
-
Since Legionella pneumophila (L. pneumophila) was identified in 1976 as the causative microorganism for severe pneumonia termed Legionnaires Disease (LD), more than 65 species belonging to the genus Legionella have been isolated worldwide[1]. Approximately 20 Legionella spp. have been documented to be pathogenic in humans, primarily leading to transient flu-like illness (Pontiac fever) or rapid and potentially fatal pneumonia (LD). As the most common cause of legionellosis, L. pneumophila is responsible for nearly 95% of global LD cases, while other non-L. pneumophila species, such as L. longbeachae, L. bozemanii, and L. micdadei, are sporadically isolated from patients, but the isolation rates vary by geography and the patient population[1-3]. In particular, L. micdadei, or Tatlockia micdadei, which was discovered by Hugh Tatlock in 1943, and was also known as the Pittsburgh pneumonia agent in the 1980s, has been recorded in a limited number of respiratory tract infections and rare cases of extra-pulmonary infections, such as secretory diarrhea, endocarditis, brain abscess, and prosthetic joint infection[3].
Legionella spp. are fastidious, aerobic bacteria and facultative intracellular parasites of human macrophages or free-living amoebae. Despite their extraordinary nutritional requirements and obligate culture conditions, dozens of Legionella spp. have been recovered and identified from a variety of environmental and clinical specimens during routine etiological surveillance or outbreak investigations in the past[1,2]. In China, the first L. micdadei infections were reported among dozens of construction workers in rural Beijing in 1990[4]. A co-infection of L. micdadei and Staphylococcus aureus was recently confirmed in a Shanghai hospital by metagenomic next-generation sequencing of a joint aspirate in a patient with arthritis[5]. However, most of the reported cases have been diagnosed with serological or molecular evidence, as few laboratories have successfully isolated L. micdadei from clinical samples.
In 2014, a clinical isolate from a bronchoalveolar lavage specimen was suspected to be Legionella spp. according to a combined diagnostic approach, including routine staining tests and microscopy, biochemical tests, the Legionella Latex test, and molecular assays[6]. The colonies agglutinated with diagnostic antisera for L. micdadei (Tianjin Biochip Corp., Tianjin, China) did not react with antisera from species other than L. pneumophila; hence, this clinical isolate was eventually designated to be L. micdadei and called strain 2014LM. Before this isolate, only three L. micdadei strains with complete genome sequences were accessible in the GenBank database, such as strains LMI, NZ2015, and NZ2016 (GenBank Accession nos: NZ_LN614830, CP020614, and CP020615), but none of these are clinical isolates from China. Therefore, the whole genome of 2014LM was sequenced using the MiSeq platform. Briefly, colonies of strain 2014LM on a Buffered Charcoal Yeast Extract (BCYE)-α plate were suspended in 0.5 mL of phosphate-buffered saline. Genomic DNA was purified using a phenol-chloroform extraction protocol. The concentration of the extracted DNA was determined on a model P330 nanophotometer (Implen, München, Germany) and was subsequently sequenced by Tianjin Novogene Bioinformatics Technology Co., Ltd. (Tianjin, China). Approximately 1,058 Mb of raw sequence data were obtained for strain 2014LM, with more than 100× coverage of a typical Legionella spp. genome. Using CLC Genomics Workbench version 11.0 (Qiagen, Aarhus, Denmark), the raw data were assembled to 3.37 Mb and consisted of 95 contigs ranging from 252 to 313,670 bp. The L50 value for the assembled genome was 7, and the N50 value was 154,003.
Next, genomic characterization and the genetic relationship between strain 2014LM and other Legionella spp. were investigated. The draft genome was annotated on the RAST (Rapid Annotation Using Subsystem Technology, version 2.0) server (
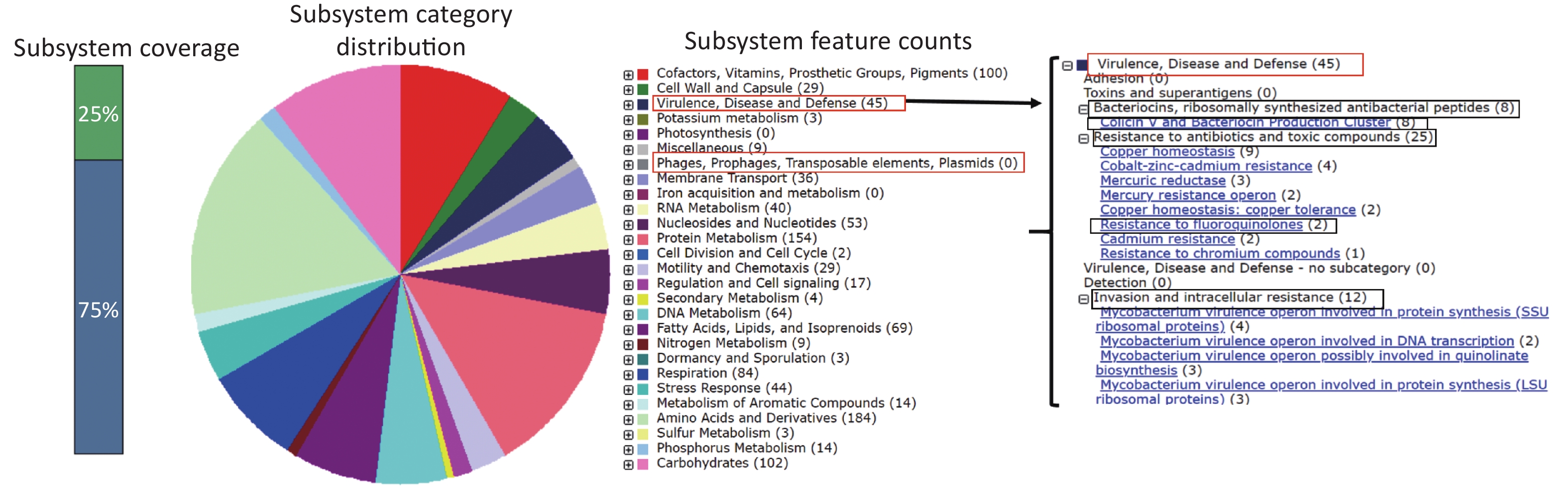
https://rast.nmpdr.org , RAST ID: 886170), which predicted 3,178 coding sequences (CDSs) categorized into 265 subsystems (Supplementary Figure S1, available in www.besjournal.com). Notably, 45 CDSs belonged to the subsystem of “Virulence, Disease, and Defense,” which consisted of 8 CDSs for “Colicin V and Bacteriocin Production Cluster,” 25 CDSs for “Resistance to antibiotics and toxic compounds,” and another 12 CDSs for “Invasion and intracellular resistance.” Two CDSs were associated with “Resistance to fluoroquinolones,” suggesting that strain 2014LM was potentially resistant to fluoroquinolones (Supplementary Figure S1). However, no CDS was identified in the subsystem of “Phages, Prophages, Transposable elements, and Plasmids.”
Figure S1. Statistical distribution of the subsystems in strain 2014LM annotated on the RAST server. Numbers in brackets indicate coding sequences (CDSs) of each subsystem.
To compare the genomic components of strain 2014LM with other L. micdadei strains and L. pneumophila strains at the (intra and inter) species levels, three L. micdadei strains, namely LMI, NZ2015, and NZ2016, and the type strain L. pneumophila, Philadelphia-1 (GenBank accession no.: AE017354), were selected as the reference strains (Supplementary Table S1, available in www.besjournal.com). The genome sizes of the five strains ranged from 3.25 to 3.40 Mb; however, the range of the GC contents for the L. micdadei strains was 40.5%–40.6%, whereas it was 38.3% for the Philadelphia-1 strain (Supplementary Table S1), suggesting genomic homogeneity within the L. micdadei strains and a genomic component difference between the two species. Based on the annotations from the RAST server, a variation in the number of CDSs was observed for each strain, i.e., 3,055 CDSs for strain LMI, 3,082 CDSs for strain NZ2015, 3,054 CDSs for strain NZ2016, and 3,210 CDSs for the Philadelphia-1 strain, respectively (Supplementary Table S1).
Strain GenBank
accession No.Isolation
sourceCountry Collection
timeSpecies Genome
Size
(bp)CDSs G+C
content
(%)Prophages 2014LM JABTVM000000000.1 Homo sapiens China 2014 L. micdadei 3.37M 3,178 40.6 7.7K LMI NZ_LN614830 Homo sapiens USA 1943 L. micdadei 3.31M 3,055 40.5 20.3K, 9.7K NZ2015 CP020614 Homo sapiens New Zealand 2015 L. micdadei 3.28M 3,082 40.5 19.8K NZ2016 CP020615 Homo sapiens New Zealand 2016 L. micdadei 3.25M 3,054 40.5 9.6K, 12.1K, 21.6K Philadelphia-1 AE017354 Homo sapiens USA 1977 L. pneumophila 1 3.40M 3,210 38.3 30.7K, 7.1K, 36.1K Table S1. Summary of general information for the reference Legionella strains in this study
Strain 2014LM and the four reference strains were isolated from clinical specimens. The profile of potential virulence genes was a critical indicator for comparing the pathogenicity of each strain. According to in silico searches of genome sequences on the VFDB (Virulence Factor of Pathogenic Bacteria) server (
http://www.mgc.ac.cn ), the L. micdadei strains and Philadelphia-1 shared ten virulence genes among the nine virulence factor classes, including Hsp60, Type IV pili, macrophage infectivity potentiator, ferrous iron transport, fur-regulated gene, iron acquisition/assimilation locus, alternative sigma factor RpoS, carbon storage regulator A, LetA/LetS two component, and RelA (Table 1, colored in red). Five virulence factors were presented in Philadelphia-1 but not in the L. micdadei strains, including MOMP, LigA, Lvg, Legiobactin, and RtxA (Table 1, colored in light blue). Two virulence genes were observed in the L. micdadei strains but not in Philadelphia-1, such as isocitrate lyase and LPS-modifying enzyme (Table 1, colored in green). The L. micdadei strains and Philadelphia-1 also contained several virulence factors consisting of multiple proteins or effectors, despite variations in diverse combinations of effectors. In general, Philadelphia-1 harbored more effector proteins than those in the L. micdadei strains, such as enhanced entry proteins, the cytochrome c maturation locus, the Dot/Icm Type IVB secretion system, the Lsp type II secretion system, and Type IVB secretion effectors (Table 1, colored in purple). Particularly, 35 Type IVB secretion effectors were identified in Philadelphia-1, in contrast to four such effectors in the L. micdadei strains. The only exception was observed for the Lvh (Legionella vir homologs) type IVA secretion system, in which the L. micdadei strains possessed slightly more effectors than Philadelphia-1 (Table 1, colored in yellow). Within L. micdadei, strain 2014LM and LMI shared identical profiles at the virulence factor class level and detailed factors. Minor differences were observed in the profiles of the two New Zealand isolates with that of strains 2014LM and LMI, mainly by the numbers of Lvh type IVA secretion systems, in which New Zealand isolates contained fewer virulence factors (Table 1). This observation is concordant with previous conclusions that many effector repertoires are conserved in L. pneumophila but are rarely detected in other Legionella spp.[7,8].Vf class Virulence factors 2014LM LMI NZ2015 NZ2016 Philadephia-1 Adherence Hsp60 + + + + + MOMP − − − − + Type IV pili + + + + + Intracellular survival LigA − − − − + Lvg − − − − + Mip + + + + + Invasion Enhanced entry proteins (3) (3) (3) (3) (4) Iron uptake Cytochrome c maturation (ccm) locus (5) (5) (5) (5) (6) Ferrous iron transport + + + + + Fur-regulated gene + + + + + Iron acquisition/assimilation locus + + + + + Legiobactin − − − − + Regulation Alternative sigma factor RpoS + + + + + Carbon storage regulator A + + + + + LetA/LetS two component + + + + + RelA + + + + + Secretion system Dot/Icm type IVB secretion system (23) (23) (23) (21) (24) Lsp type II secretion system (8) (8) (8) (8) (11) Lvh (Legionella vir homologs)
type IVA secretion system(11) (11) (8) − (10) Type IVB secretion effectors (4) (4) (4) (4) (35) Toxin RtxA − − − − + Lipid and fatty acid metabolism Isocitrate lyase (Mycobacterium) + + + + − Endotoxin LPS-modifying enzyme (Bordetella) + + + + − Note. The presence of specific virulence genes in reference genomes is marked as “+”, while absence as “−”. Numbers in the table indicate diverse virulence genes, the detailed combination of genes is listed as follows: Enhanced entry proteins: 3 (enhA, enhB, enhC); 4 (enhA, enhB, enhC, lidL). Cytochrome c maturation (ccm) locus: 5 (ccmA, ccmB, ccmC, ccmE, ccmF); 6 (ccmA, ccmB, ccmC, ccmD, ccmE, ccmF). Dot/Icm type IVB secretion system: 23 (dotA, dotB, dotC, dotD, icmB/dotO, icmC/dote, icmD/dotP, icmE/dotG, icmF, icmH/dotU, icmJ/dotN, icmK/dotH, icmL/dotI, icmM/dotJ, icmN/dotK, icmO/dotL, icmP/dotM, icmQ, icmS, icmT, icmW, icmX, ivgA); 21 (dotA, dotB, dotC, dotD, icmB/dotO, icmC/dote, icmD/dotP, icmE/dotG, icmF, icmH/dotU, icmJ/dotN, icmK/dotH, icmL/dotI, icmN/dotK, icmO/dotL, icmP/dotM, icmQ, icmS, icmT, icmW, ivgA); 24 (dotA, dotB, dotC, dotD, icmB/dotO, icmC/dotE, icmD/dotP, icmE/dotG, icmF, icmG/dotF, icmH/dotU, icmJ/dotN, icmK/dotH, icmL/dotI, icmM/dotJ, icmN/dotK, icmO/dotL, icmP/dotM, icmQ, icmR, icmS, icmV, icmW, icmX). Lsp type II secretion system: 8 (lspD, lspE, lspF, lspG, lspI, lspJ, lspK, lspL); 11 (lspC, lspD, lspE, lspF, lspG, lspH, lspI, lspJ, lspK, lspL, lspM). Lvh (Legionella vir homologs) type IVA secretion system: 11 (lvhB2, lvhB3, lvhB4, lvhB5, lvhB6, lvhB7, lvhB8, lvhB9, lvhB10, lvhB11,lvhD4); 8 (lvhB4, lvhB5, lvhB6, lvhB8, lvhB9, lvhB10, lvhB11, lvhD4); 10 (lvhB2, lvhB4, lvhB5, lvhB6, lvhB7, lvhB8, lvhB9, lvhB10, lvhB11, lvhD4). Type IVB secretion effectors: 4 (lepB, sdcB, sdhA, vipF); 35 (drrA/sidm, laiE, lepA, lepB, lidA, ralF, sdbA, sdbB, sdbC, sdcA, sdeA/laiA, sdeB/laiB, sdeC/laiC, sdeD/laiF, sdhA, sdhB, sidA, sidB, sidC, sidD, sidE/laiD, sidF, sidG, sidH, vipA, vipD1, vipD2, vipD3, vipE, vipF, wipA, wipB, wipC, ylfA, ylfB). Table 1. Summary of virulence factors predicted in the VFDB database
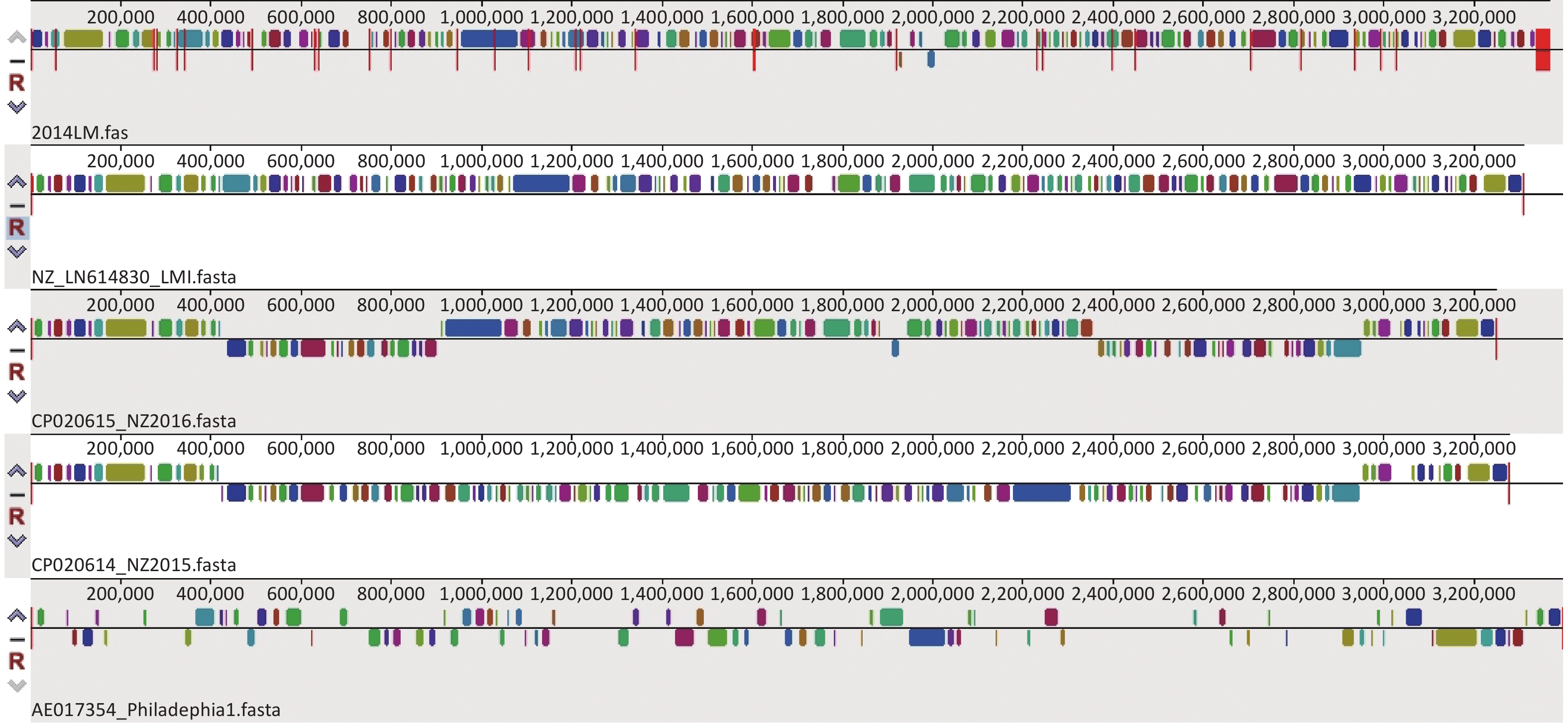
MAUVE software is a popular genome comparison tool for large genomes. The multiple genome alignment results from MAUVE software (version 20150226) for the five strains indicated that the four L. micdadei strains shared similar locally collinear blocks (LCBs) in terms of size, number, and arrangement, but these LCBs differed from those of the Philadelphia-1 strain, suggesting that the L. micdadei genomes are highly conserved with relatively identical genome sizes and contents, and there are considerable interspecies genomic differences between Legionella spp. (Figure 1). According to the MAUVE alignment results, strain 2014LM contained a distinct genome structure from the Philadelphia-1 strain, which was recommended by the RAST database as one of the closest neighbors to strain 2014LM. Such inaccurate assignments probably occurred because only four reference L. pneumophila strains are available in that database.
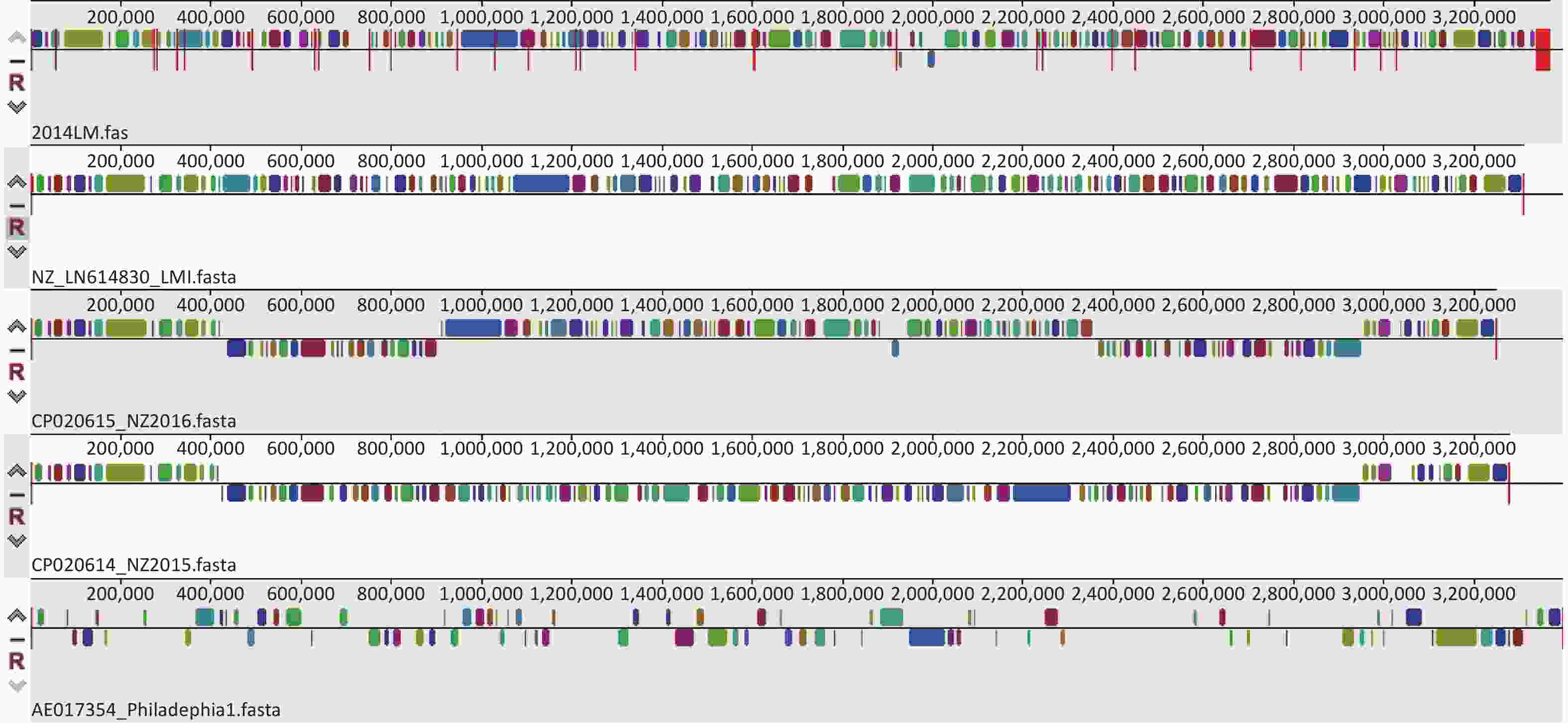
Figure 1. Multiple genome alignment of five Legionella strains using MAUVE. Colored blocks are locally collinear blocks (LCBs) representing diverse conservative regions of the reference strains. Blocks above the center line indicate regions coded in the forward direction, while blocks below the line indicate coding in the reverse direction. Blanks represent nonconserved regions.
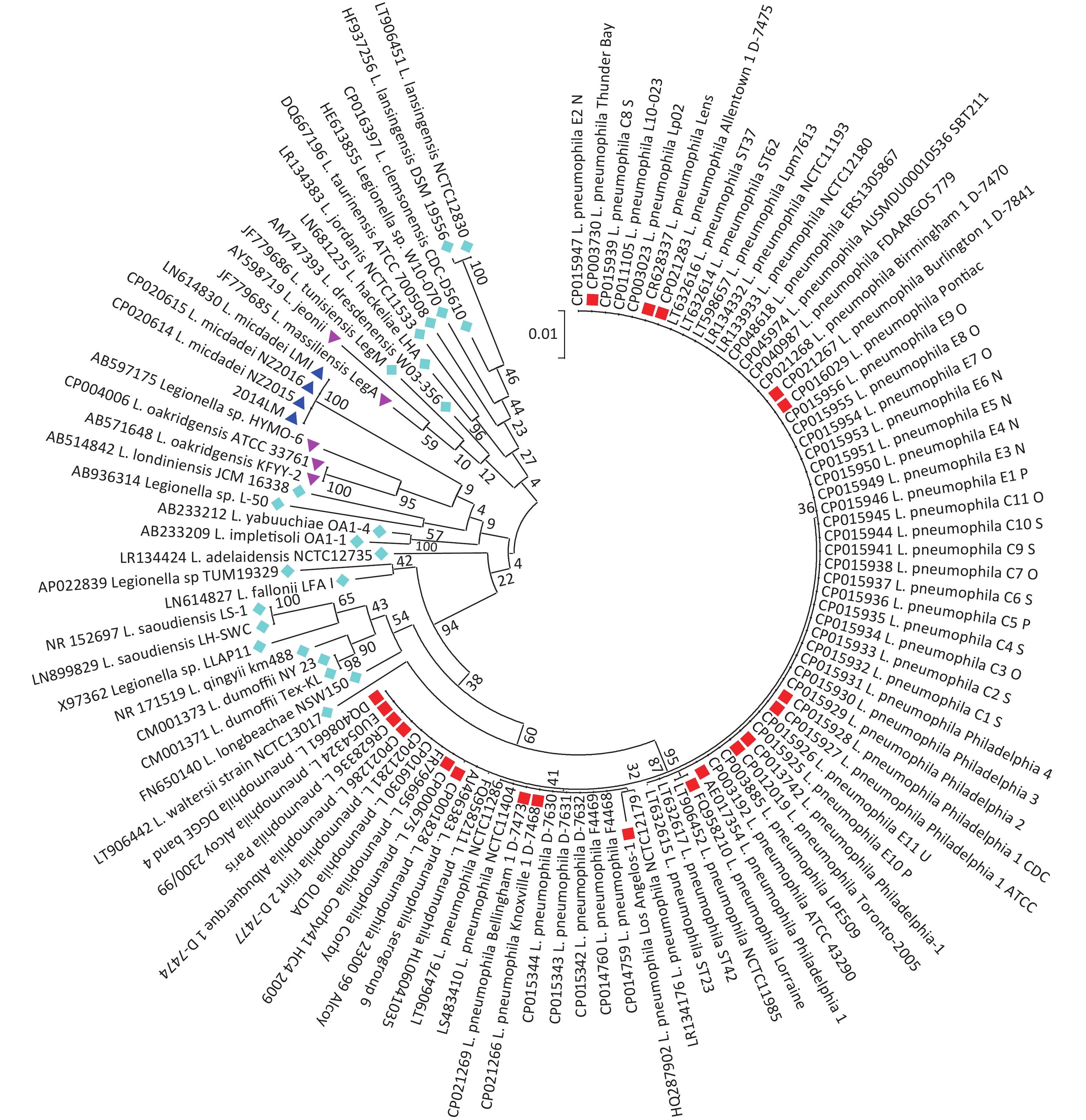
The 16S rDNA sequence is well-known for its ability to differentiate bacterial strains at the genus or species level. The full-length 16S rDNA of strain 2014LM was searched on the NCBI BLAST server, and 263 aligned sequences were downloaded. Notably, the other three L. micdadei strains ranked on top of the hit list, showing 100% coverage. After removing duplicate copies from dozens of strains, the 16S rDNA sequences of the remaining 107 strains were aligned and analyzed with MEGA software (version 7.0.21), which yielded a maximum likelihood tree after bootstrapping with 1,000 replications (Figure 2, Supplementary Table S2, available in www.besjournal.com). Seventy-four L. pneumophila strains clustered in the main branch, while the 33 non-LP strains (including the four L. micdadei strains and nearly 20 rare Legionella spp.) formed another distinct branch, suggesting interspecific genetic heterogenicity among Legionella spp. and closer genetic relationships within the non-LP strains. In particular, the four L. micdadei strains belonged to a unique sub-branch flanked by several non-LP species, such as L. oakridgensis and L. massiliensis. Thus, the phylogenetic tree demonstrated considerable genetic diversity within the genus Legionella and a relatively distant genetic relationship between the L. pneumophila strains and the L. micdadei strains. The phylogenetic tree also indicated that the 16S rDNA sequence is a reliable tool for identifying Legionella spp.
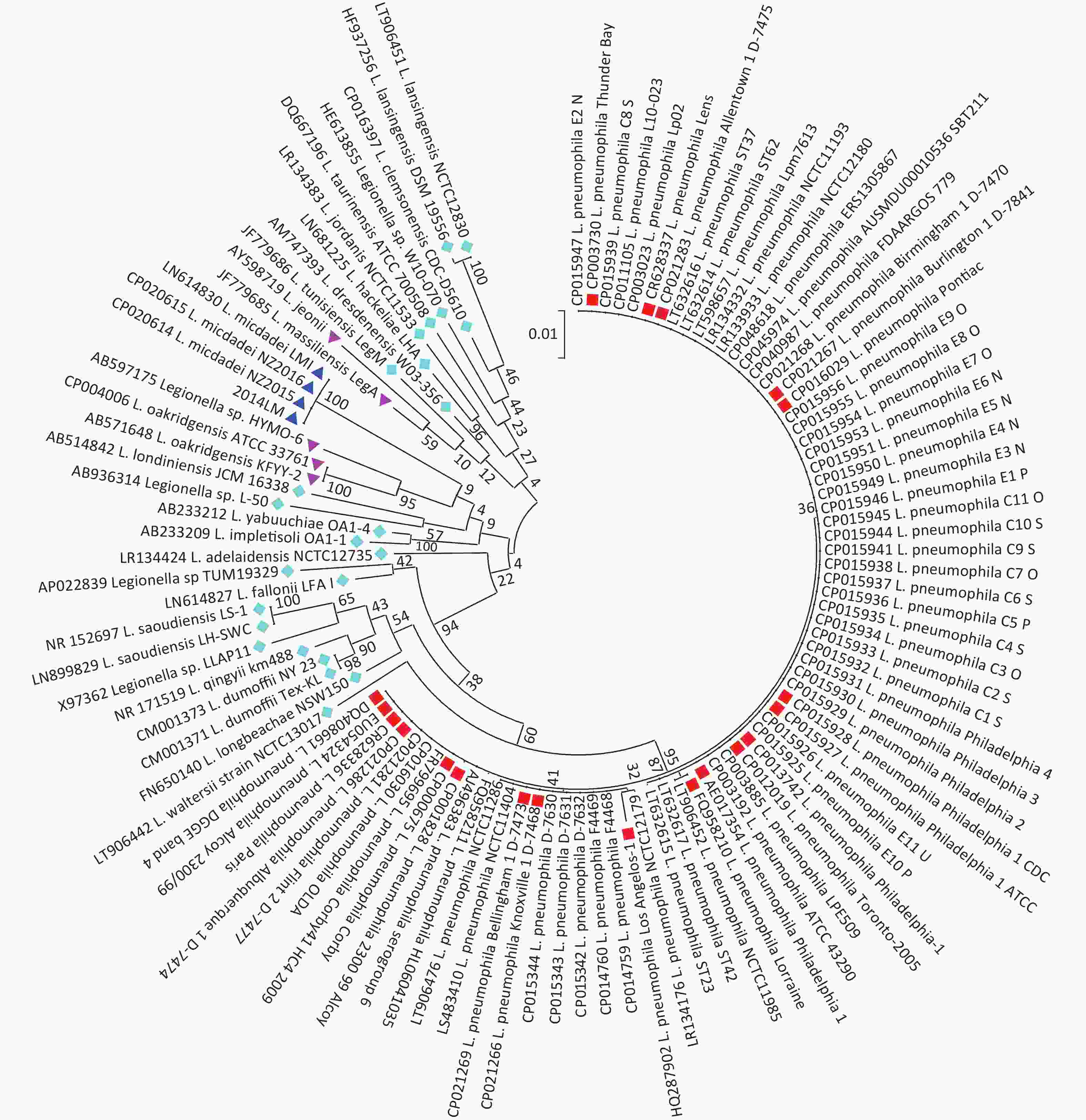
Figure 2. Phylogenetic tree of the 107 Legionella strains based on the full-length 16S rRNA gene. The scale bar on the tree refers to a phylogenetic distance of 0.01 nucleotide substitutions per site. Numbers on the branches represent bootstrap percentages after 1,000 replications to construct the ML tree.




Previous comparative genomic analyses indicated that the Legionella genomes are highly dynamic due to a large mobilome[7]. The mobilome is comprised of diverse genomic elements, such as plasmids, genomic islands, and transposons, which move between genomes. A putative complete prophage had been reported in L. micdadei strain LMI, while incomplete phage DNA has also been observed on the Phaster server (
https://phaster.ca ) in three genomic regions of strain NZ2015 and two regions of strain NZ2016[9]. However, when the genomic sequences of the five strains were searched on the Phaster sever, an incomplete 7.7 kb prophage was identified for strain 2014LM, while one to three incomplete prophages were observed for the other four strains, with varying sizes between 7.1 and 36.1 kb (Supplementary Table S1). The predicted prophage score for the Philadelphia-1 strain was 50–60, whereas the potential prophages in the L. micdadei strains scored 20–40. The Phaster search result was inconsistent with previous reports about strains LMI and NZ2015, as there were three incomplete prophages in LMI instead of one complete prophage, while there was one incomplete prophage rather than three genomic regions in NZ2015[9]. Such inconsistency probably occurred due to updating of the Phaster database. Therefore, the difference in prophage components among the four L. micdadei strains supported previous findings that variations in the mobilome may account for the geographically distinct L. micdadei strains[7,9].Another critical feature of a pathogenic microbe is its spectrum of antimicrobial resistance, which is necessary for clinicians to provide timely and appropriate treatment, although antibiotic resistance in Legionella infections has historically not been a concern[10]. As obligate culture conditions are required for Legionella spp., it takes up to 14 days to isolate a clinical strain and another 48 hours for routine in vitro antimicrobial susceptibility tests; these time-consuming procedures are unlikely to be useful in clinical practice and may underestimate antibiotic resistance in Legionella spp.[10]. In this study, an in silico screen of the CARD (The Comprehensive Antibiotic Resistance Database,
https://card.mcmaster.ca/ ) database revealed that the five strains were positive for adeF, which is potentially resistant to tetracyclines and fluoroquinolones. According to current guidelines, either fluoroquinolone or macrolide is recommended as the first-line therapy for LD[10]. Strain 2014LM carried fluoroquinolone resistance, which is consistent with the RAST annotation result; however, it might be susceptible to macrolides. Comprehensive in silico prediction of the resistome using genomic or metagenomic data will provide a rapid, precise, and reliable option for such slow-growing microbes in clinical practice.In conclusion, the genomic characterization of the first sequenced clinical L. micdadei isolate in China was investigated in the present study. Strain 2014LM shared an identical genomic structure, possessed a similar virulence gene profile with reference to the L. micdadei strains, and contained potential resistance to tetracyclines and fluoroquinolones. Comparative genomics and phylogenetic analyses demonstrated high intraspecies genetic homogeneity within the L. micdadei strains and relatively closer genetic relationships between the L. micdadei strains and the non-LP strains. Therefore, genomic evidence of this less common bacterial pathogen may contribute to exploring global genetic diversity in Legionella. Extensive comparative genomics of emerging L. micdadei and other non-LP isolates will expand our understanding of the pathogenic mechanisms of Legionella spp. and their global genetic diversity.
Conflict of interest The authors declare no conflicts of interest in the present study.
-
Accession No Species Strain JABTVM000000000 L. micdadei 2014LM CP020614 L. micdadei NZ2015 CP020615 L. micdadei NZ2016 LN614830 L. micdadei LMI AB233209 L. impletisoli OA1-1 AB233212 L. yabuuchiae OA1-4 LR134424 L. adelaidensis NCTC12735 AE017354 L. pneumophila Philadelphia_1 CP000675 L. pneumophila Corby FR799695 L. pneumophila Corby41_HC4_2009 CP001828 L. pneumophila 2300_99_Alcoy CP014759 L. pneumophila F4468 CP014760 L. pneumophila F4469 CP015342 L. pneumophila D-7632 CP015343 L. pneumophila D-7631 CP015344 L. pneumophila D-7630 CP021266 L. pneumophila Knoxville_1_D-7468 CP021269 L. pneumophila Bellingham_1_D-7473 LS483410 L. pneumophila NCTC11404 LT906476 L. pneumophila NCTC11286 FQ958211 L. pneumophila HL06041035 CP003192 L. pneumophila ATCC_43290 CP003885 L. pneumophila LPE509 CP012019 L. pneumophila Toronto-2005 CP013742 L. pneumophila Philadelphia-1 CP015925 L. pneumophila E10_P CP015926 L. pneumophila E11_U CP015927 L. pneumophila Philadelphia_1_ATCC CP015928 L. pneumophila Philadelphia_1_CDC_ CP015929 L. pneumophila Philadelphia_2 CP015930 L. pneumophila Philadelphia_3 CP015931 L. pneumophila Philadelphia_4 CP015932 L. pneumophila C1_S CP015933 L. pneumophila C2_S CP015934 L. pneumophila C3_O CP015935 L. pneumophila C4_S CP015936 L. pneumophila C5_P CP015937 L. pneumophila C6_S CP015938 L. pneumophila C7_O CP015941 L. pneumophila C9_S CP015944 L. pneumophila C10_S CP015945 L. pneumophila C11_O CP015946 L. pneumophila E1_P CP015949 L. pneumophila E3_N CP015950 L. pneumophila E4_N CP015951 L. pneumophila E5_N CP015953 L. pneumophila E6_N CP015954 L. pneumophila E7_O CP015955 L. pneumophila E8_O CP015956 L. pneumophila E9_O CP016029 L. pneumophila Pontiac CP021267 L. pneumophila Burlington_1_D-7841 CP021268 L. pneumophila Birmingham_1 D-7470 CP040987 L. pneumophila FDAARGOS_779 CP045974 L. pneumophila AUSMDU00010536_SBT211 CP048618 L. pneumophila ERS1305867 LR133933 L. pneumophila NCTC12180_ LR134332 L. pneumophila NCTC11193 LT598657 L. pneumophila Lpm7613 LT632614 L. pneumophila ST62 LT632616 L. pneumophila ST37 CP021283 L. pneumophila Allentown_1_D-7475 CR628337 L. pneumophila Lens LT632617 L. pneumophila ST42 CP016030 L. pneumophila OLDA CP021281 L. pneumophila Flint_2_D-7477 CP021286 L. pneumophila Albuquerque_1_D-7474 CR628336 L. pneumophila Paris EU054324 L. pneumophila Alcoy 2300/99 LT632615 L. pneumophila ST23 FQ958210 L. pneumophila Lorraine LT906452 L. pneumophila NCTC11985 LR134176 L. pneumophila NCTC12179 CP003023 L. pneumophila Lp02 CP011105 L. pneumophila L10-023 CP015939 L. pneumophila C8_S CP015947 L. pneumophila E2_N CP003730 L. pneumophila Thunder Bay DQ408661 L. pneumophila DGGE band 4 HQ287902 L. pneumophila Los Angelos-1 AJ496383 L. pneumophila serogroup 6 LT906442 L. waltersii NCTC13017 LN614827 L. fallonii LFA_I CM001371 L. dumoffii Tex-KL CM001373 L. dumoffii NY 23 NR_171519 L. qingyii km488 FN650140 L. longbeachae NSW150 LN899829 L. saoudiensis LH-SWC NR_152697 L. saoudiensis LS-1 AP022839 Legionella_sp. TUM19329 X97362 Legionella sp. LLAP11 AM747393 L. dresdenensis W03-356 JF779685 L. massiliensis LegA JF779686 L. tunisiensis LegM DQ667196 L. taurinensis ATCC 700508 CP016397 L. clemsonensis CDC-D5610 HE613855 Legionella sp. W10-070 HF937256 L. lansingensis DSM 19556 LT906451 L. lansingensis NCTC12830 LN681225 L. hackeliae LHA_ LR134383 L. jordanis NCTC11533 AB571648 L. oakridgensis KFYY-2 CP004006 L. oakridgensis ATCC_33761___ AB597175 Legionella sp. HYMO-6 AB936314 Legionella sp. L-50 AB514842 L. londiniensis JCM 16338 AY598719 L. jeonii Table S2. List of Legionella spp. strains in phylogenetic analysis
HTML
 22286Supplementary Materials.pdf
22286Supplementary Materials.pdf
|

|

 Quick Links
Quick Links
 DownLoad:
DownLoad: