
-
Thalassemia is an autosomal recessive disease involving moderate or severe hemolytic anemia caused by insufficient or absent globin chains. It is widespread in the Mediterranean, Southeast Asia and southern China[1]. The most common forms are α-thalassemia (OMIM: #604131) and β-thalassemia (OMIM: #613985), which affect the synthesis of the α- and β-globin subunits, respectively[2]. An estimated 1%–5% of the global population carries a genetic thalassemia mutation[3]. The frequency of thalassemia gene carriage in southern China is as high as 3%–24%[4]. People carrying thalassemia variants are concentrated in Guangdong, Guangxi, and Hainan Provinces in China[5, 6]. Notably, as a consequence of recent massive population migrations, thalassemia is no longer restricted to traditional high-incidence regions and is now a relatively common clinical problem in areas including Hunan, Jiangxi, and Fujian. The genetic spectrum of thalassemias in Hunan Province shows high complexity and diversity and remains to be clearly elucidated. Hence, we performed a large-scale next-generation sequencing survey of reproductive age couples covering 42 districts and counties in 14 cities in Hunan Province.
-
A cross-sectional study was performed among couples of reproductive age receiving routine health examinations between June 2020 and April 2021 in 14 cities of Hunan Province. One or both members of each couple were registered in Hunan Province.
Multi-stage stratified random sampling was used. The total required sample size was summed after estimation of the sample size for 14 cities. The number of samples for each city was calculated with a formula[7]. The thalassemia carrier rate of each city was estimated on the basis of previous studies and the thalassemia carrier rate of neighboring provinces[8-11]. The number of samples was increased 15% for each city to avoid data deficiencies (Supplementary Table S1, available in www.besjournal.com).
City Total population* Thalassemia carrier rate, % Samples Changsha 6,192,142 5 2,098 Zhuzhou 5,036,241 8 1,212 Xiangtan 6,720,139 5 2,121 Hengyang 5,223,156 8 918 Shaoyang 8,015,293 8 1,272 Yueyang 7,859,248 3 3,302 Changde 4,131,011 3 3,572 Zhangjiajie 4,324,888 4 2,492 Yiyang 5,478,790 4 2,649 Chenzhou 4,658,278 11 904 Yongzhou 3,026,515 12 810 Huaihua 6,045,994 8 1,288 Loudi 2,904,968 5 2,083 Xiangxi 1,687,625 8 1,225 Total 71,304,288 25,946 Note. *Population data are from the sixth census in China. Table S1. Sampling distribution of 14 cities in Hunan Province
The required samples of 14 cities were distributed to districts and counties according to their proportion of population. The population ratio of districts to counties in the entire province was approximately 1:2. One district and two counties were randomly chosen in each city, and a total of 42 districts and counties were included in the study. Carrier rate ratio of each city was calculated by carrier rate of each city multiplying by their population rate (Carrier rate ratio = Carrier rate × population rate). Total carrier rate was equal to the sum of the carrier ratio of all cities.
A total of 25,946 individuals of reproductive age receiving routine health examinations between June 2020 and April 2021 were included in this epidemiological survey of thalassemia in Hunan Province. All participants were collected through a random sampling method and came from 14 cities across Hunan Province. The ages of the participants ranged from 20 to 42 years. Informed written consent was provided by all participants after a clear description of the study objectives was provided. This study was approved by the medical ethics committee of the Hunan Provincial Maternal and Child Health Care Hospital (202030).
-
Blood samples (3 mL) from all participants were collected in EDTA tubes. Complete blood cell count, hemoglobin electrophoresis and molecular parameters were assessed simultaneously. Hematological parameters were determined with an automated hematology analyzer, and hemoglobin analysis was performed with capillary electrophoresis (Sebia, France and Helena, USA).
-
DNA Extraction Genomic DNA was extracted from whole blood with a Q Kingfisher Flex system (Thermo Scientific, Rockford, IL) and isolated with a GenMag Nucleic Acid Isolation kit (Magnetic bead method) (GenMagBio, Beijing, China). A NanoDrop-8,000 spectrophotometer (Thermo Scientific, Waltham, MA, USA) was used to quantify the concentrations of DNA samples.
Library Preparation and NGS Sequencing For the variants of the α and β globin genes (HBA1, HBA2, and HBB), five α-thalassemia associated deletions (-α3.7, -α4.2, --SEA, --FIL, and --THAI) and three β-globin gene deletions [Chinese Gγ (Aγδβ)0-thal, Southeast Asia HPFH(SEA-HPFH), and 1357 bp deletion], a tag sequence was introduced for identification, and the sequences were amplified and enriched through multiplex PCR with primers described in a patent [patent ID CN108796054A] and published article[12]. The PCR products of samples (≤ 96) were pooled for library preparation. For sequencing and library identification, the DNA sequences were supplemented with a linker sequence in each library. The detailed experimental protocol and bioinformatic analysis methods were as previously described [13]. We used a next-generation sequencing library protocol for library construction, including purified genomic DNA (Qiagen DNA Purification kit, Hilden, Germany), DNA quantification (NanoDrop 8,000 UV-vis spectrophotometer; Thermo Fisher Scientific, Waltham, MA, USA), DNA fragmentation (excluding CNV amplicons), blunt-ended fragmentation (Enzymatics kits, Qiagen, Hilden, Germany), 3’-dA overhang, paired-end adapter ligation, DNA fragment separation (CNVs not included) and size selection with magnetic beads. Finally, sequencing was performed with the paired-end tag (PE100) on a MGISEQ-2,000 chip. Positive NGS results were validated with Sanger sequencing.
Bioinformatic Analyses A bioinformatic pipeline focused on detecting Hb gene deletions and point mutations was developed. First, we filtered low quality reads and reads with sequencing adaptor contamination, and classified the clean data by using adapter information (index primer). Then, we aligned the clean data to the human genome reference sequence (hg19) in BWA software[14] and generated the final bam files with SAMtools. Each consensus sequence’s length, coverage and depth were recorded with ReSeqTools[13]. Finally, the HbVar[15] and IthaGenes[16] databases were used to annotate the detected mutation types.
α-globin Triplication The NGS suggested high risk of suspected triplication among participants, as further confirmed by an anti-3.7/4.2 multiplex-PCR method[17]. The phenotypic data were assessed with factorial ANOVA (factors including RBC, HB, MCV, MCH, Hb_ A, Hb_ A2, and HBF), and the P value indicated no statistical difference (P > 0.05).
-
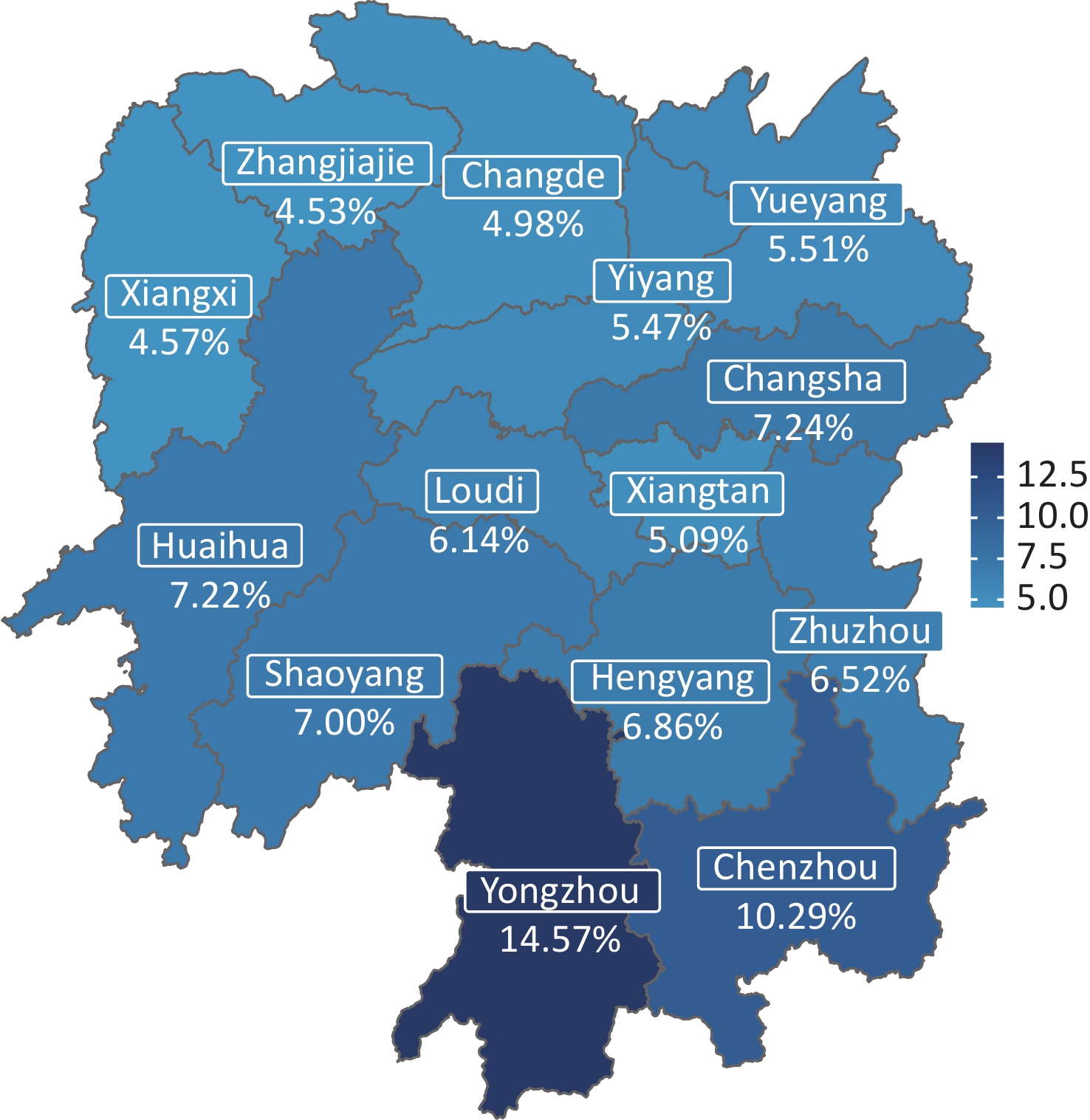
The overall carrier rate of thalassemia in Hunan Province was 7.1%. The carrier rates among the 14 cities varied between 4.53% and 14.57% (Figure 1, Table 1). The two regions with the highest carrier rate of thalassemia were Yongzhou (14.57%) and Chenzhou (10.29%), which are bordered by Guangxi Province and Guangdong Province, respectively. Changsha, the capital city, which is located in the center of Hunan Province, ranked third, with a carrier rate of 7.24%. The lowest carrier rates were detected in Zhangjiajie (4.53%) and Xiangxi Prefecture (4.57%), in the northwestern part of the province.
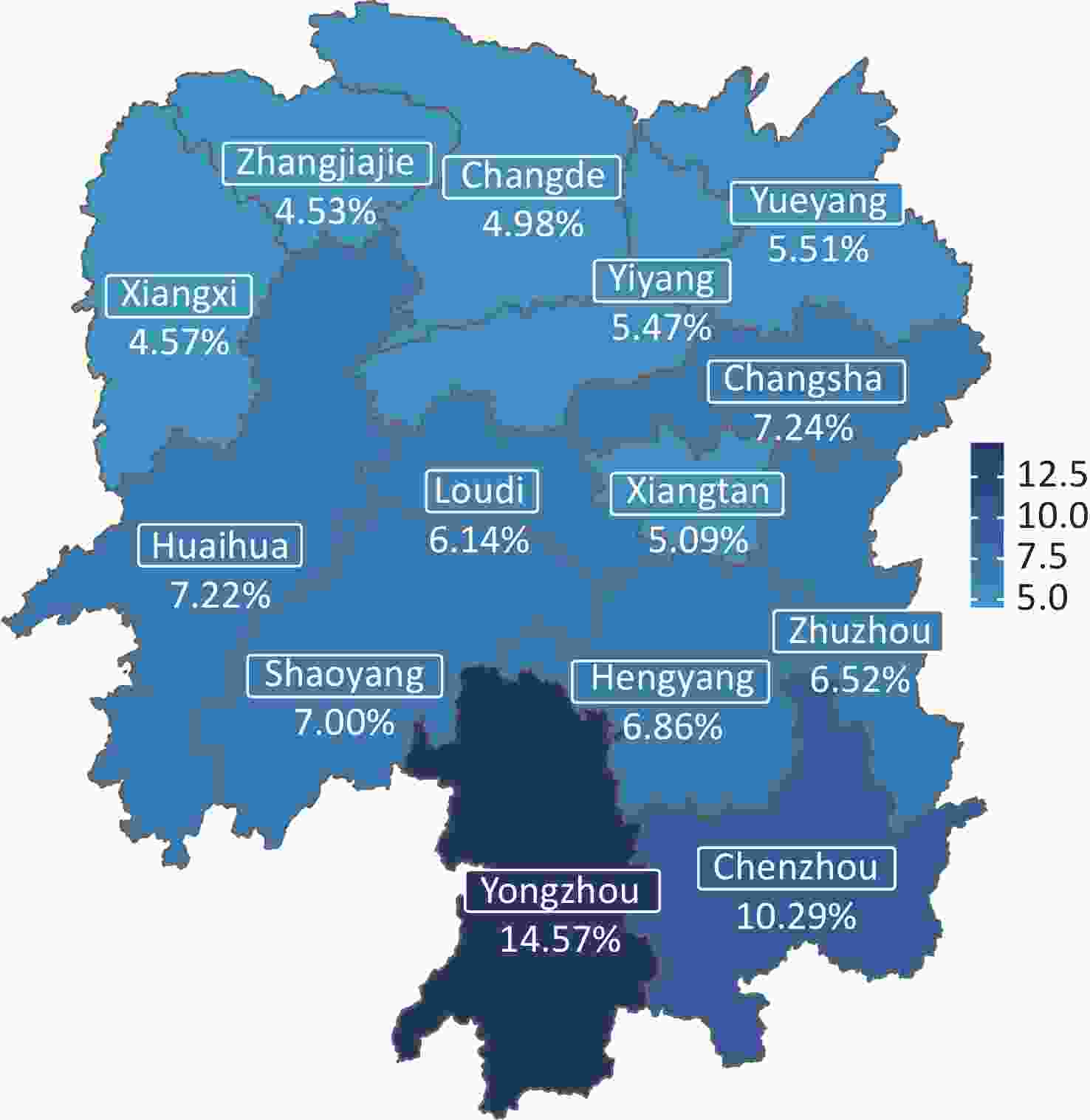
Figure 1. Geographical distribution of thalassemia carriers in Hunan Province.
Region Samples Thalassemia carriers, n (%) Population,
n (%)Carrier rate ratio*, % Total α-thalassemia β-thalassemia α- and
β-thalassemiaTotal α-thalassemia β-thalassemia α- and
β-thalassemiaYongzhou 810 118 (14.57) 75 (9.26) 41 (5.06) 2 (0.25) 5,194,275
(7.55)1.10 0.70 0.38 0.02 Chenzhou 904 93 (10.29) 66 (7.30) 26 (2.88) 1 (0.11) 4,583,531
(6.66)0.69 0.49 0.19 0.01 Changsha 2,098 152 (7.24) 118 (5.62) 32 (1.53) 2 (0.10) 6,512,131
(9.46)0.69 0.53 0.14 0.01 Huaihua 1,288 93 (7.22) 72 (5.59) 21 (1.63) 0 (0.00) 5,092,480
(7.40)0.53 0.41 0.12 0.00 Shaoyang 1,272 89 (7.00) 54 (4.25) 33 (2.59) 2 (0.16) 7,927,558
(11.52)0.81 0.49 0.30 0.02 Hengyang 918 63 (6.86) 40 (4.36) 21 (2.29) 2 (0.22) 7,902,022
(11.48)0.79 0.50 0.26 0.03 Zhuzhou 1,212 79 (6.52) 49 (4.04) 27 (2.23) 3 (0.25) 3,898,444
(5.66)0.37 0.23 0.13 0.01 Loudi 2,083 128 (6.14) 78 (3.74) 46 (2.21) 4 (0.19) 4,323,502
(6.28)0.39 0.24 0.14 0.01 Yueyang 3,302 182 (5.51) 129 (3.91) 51 (1.54) 2 (0.06) 5,648,840
(8.21)0.45 0.32 0.13 0.00 Yiyang 2,649 145 (5.47) 103 (3.89) 42 (1.59) 0 (0.00) 4,307,933
(6.26)0.34 0.24 0.10 0.00 Xiangtan 2,121 108 (5.09) 66 (3.11) 39 (1.84) 3 (0.14) 2,888,294
(4.20)0.21 0.13 0.08 0.01 Changde 3,572 178 (4.98) 132 (3.70) 44 (1.23) 2 (0.06) 6,225,913
(9.05)0.45 0.33 0.11 0.01 Xiangxi 1,225 56 (4.57) 41 (3.35) 14 (1.14) 1 (0.08) 2,845,811
(4.13)0.19 0.14 0.05 0.00 Zhangjiajie 2,492 113 (4.53) 86 (3.45) 27 (1.08) 0 (0.00) 1,478,149
(2.15)0.10 0.07 0.02 0.00 Total 25,946 1,597 (6.16) 1,109 (4.27) 464 (1.79) 24 (0.09) 68,828,883
(100)7.10 4.83 2.15 0.12 Note. *Carrier rate ratio = Carrier rate × population rate. Total carrier rate was equal to the sum of the carrier ratio of all cities. Table 1. Carrier rate of α-thalassemia, β-thalassemia, and composite α- and β-thalassemia among 14 cities in Hunan Province
The carrier rates of α-thalassemia, β-thalassemia, and α combined with β-thalassemia were 4.83%, 2.15% and 0.12%, respectively (Table 1). The top three regions with high α-thalassemia carrier rates were Yongzhou (9.26%), Chenzhou (7.30%) and Changsha (5.62%). β-thalassemia was encountered primarily in Yongzhou (5.06%), Chenzhou (2.88%) and Shaoyang (2.59%).
-
In a total of 25,946 individuals screened for thalassemia, 1,597 were found to be thalassemia carriers, of whom 1,109 were α-thalassemia carriers, 464 were β-thalassemia carriers, and 24 others were carriers of both α- and β-thalassemia. Among α-thalassemia carriers, 28 different genotypes were detected, and -α3.7/αα was the most frequent genotype, accounting for more than half (50.23%).
Other highly prevalent genotypes of α-thalassemia were --SEA/αα, -α4.2/αα, αWSα/αα, αCSα/αα, and αQSα/αα. Overall, these six genotypes accounted for 96.30% of all α-thalassemia genotypes (Table 2). Rare α-globin genotypes were also identified and found to account for 2.43% of all genotypes (27/1,109). The top three uncommon genotypes were αCD30α/αα (0.45%), αCD108α/αα (0.45%), and αCD61α/αα (0.36%). We identified 25 different β-thalassemia genotypes in 464 participants. The three most frequent mutations were seen in genotypes βIVS-II-654/βN (28.23%), βCD41-42/βN (27.37%), and βCD17/βN (13.36%). Thirteen rare β-globin genotypes were identified, which accounted for 6.68% (31/464) of the total, of which β-50/βN accounted for the highest proportion (2.37%). Among the 24 participants who were carriers for both α and β-thalassemia, 83.3% (20/24) of the genotypes were caused by coinheritance of common α-globin gene deletions (-α3.7/αα, --SEA/αα, or -α4.2/αα) with a β-globin gene point mutation (Table 2).
α-thalassemia β-thalassemia α+β thalassemia Genotype N % Genotype N % Genotype N % -α3.7/αα 557 50.23 βIVS-II-654/βN 131 28.23 -α3.7/αα; βCD41-42/βN 4 16.67 --SEA/αα 312 28.13 βCD41-42/βN 127 27.37 -α3.7/αα; βIVS-II-654/βN 3 12.50 -α4.2/αα 108 9.74 βCD17/βN 62 13.36 --SEA/αα; βCD41-42/βN 2 8.33 αWSα/αα 55 4.96 βCD71-72/βN 22 4.74 -α3.7/αα; β-28/βN 1 4.17 αCSα/αα 21 1.89 βCD26/βN 21 4.53 -α3.7/αα; β5'UTR+43 to +40/βN 1 4.17 αQSα/αα 15 1.35 β-28/βN 20 4.31 -α3.7/αα; βCD17/βN 1 4.17 αCD30α/αα 5 0.45 β5'UTR+43 to +40/βN 17 3.66 -α3.7/αα; βCD71-72 /βN 1 4.17 αCD108α/αα 5 0.45 βCD27-28/βN 12 2.59 -α4.2/αα; βCD17/βN 1 4.17 αCD61α/αα 4 0.36 β-50/βN 11 2.37 -α4.2/αα; βIVS-II-654/βN 1 4.17 Hb Phnom Penh 3 0.27 βCD43/βN 10 2.16 --SEA/αα; β-28/βN 1 4.17 -α3.7/--SEA 3 0.27 β-29/βN 7 1.51 --SEA/αα; β-29/βN 1 4.17 αWSα/-α3.7 3 0.27 SEA-HPFH 4 0.86 --SEA/αα; β5'UTR+43 to +40 /βN 1 4.17 αIVS-I-38α/αα 2 0.18 β-31/βN 3 0.65 --SEA/αα; βCD26/βN 1 4.17 -α3.7/-α3.7 2 0.18 βCD14-15/βN 3 0.65 --SEA/αα; βIVS-II-654/βN 1 4.17 αCAP+29α/αα 1 0.09 βCAP+8/βN 2 0.43 αWSα/αα; βIVS-II-654 /βN 1 4.17 Hb Agrinio 1 0.09 ChineseGgamma 2 0.43 αCSα/αα; βCD17 /βN 1 4.17 Hb Cervantes 1 0.09 βIVS-II-848/βN 2 0.43 αCSα/αα; βIVS-II-654 /βN 1 4.17 Hb Iberia 1 0.09 β5'UTR+43 to +40/βIVS-II-654 1 0.22 αCD30α/αα; βIVS-II-654/βN 1 4.17 αWSα/αWSα 1 0.09 β-56/βN 1 0.22 αIVS-I-117α/αα 1 0.09 β-72/βN 1 0.22 αCD31α/αα 1 0.09 βnt-77/βN 1 0.22 -α3.7/-α4.2 1 0.09 βCD20-21/βN 1 0.22 -α4.2/--SEA 1 0.09 Hb Knossos 1 0.22 --THAI/αα 1 0.09 βIVS-II-643/βN 1 0.22 αQSα/-α3.7 1 0.09 βpolyA/βN 1 0.22 -α4.2 1 0.09 αCSα/-α4.2 1 0.09 αWSα/-α4.2 1 0.09 Total 1,109 100 Total 464 100 Total 24 100 Note. *HGVS names of all mutations are shown in Supplementary Table S2 (available in www.besjournal.com). Table 2. Distribution of α- and β-thalassemia genotypes in Hunan Province
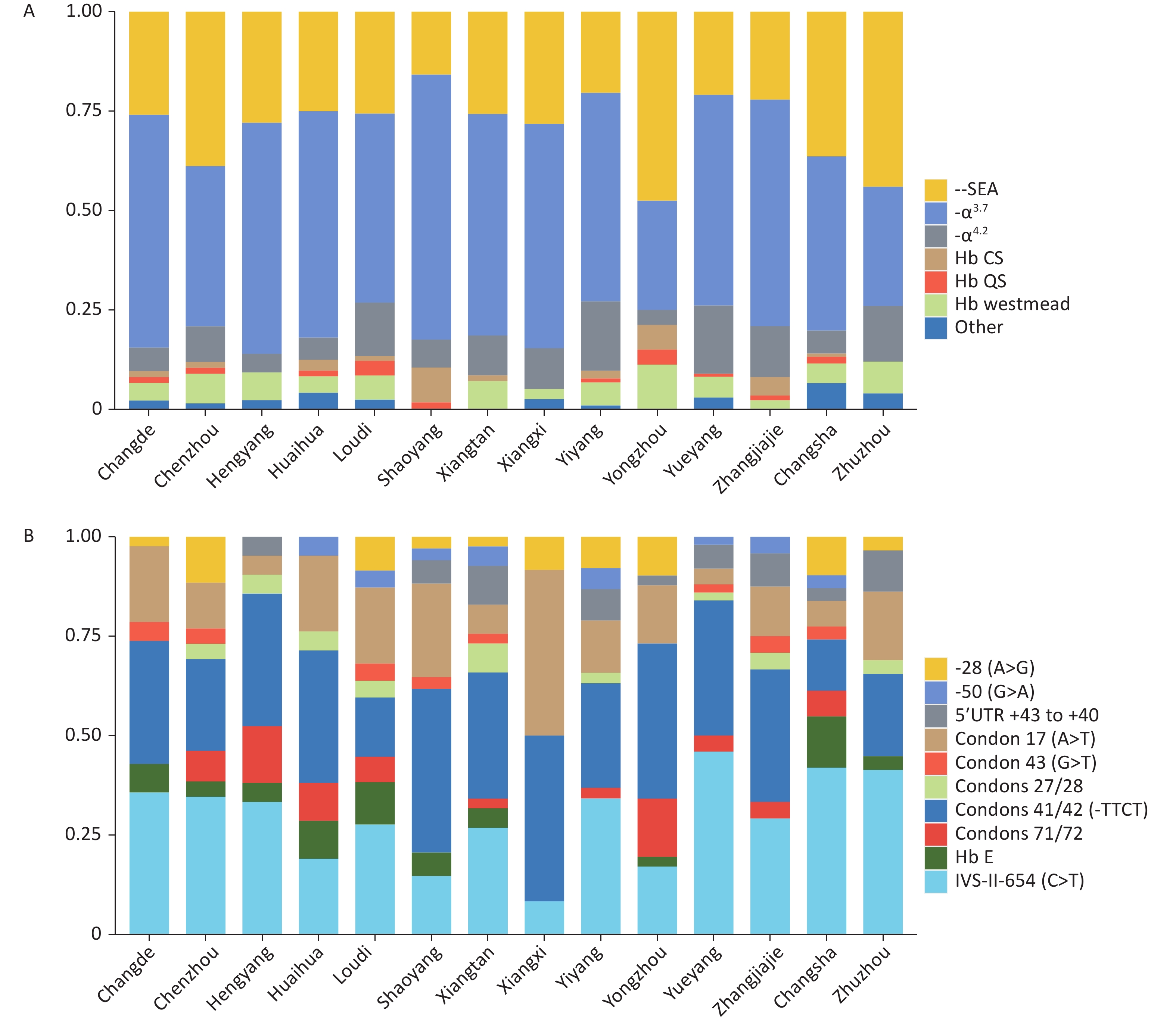
Significant differences in genotype composition were observed among cities in Hunan Province. -α3.7 was the main α-thalassemia genotype in most cities. --SEA was the main genotype in Yongzhou and Zhuzhou, accounting for 47.50% and 44%, respectively (Figure 2A). βIVS-II-654 and βCD41-42 were the most common β-thalassemia genotypes in the entire province (Figure 2B).
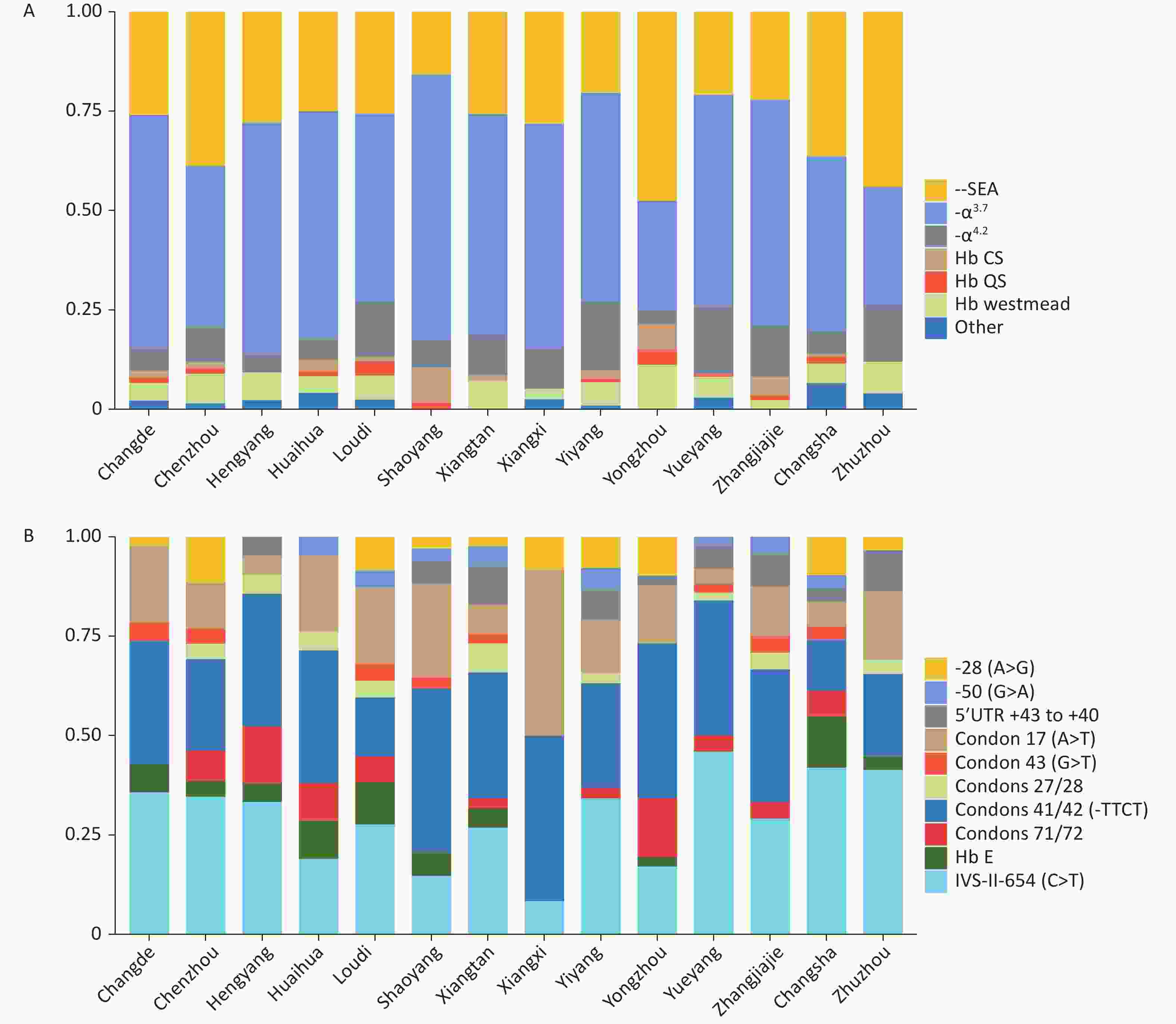
Figure 2. Allele frequencies of thalassemia in different regions of Hunan Province. (A) Allele frequency of α-thalassemia among 14 cities in Hunan Province. (B) Allele frequency of common mutations of β-thalassemia among 14 cities in Hunan Province.
-
A total of 127 participants were carriers of abnormal hemoglobin variants, with a rate of 0.49%. Hb New York (22.83%), Hb Hekinan II (13.39%), Hb Owari (α1) (7.87%), Hb J-Bangkok (β) (5.51%) and Hb Zengcheng (β) (5.51%) had the highest incidence among the abnormal hemoglobin variants in Hunan Province (Supplementary Table S3, available in www.besjournal.com).
Mutation type Number Ratio, % Mutation type Number Ratio, % Hb Zambia 1 0.79 Hb J-Bangkok 7 5.51 Hb Agrinio 1 0.79 Hb Knossos 1 0.79 Hb Athens-GA 1 0.79 Hb Manitoba II 1 0.79 Hb Broomhill 4 3.15 Hb Mantes-La-Jolie 1 0.79 Hb Cervantes 1 0.79 Hb Nevers 1 0.79 Hb Fuchu-I 1 0.79 Hb New York 29 22.83 Hb Fulwood 1 0.79 Hb Owari 10 7.87 Hb G-Coushatta 6 4.72 Hb Penang 2 1.57 Hb G-Honolulu 3 2.36 Hb Phnom Penh 3 2.36 Hb Gorwihl 1 0.79 Hb Port Phillip 1 0.79 Hb Groene Hart 3 2.36 Hb Pretoria(α2) 1 0.79 Hb G-taipei 3 2.36 Hb Q-Thailand 3 2.36 Hb G-Waimanalo 1 0.79 Hb Shenyang 1 0.79 Hb Hachioji 3 2.36 Hb Shizuoka 1 0.79 Hb Hamilton 1 0.79 Hb Tokoname 1 0.79 Hb Handsworth 1 0.79 Hb Ty Gard 1 0.79 Hb Haringey 1 0.79 Hb Yaounde 1 0.79 Hb Hekinan II 17 13.39 Hb Zengcheng 7 5.51 Hb I 2 1.57 Hb Zurich-Albisrieden 1 0.79 Hb Iberia 1 0.79 Hb Zurich-Langstrasse 1 0.79 Table S3. Distribution of abnormal hemoglobin mutations
-
Triplication of the α-globin gene (αααanti3.7 and αααanti4.2) was detected in 71 participants of 3,569 randomly selected samples, with an estimated rate of 1.99%. They included 34 with αα/αααanti4.2 and 37 with αα/αααanti3.7; in the latter group, one participant had combined -α3.7 deletion (αααanti3.7/-α3.7), and two participants had β-thalassemia. The phenotypic characteristics of all cases are shown in Table 3. (Supplementary Table S4, available in www.besjournal.com).
Genotype Number Hematological phenotype RBC (× 1012/L) Hb (g/L) MCV (fL) MCH (pg) Hb A (%) Hb A2 (%) Hb F (%) αααanti3.7/αα, βN/βN 34 4.63 ± 0.53 143.03 ± 20.13 91.84 ± 5.49 30.84 ± 2.26 97.03 ± 0.64 2.70 ± 0.25 0.27 ± 0.60 αααanti4.2/αα, βN/βN 34 4.75 ± 0.47 146.15 ± 15.19 91.34 ± 3.72 30.80 ± 1.35 97.06 ± 0.49 2.77 ± 0.22 0.17 ± 0.42 αααanti3.7/-α3.7, βN/βN 1 4.33 131 88.2 30.3 97.3 2.4 0.3 αααanti3.7/αα, βN/βCD14-15 1 5.36 109 64 20.3 93.7 5.5 0.8 αααanti3.7/αα, βN/βIVS-II-654 1 5.62 114 67.5 20.2 92.6 4.9 2.5 Table 3. Hematological phenotypic analysis of α triplication
-
Herein, we conducted a large-scale, large-sample, province-wide study. The overall carrier rate of thalassemia was 7.1%, including 4.83% for α-thalassemia, 2.15% for β-thalassemia, and 0.09% for α- and β-thalassemia (Table 1). These values are lower than the reported carrier rates in the six provinces in South China, Guangdong, Guangxi, Hainan, Yunnan, Jiangxi, and Guizhou[8, 18]. The carrier rates of thalassemia in Hunan and Fujian were similar[10]. However, the current values were 1.74 times higher than Hunan Province’s overall carrier rate first reported in 2007 (4.13%). The carrier rate of thalassemia in Changsha (7.24%) was higher than that in a previous study (4.54%)[19]. Our study more accurately reflects the carrier rate of thalassemia in various areas, because we performed genetic testing with the NGS method on all recruited individuals, whereas previous studies have used traditional testing methods in individuals with positive hematologic phenotypes.
Our results also indicated a higher carrier rate of thalassemia in southern than northern Hunan Province, in agreement with the general geographical distribution of thalassemia in China. The higher carrier rate in Yongzhou (14.57%) and Chenzhou (10.29%), located in the southern region, was partly due to their geographical proximity to Guangdong and Guangxi; people in this area migrate to and from Guangdong and Guangxi (Table 1). In our study, the carrier rate of thalassemia in the Chenzhou region (10.29%) was close to that in a 2018 study (10.78%)[15].
Previous research has revealed that, in contrast to --SEA/αα, which is most common in Guangxi, Guangdong and Yunnan [20,21], -α3.7/αα is the most common α-thalassemia genotype in Hunan[18, 22]. In addition to the six common genotypes (-α3.7/αα, --SEA/αα, -α4.2/αα, αWSα/αα, αCSα/αα, and αQSα/αα), we identified 13 rare genotypes of α-globin genes, accounting for 2.43% of all genotypes (27/1,109), including CD108 (ACC>AAC), CAP +29(G>C), Hb Agrinio and Hb Cervantes, which had not previously been reported in China. Among 489 β-thalassemia carriers, 25 genotypes were identified, and the most common mutations were βIVS-II-654/βN (28.23%), βCD41-42/βN (27.37%), and βCD17/βN (13.36%). The top three β-thalassemia mutations in Hunan were similar to those in the neighboring Guizhou Province but differed from those in Guangdong, Guangxi, Hainan, and Yunnan[2].
In contrast to PCR-RDB method, 13 additional rare genotypes of β-globin genes has been identified by NGS method accounting for 6.68% of all genotypes. β-50/βN (2.37%) was the most prevalent rare genotype, and CAP+8 (C>T), IVS-II-848 (C>T), -56 (G>C), beta nt-77 (G>C), codon 20/21 (-TGGA), and Hb Knossos had not previously been reported in China. These results indicated that the type and frequency of thalassemia gene mutations in Hunan Province have clear regional specificity, and that screening and detection of rare gene mutations should be emphasized.
We found 127 cases of abnormal hemoglobin mutation in 25,946 individuals, with a carrier rate of 0.49%, a value higher than previously reported in Guangdong (0.358%) and Fujian (0.26%)[10, 16, 19, 22-24]. We detected 40 different abnormal hemoglobin variants; this is the first report of simultaneous detection of a wide range of hemoglobin variants in China. Most detected variants were apparently silent and did not affect hemoglobin function and stability, or cause clinical effects. However, these findings enrich the known genetic landscape of hemoglobinopathies in Hunan Province.
The carrier rate of α-globin triplication in Hunan Province was 1.99%, a rate higher than reported previously in five southern provinces[13,18,20]. We observed no significant abnormalities in the hematological phenotype of the simple triad in our study, in agreement with previous research[21].
According to many reports, coinheritance of α-globin gene triplication with β-thalassemia mutations may lead to moderate to severe anemia, and some patients may require intermittent blood transfusion therapy[25]. However, in this study, two cases of heterozygous β-thalassemia combined with the α-triad showed only mild anemia. A related study in Hong Kong, China has also reported two cases with β0 mutation combined with α triplication showing a phenotype of only mild thalassemia[26]. An Iranian study of 67 cases with similar genotypes has reported that only four cases had hemoglobin lower than 90 g/L, and no cases required blood transfusion[27]. These results suggest that the clinical phenotypes of people with α-globin triplication combined with heterozygous β-thalassemia are quite diverse, and therefore, must be fully elucidated in clinical work.
In conclusion, this large-scale, large-sample and province-wide study is the first comprehensive investigation of the spectrum of thalassemia mutations in Hunan Province. Our findings showed the great complexity and diversity of thalassemia gene mutations in the Hunan population. The findings should be important for genetic counseling and the prevention of severe thalassemia in this region.
-
α-thalassemia mutation β-thalassemia mutation Common name HGVS name Common name HGVS name -α3.7/ -α3.7/ IVS-II-654 (C>T) HBB:c.316-197C>T --SEA/ --SEA/ Codons 41/42 (-TTCT) HBB:c.126_129delCTTT -α4.2/ -α4.2/ Codon 17 (A>T) HBB:c.52A>T αWSα/ HBA2:c.369C>G Codons 71/72 (+A) HBB:c.216_217insA αCSα/ HBA2:c.427T>C -28 (A>G) HBB:c.-78A>G αQSα/ HBA2:c.377T>C Hb E HBB:c.79G>A Alpha2 Codon 30 del GAG HBA2:c.91_93delGAG 5’UTR+43 to +40 (-AAAC) HBB:c.-11_-8delAAAC Codon 108 (ACC>AAC) HBA2:c.326C>A Codons 27/28 (+C) HBB:c.84_85insC Codon 61 (AAG>TAG) HBA2:c.184A>T -50 (G>A) HBB:c.-100G>A Hb Phnom Penh HBA1:c.354_355insATC Codon 43 (G>T) HBB:c.130G>T CAP +29 (G>C) HBA1:c.-9G>C -29 (A>G) HBB:c.-79A>G Hb Agrinio HBA2:c.89T>C SEA-HPFH SEA-HPFH Hb Cervantes HBA2:c.356C>T -31 (A>C) HBB:c.-81A>C Hb Iberia HBA2:c.313T>C Codons 14/15 (+G) HBB:c.45_46insG IVS-I-117 (G>A) HBA2:c.96-1G>A CAP +8 (C>T) HBB:c.-43C>T α2 Codon 31 (AGG>AAG) HBA2:c.95G>A ChineseGgamma ChineseGgamma --THAI/ --THAI/ IVS-II-848 (C>T) HBB:c.316-3C>T -56 (G>C) HBB:c.-106G>C -72 (T>A) HBB:c.-122T>A beta nt-77 (G>C) HBB:c.-127G>C Codon 20/21 (-TGGA) HBB:c.62_65delTGGA Hb Knossos HBB:c.82G>T Poly A (A>G) HBB:c.*111A>G Table S2. HGVS names of all mutations
Hematological phenotype Df Sum Sq Mean Sq F value Pr (>F) RBC 1.00 0.25 0.25 0.94 0.34 HB 1.00 0.03 0.03 0.11 0.74 MCV 1.00 0.04 0.04 0.16 0.69 MCH 1.00 0.64 0.64 2.46 0.12 Hb_A 1.00 0.02 0.02 0.08 0.78 Hb_A2 1.00 0.18 0.18 0.71 0.40 HBF 1.00 0.15 0.15 0.56 0.46 Residuals 60.00 15.69 0.26 NA NA Table S4. Analysis of hematological phenotypes by factorial ANOVA
HTML
Population Samples and Statistical Analysis
Hematological Analyses
Genetic Analysis
Carrier Rate of Thalassemia in Hunan Province
Mutation Spectrum of α- and β-thalassemia Genes in Hunan
Results of Structural Hemoglobin Variants
Detection of α Triplication and Analysis of Hematological Phenotype
CONFLICTS OF INTEREST The authors declare no conflicts of interest.
&These authors contributed equally to this work.
 22189Supplementary Materials.pdf
22189Supplementary Materials.pdf
|

|

 Quick Links
Quick Links
 DownLoad:
DownLoad: