverse effects of TCS exposure on renal function. The current results support the hypothesis that Akt-caspase-3 is an essential factor in HRGEC apoptosis. These findings should be confirmed in future studies. This study provides a theoretical basis for further clarifying the nephrotoxic characteristics of TCS.
No potential conflicts of interest are disclosed.
MA Yan conceived the design, and CHEN Chen and HUO Jun Sheng supervised the study. WANG Jing Bo, CHENG Jia Li, and SHEN Shi performed molecular biology experiments. CHEN Xi wrote the manuscript.
HTML
 21388Supplementary Materials.pdf
21388Supplementary Materials.pdf
|

|

 Quick Links
Quick Links
 DownLoad:
DownLoad: