
-
Cellular senescence is a state of irreversible cell cycle arrest induced by various cytotoxic factors including telomere dysfunction, ionizing radiation (IR), oxidative stress, oncogene overexpression, and genotoxic agents[1]. Senescence was originally defined in normal human diploid fibroblasts but similar phenotypes have been observed in various cell types, including tumor cells, particularly in apoptosis-defective contexts[2]. Although the induction of senescence in cancer cells has been considered a promising alternative therapeutic strategy for its tumor-suppressing properties[3], senescent cells secrete numerous soluble factors such as cytokines, chemokines, growth factors, and proteases (termed the senescence-associated secretory phenotype, SASP[4,5]), which exert anti-tumorigenic and/or pro-tumorigenic effects by altering the tumor microenvironment and local immune response[5,6]. Notably, accumulating evidence shows that therapy-induced senescent tumor cells are capable of reversing senescence via genome reorganization or uptake by neighboring cells, conferring stemness and enhancing cell survival and tumor aggressiveness[7,8]. Therefore, a two-step senescence-focused anticancer therapeutic strategy involving senescence-eliciting therapy followed by the elimination of senescent cells with senolytics has recently been proposed[9,10].
The precise mechanisms underlying the induction and development of senescence are not fully understood. A prevailing model suggests that all senescence-inducing stimuli ultimately activate a DNA damage response, which in turn activates both the checkpoint kinase-p53 and p38-MAPK pathways[11]. Activated p53 transcriptionally upregulates p21, which suppresses cyclin dependent kinase 2 (Cdk2)-mediated retinoblastoma (Rb) inactivation and consequently prevents S phase entry. The p38-MAPK pathway upregulates p16 and prevents Cdk4/6-mediated Rb phosphorylation. Therefore, this model proposes that senescent cells are arrested at G1 phase through Rb-mediated inhibition of E2F-dependent transcription. However, substantial evidence has revealed that replicative senescence and DNA damage-induced premature senescence can be triggered and developed in cells arrested at G2 phase[12-14], which is different from the classical senescence model. Furthermore, recent studies have suggested that “mitotic skip” is involved in tetraploid senescence, particularly when induced by DNA damage, in which p53 activation during G2 phase plays a key role[15,16]. Therefore, the specific mechanisms and phases in which cells exit the cell cycle and undergo senescence remain largely unknown.
In normal proliferating mammalian cells, mitotic progression is mediated by key mitotic kinases, including Cdk1, polo-like kinase 1 (Plk1), and Aurora kinases (Aurora A/B). The cyclin B/Cdk1 complex is the master regulator of mitotic entry and its inhibition is fundamental for anaphase onset[17]. Cells in which Plk1 is inhibited arrest in prometaphase, with defects in spindle assembly and chromosome alignment[18]. Aurora kinases are overexpressed in a wide range of human cancers and play crucial roles in cell proliferation[19]. Aurora A, in particular, is primarily associated with centrosome maturation, bipolar spindle assembly, and chromosome separation[19]. The protein level of Aurora A is low during G1/S phase but increases in G2, with both function and expression peaking in early M phase[20]. Aurora A activation is considered a key regulator of G2-M transition progress, and promotes mitotic entry by controlling the activation of cyclin B/Cdk1[21,22]. Aurora A also directly phosphorylates and activates Plk1 in G2 phase[23,24].
Mitotic regulators decrease quickly in response to DNA damage in cells with functional p53[13-15], and the loss of mitotic regulators is considered a fundamental feature of senescence[25]. Although the loss of mitotic regulators is thought to be the cause, rather than the consequence, of senescence, the role of these mitotic regulators in senescence induction is not completely understood, especially in the process through which G2-arrested cells enter senescence. Here, we demonstrate that p21 overexpression, in combination with Aurora A kinase decline, appears to be the key event for mitosis skip-mediated senescence entry of G2-arrested cells damaged by IR.
-
Human malignant melanoma A375 cells were purchased from the Cell Bank of Shanghai Institute of Biochemistry and Cell Biology. Human uveal melanoma 92-1 cells (92-1) were obtained from our laboratory. A375 cells and 92-1 cells were cultured in RPMI-1640 medium (Gibco, NY, USA) supplemented with 10% fetal bovine serum (FBS; Gibco), 100 μg/mL streptomycin, and 100 units/mL penicillin in a humidified atmosphere with 5% CO2. The cells were seeded in 35 mm culture dishes (2 × 105 cells per dish) or 60 mm culture dishes (5 × 105 cells per dish) and incubated for 48 h to approximately 70% confluence. Cells were irradiated at room temperature with X-rays generated using a Faxitron RX-650 cabinet irradiator (Faxitron, Tucson, USA) at a dose rate of 1 Gy/min (100 kVp, 5 mA). The cells exposed to 5 Gy were maintained at room temperature for less than 15 min during irradiation. Unirradiated control (Ctrl) cells were transported to the radiation laboratory in parallel, but without irradiation. All cells were cultured in dishes or flasks and sealed with parafilm to prevent contamination during irradiation.
-
The cultured cells were digested with 0.25% trypsin, washed once with PBS, and fixed with 70% cold ethanol. The fixed cells were centrifuged at 1,500 rpm for 3 min, rinsed twice with PBS, and resuspended in PBS containing 150 µg/mL RNase A and 5 µg/mL propidium iodide (PI). Finally, the stained cells were analyzed for DNA content using a FlowSight flow cytometer (Merck Millipore, Darmstadt, Germany), and the data were analyzed using FlowJo software (v6.0, Tree Star, Ashland, USA).
-
Immunoblotting was performed as previously described[14]. Briefly, cells were seeded on sterile coverslips in 35 mm culture dishes, cultured for 48 h, irradiated, and incubated for the indicated times. The irradiated cells were fixed with 4% paraformaldehyde for 10 min at room temperature, followed by incubation in chilled methanol for 20 min at –20 °C, and permeabilization with 0.5% Triton X-100 in PBS for 10 min. Nonspecific binding sites were blocked with 5% non-fat-dried milk in PBS for 2 h at room temperature before incubation with anti-Ki67 rabbit polyclonal (Abcam, Cambridge, UK), anti-γH2AX (phosphor S139) mouse monoclonal (Abcam), or anti-histone H3 (phosphor S10, pS10 H3; Abcam) rabbit monoclonal primary antibodies. The membranes were then incubated with anti-mouse or anti-rabbit secondary antibodies (Invitrogen, Carlsbad, CA, USA) conjugated to Alexa Fluor 488/594 for 1 h. The cell nuclei were counterstained with 4’,6-diamidino-2-phenylindole (DAPI; Molecular Probes, Eugene, OR, USA). Images were obtained using a Zeiss LSM 700 Meta-laser scanning confocal microscope (Zeiss, Oberkochen, Germany).
-
Small interfering RNAs (siRNAs) against p21 (sip21) and negative control siRNAs (NC) were purchased from RiboBio (Guangzhou, China). siRNAs were introduced into the cells at a final concentration of 100 nmol/L using the Lipofectamine 2000 reagent (Invitrogen), following the manufacturer’s instructions. The knockdown efficiency of the siRNAs was evaluated using western blot analysis.
-
For live-cell imaging assays, cells were grown on a Lab-Tek II chambered cover glass (Thermo Scientific, Waltham, USA) in RPMI-1640 medium. Fluorescent ubiquitination-based cell cycle indicator (FUCCI; Molecular Probes) reagents were added to the medium according to the manufacturer’s instructions. Time-lapse images were captured every 15 min for 72 h using green emission filters (494/20 and 536/40). Images were obtained using a DeltaVision Elite (Applied precision) microscope maintained at 37 °C, equipped with a 10×0.4 NA or 20×0.75 NA lens (Olympus, Tokyo, Japan) and cooled Cool Snap CCD camera.
-
SA-β-Gal staining was performed as previously described[14]. Briefly, cells were exposed to X-rays and stained using a senescence-associated β-galactosidase staining kit (Beyotime, Shanghai, China), following the manufacturer’s protocol. Senescent (SA-β-Gal positive) cells were identified using a light microscope and the percentage of SA-β-Gal positive cells was calculated, based on counts of more than 500 cells per experimental group.
-
Cells were lysed using radioimmunoprecipitation assay (RIPA) buffer (Beyotime, Shanghai, China). Samples were centrifuged at 10,000 g for 15 min at 4 °C, and total protein concentrations from supernatants were determined using the BCA protein assay kit (Bio-Rad, Hercules, USA). Thereafter, the proteins were resolved by SDS-PAGE and transferred onto polyvinylidene difluoride (PVDF) membranes (Merck Millipore). The membranes were blocked for 2 h in 5% non-fat dry milk and incubated with primary antibodies for 2 h. Next, the membranes were washed three times with PBS containing 0.1% Tween-20, incubated with an HRP-conjugated secondary antibody for 1.5 h at room temperature, and washed with PBS containing 0.1% Tween-20. Protein bands were visualized using an enhanced chemiluminescence system (Merck Millipore) and exposed to X-ray medical film (Kodak, Tokyo, Japan). GAPDH was used as a loading control. The following primary antibodies were used in this study: anti-p53 (Abcam, Cambridge, UK), anti-p21 (Santa Cruz Biotechnology, Dallas, TX, USA), anti-pS10 H3 (Abcam, Cambridge, UK), and anti-GAPDH (Santa Cruz Biotechnology).
-
Flow cytometry was performed to determine the levels of early-stage apoptosis, late-stage apoptosis, and necrosis using the Annexin V-FITC/PI kit (BD Biosciences, San Jose, USA) according to the manufacturer’s protocol. Briefly, the cultured cells were digested with 0.25% trypsin, washed once with cold PBS, and resuspended in binding buffer at concentrations between 1 × 106 and 1 × 107 cells/mL. FITC-labeled Annexin V (5 μL) and PI (5 μL) were added to 100 μL of the cell suspension. After incubation for 15 min at room temperature, the number of stained cells was immediately assessed using a FlowSight flow cytometer (Merck Millipore), and at least 10,000 gated events were acquired from each sample. The total apoptotic rate was calculated as the sum of early and late apoptotic rates. Data were analyzed using the IDEAS Application (Merck Millipore).
-
For direct comparisons, statistical significance was calculated using the unpaired Student’s t-test. For multigroup comparisons, a two-way analysis of variance (ANOVA) was performed. P values were represented as: *P < 0.05; **P < 0.01; ***P < 0.001. All experiments were repeated at least three times and the values shown on graphs represent the means ± standard error of the mean (SEM).
-
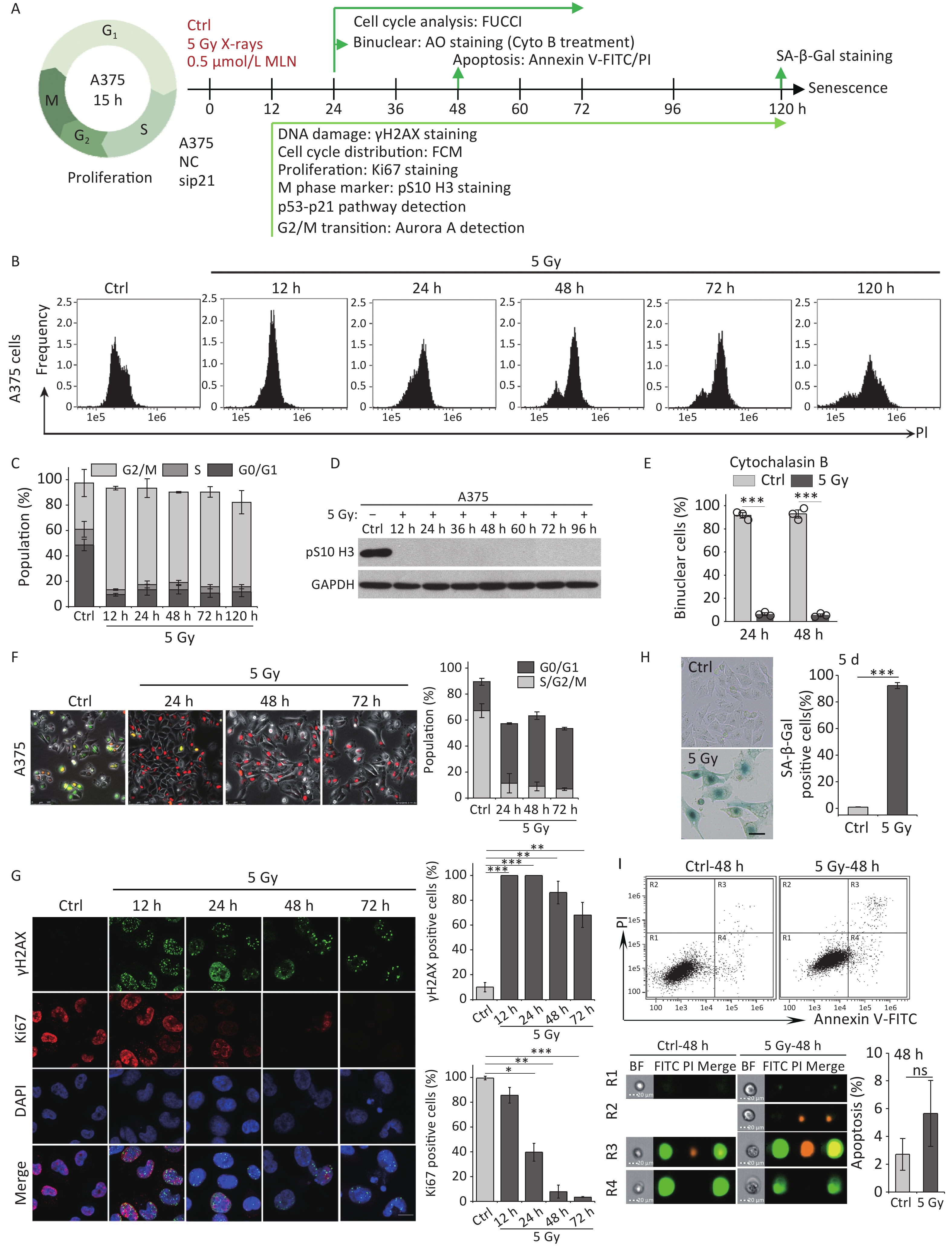
Our previous studies have shown that high-dose IR treatment induces long-term G2 phase arrest in human uveal melanoma 92-1 cells[13,14,26]. In the current study, we designed experiments to explore the fate of G2 phase-arrested cells (Figure 1A). First, we confirmed that A375 cells and 92-1 cells exposed to 5 Gy X-rays irradiation exhibit long-term G2 arrest. As shown in Figure 1B and 1C, tetraploid cells accumulated quickly among A375 cells exposed to IR, and approximately 80% of the irradiated cells were arrested at the G2/M phase at 12 h post-irradiation. This proportion remained at approximately 66.5% at 5 days post-irradiation, the proportion of cells in G2/M phase among the control cells was only 36.5% (Figure 1C). A similar phenomenon was observed in 92-1 cells (Supplementary Figure S1A and S1B, available in www.besjournal.com). These data implied a significant accumulation of tetraploid cells after irradiation. Tetraploid cells may be in G2 or M phase. To distinguish M from G2 phase in these cells, we examined irradiated A375 cells for changes in pS10 H3, a mitotic marker. The results showed that pS10 H3 expression was lost after exposure to IR (Figure 1D). In addition, to confirm whether IR treatment impedes mitotic entry, irradiated A375 cells were treated with cytochalasin B, an inhibitor of actin polymerization that prevents cytokinesis. The proportion of binuclear cells was calculated, and the results showed that approximately 90% of A375 control cells were binuclear after cytochalasin B exposure, while this proportion was approximately 5.5% in cells treated with 5 Gy of X-rays at 24 h or 48 h post-irradiation (Figure 1E), implying that IR treatment arrested cells at the G2 phase and impeded mitosis entry. Together, these results suggest that tetraploid A375 cells induced by IR treatment are cells arrested at the G2 phase.
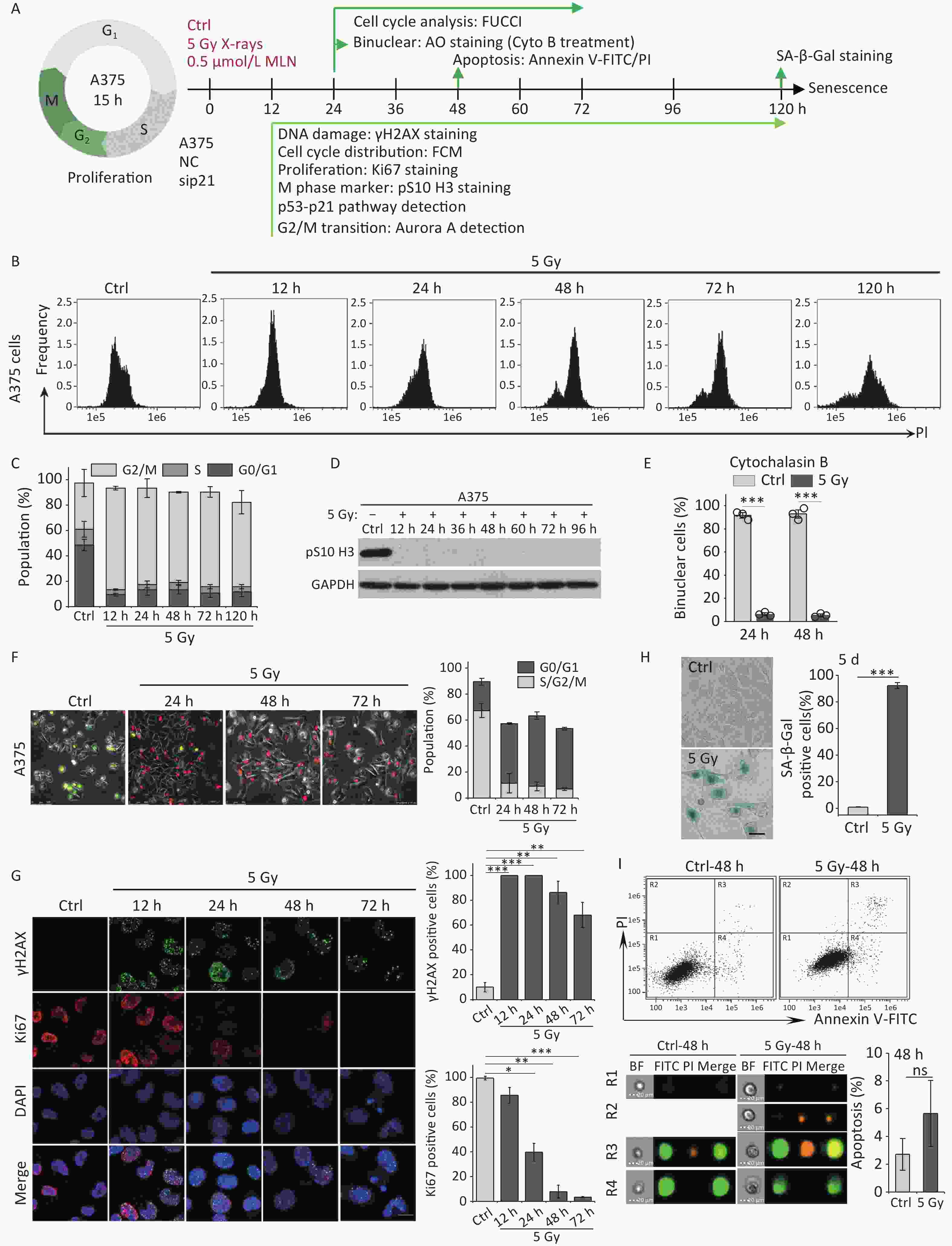
Figure 1. IR induces G2 arrest, mitosis skipping, and senescence in A375 cells. (A) Experimental design indicating main molecular targets and features of biological events on the experimental timeline; AO: acridine orange. (B) A375 cells were exposed to 5 Gy of X-rays, stained with PI, and analyzed by flow cytometry at the indicated time points. (C) The cell cycle distribution of cells in (B). (D) The protein level of mitosis marker pS10 H3 was examined by western blot analysis in A375 control (Ctrl) cells and cells exposed to 5 Gy of X-rays. Protein was harvested at the indicated time points after IR treatment and GAPDH was used as a loading control. (E) The percentage of binucleated cells among A375 cells treated with cytochalasin B only (Ctrl) or cytochalasin B together with 5 Gy of X-rays (5 Gy X-rays) at 24 h or 48 h post-irradiation. (F) A375 cells infected with FUCCI lentiviruses expressing Cdt1-RFP (red) and geminin-GFP (green) were irradiated with 5 Gy of X-rays. After 1 d of irradiation, representative micrographs at the indicated time points were captured (left) and the cell population was counted (right). Scale bar, 100 µm. (G) Representative images of γH2AX foci or Ki67-positive nuclei in A375 control (Ctrl) cells or irradiated cells (5 Gy of X-rays) at the indicated time points post-irradiation (left), and the percentage of γH2AX foci or Ki67-positive cells (right). Scale bar, 10 µm. (H) Representative images of control (Ctrl) cells or cells exposed to 5 Gy of X-rays and stained for SA-β-Gal at 5 d after treatment (left), and the calculated data (right). Scale bar, 100 µm. (I) Flow cytometric analysis of apoptotic cells in A375 control cells (Ctrl) and cells exposed to 5 Gy of X-rays at 48 h post-irradiation (upper panel). Representative images of single cell in different gating regions detected by flow cytometry (lower left panel). Quantification of apoptotic cells (lower right panel). ns, not significant; ***P < 0.001, compared to Ctrl.
However, separating the tetraploid cells from the remaining diploid cells poses a technical challenge: the two populations cannot be distinguished by DNA content alone because diploid cells in the G2 phase have the same DNA content (4n) as tetraploid cells arrested at the G1 phase. To overcome this, we combined DNA content analysis with the FUCCI reporter system[27]. FUCCI consists of two fluorescent proteins whose expression alternates based on the cell cycle position; Cdt1-RFP is expressed in the G1 phase, whereas germinin-GFP is expressed in the S/G2/M phases. Thus, S/G2/M diploid cells (green) can be efficiently separated from G1 tetraploid cells (red) using fluorescence microscopy. In addition, live-cell imaging allowed us to determine changes in the nucleus that indicate entry into mitosis, such as mitotic rounding and nuclear envelope breakdown. Using A375 cells transduced with lentiviruses expressing Cdt1-RFP and geminin-GFP proteins (A375-FUCCI cell line), we determined whether these G2-arrested cells induced by X-rays remained in the G2 phase using live-cell imaging of control and irradiated A375-FUCCI cells over a 3-day period. In contrast to control cells, cells arrested at the G2 phase degraded geminin and accumulated Cdt1 without mitotic entry 24 h after X-rays treatment (Figure 1F). Long-term observation confirmed that more than 50% of these cells expressed Cdt1 (red) and ceased cycling during the 3-day observation time; in contrast, only approximately 9% of control cells expressed germinin-GFP (Figure 1F). Combined with the results of DNA content detection (Figure 1B and 1C), these data indicate that mitosis skipping occurs in IR-exposed cells, resulting in the accumulation of tetraploid G1 cells.
Next, we explored the fate of these mitotic-skip cells in irradiated A375 cells. The proliferative potential of these cells was evaluated using Ki67 immunostaining, a marker widely used to detect cell proliferation[28]. As shown in Figure 1G, X-rays treatment induced severe DNA damage (measured by γH2AX foci) and abolished cell proliferation (loss of Ki67 staining) at 48 h post-irradiation. The morphological changes exhibited by irradiated A375 cells, including a flattened and enlarged shape (Figure 1H), implied that senescence was induced in response to DNA damage elicited by IR. Moreover, the results of an SA-β-Gal staining assay implied that these tetraploid G1 cells entered senescence at 5 d post-irradiation (Figure 1H). We also analyzed cellular apoptosis by flow cytometry with Annexin V-FITC/PI staining. However, no distinct apoptotic cells were detected in irradiated A375 cells (Figure 1I). Collectively, these results indicate that G2-arrested cells predominantly undergo senescence after mitosis skipping and entry into G1 as tetraploid cells.
-
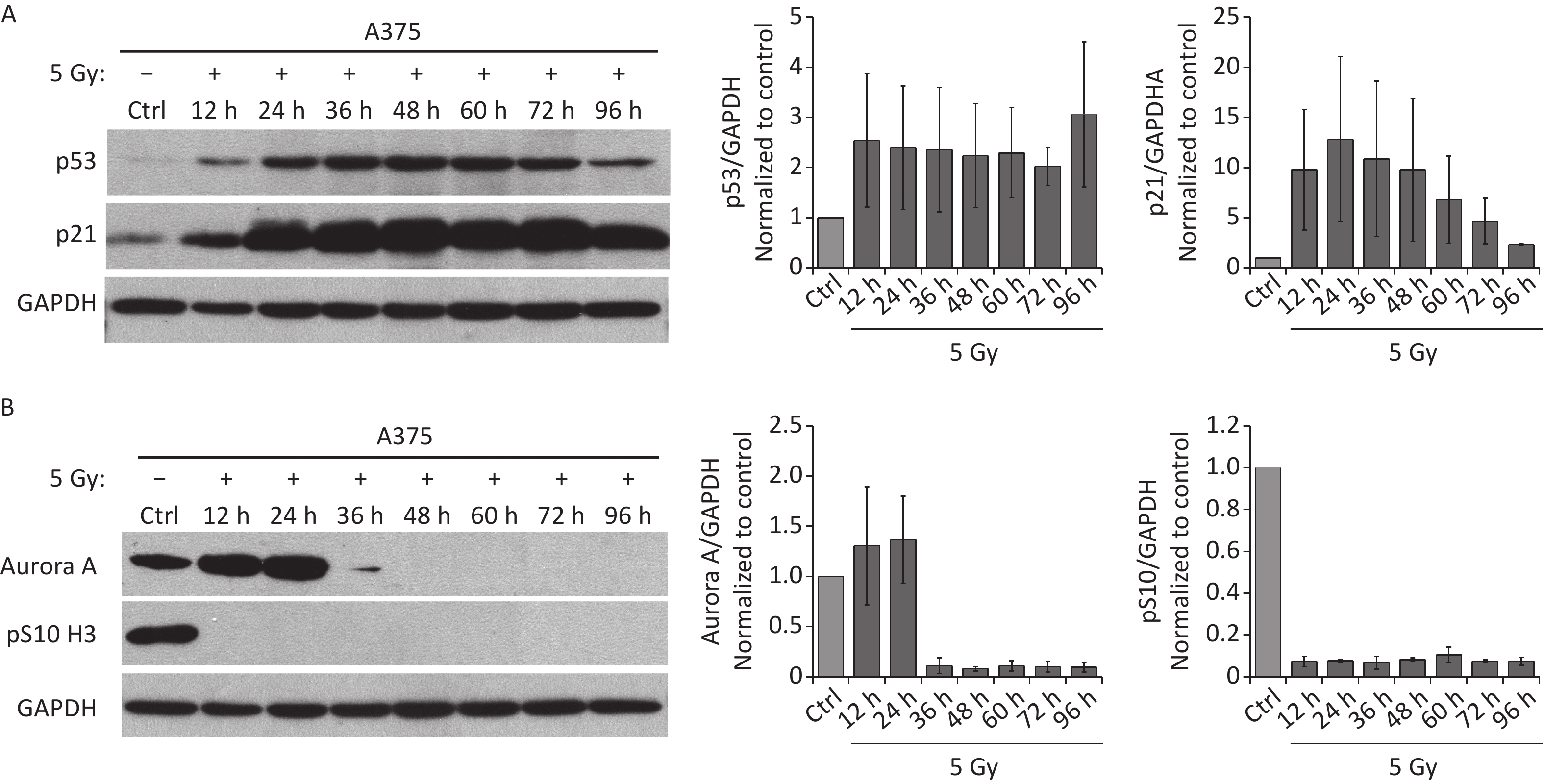
Because the p53/p21 pathway plays a central role in senescence induction[11], we measured the protein levels of p53 and p21 in irradiated A375 cells at different time points after treatment with 5 Gy of X-rays. As shown in Figure 2A, p53, and p21 protein levels increased quickly and were maintained at high levels until 3 d after irradiation, implying that the p53/p21 pathway was activated by IR in A375 cells. We next investigated whether the functional p53/p21 pathway is responsible for mitosis skip and senescence induction in tetraploid G1 cells. As a major downstream target of p53, p21 induction by various senescence-inducing stimuli results in premature activation of APC/C(Cdh1)[29], which is responsible for premature degradation of mitotic regulators[15]. In addition, several key factors essential for the G2-M transition, including Cdk1, cyclin B1, Plk1, and Aurora A significantly decline after IR exposure[13-15]. Loss of these mitotic regulators has been attributed to the induction of mitotic skipping in response to DNA damage. Because Aurora A activation triggers mitotic entry[20], we focused on whether p21 upregulation could alter Aurora A expression in irradiated A375 cells. As expected, the protein level of Aurora A decreased and was lost at 36 h after treatment with X-rays (Figure 2B), accompanied by G2 arrest and skipping of mitosis in response to IR. These results imply that Aurora A reduction is responsible for mitotic skipping and G2 senescence in response to DNA damage.
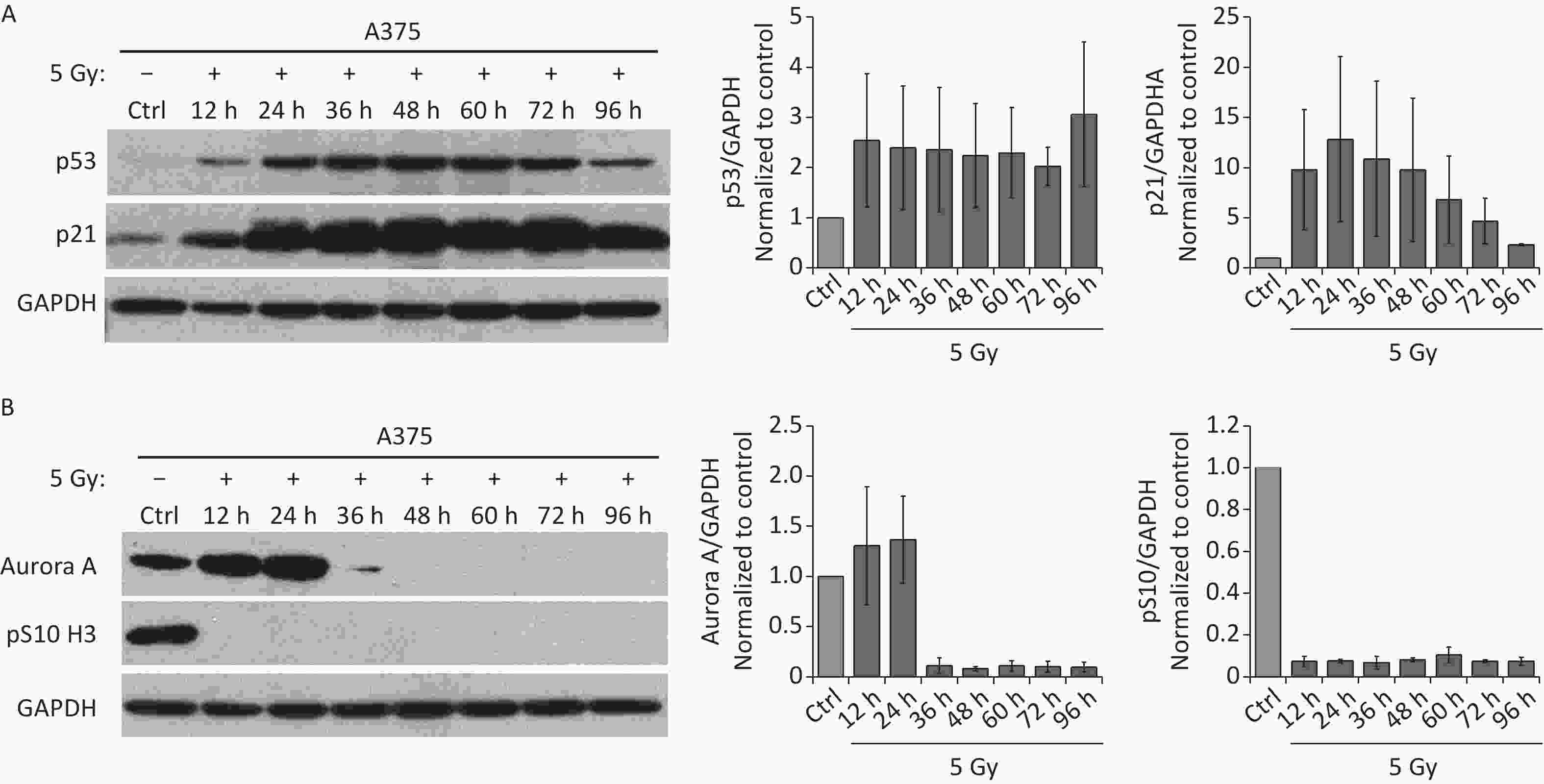
Figure 2. IR activates p53-p21 pathway and reduces Aurora A protein expression. (A–B) Western blot analysis (left) and densitometric quantification (right) of p53, p21 (A), Aurora A, and pS10 H3 (B) expression (normalized to GAPDH) in lysates of A375 control (Ctrl) cells or cells treated with 5 Gy of X-rays. Protein was harvested at the indicated time points post-irradiation and GAPDH was used as loading control.
-
To uncover the molecular basis underlying mitotic skip, we explored the relationship between activation of the p53/p21 pathway and mitosis entry. In irradiated A375 cells, p21 expression was markedly upregulated within a short time (Figure 2A), and Aurora A expression declined quickly over the same time period (Figure 2B). Notably, p21 depletion by siRNA strongly rescued the IR-induced reduction in Aurora A levels (Figure 3A and 3B). In addition, in these p21-depleted cells, a fraction of the irradiated cells entered mitosis and showed elevated pS10 H3 expression (Figure 3B and 3C). In contrast, p21 depletion did not affect the DNA repair ability of irradiated A375 cells (Figure 3C), suggesting that p21 upregulation contributed to mitosis skipping in response to IR via Aurora A reduction. Moreover, live-cell imaging results showed that a large fraction of cells did not switch from geminin to Cdt1 in response to IR; however, abnormal mitosis occurred in p21-depleted A375 cells (Figure 3D). In contrast, cells transfected with the negative siRNA left the cell cycle quickly and underwent mitosis skipping (degradation of geminin and accumulation of Cdt1 without mitosis entry) in response to IR (Figure 3D). Interestingly, an apoptosis-like phenotype was observed in p21-depleted A375 cells treated with 5 Gy of X-rays (Figure 3D). To confirm whether the cells that re-entered M phase underwent apoptosis, we further analyzed cellular apoptosis 48 h post-irradiation using the Annexin V-FITC/PI staining assay. The results showed that the proportion of apoptotic cells increased sharply in p21-depleted A375 cells in response to IR (Figure 3E). Taken together, these results suggest that p21 is responsible for Aurora A degradation and the induction of cellular senescence in G2-arrested cells.
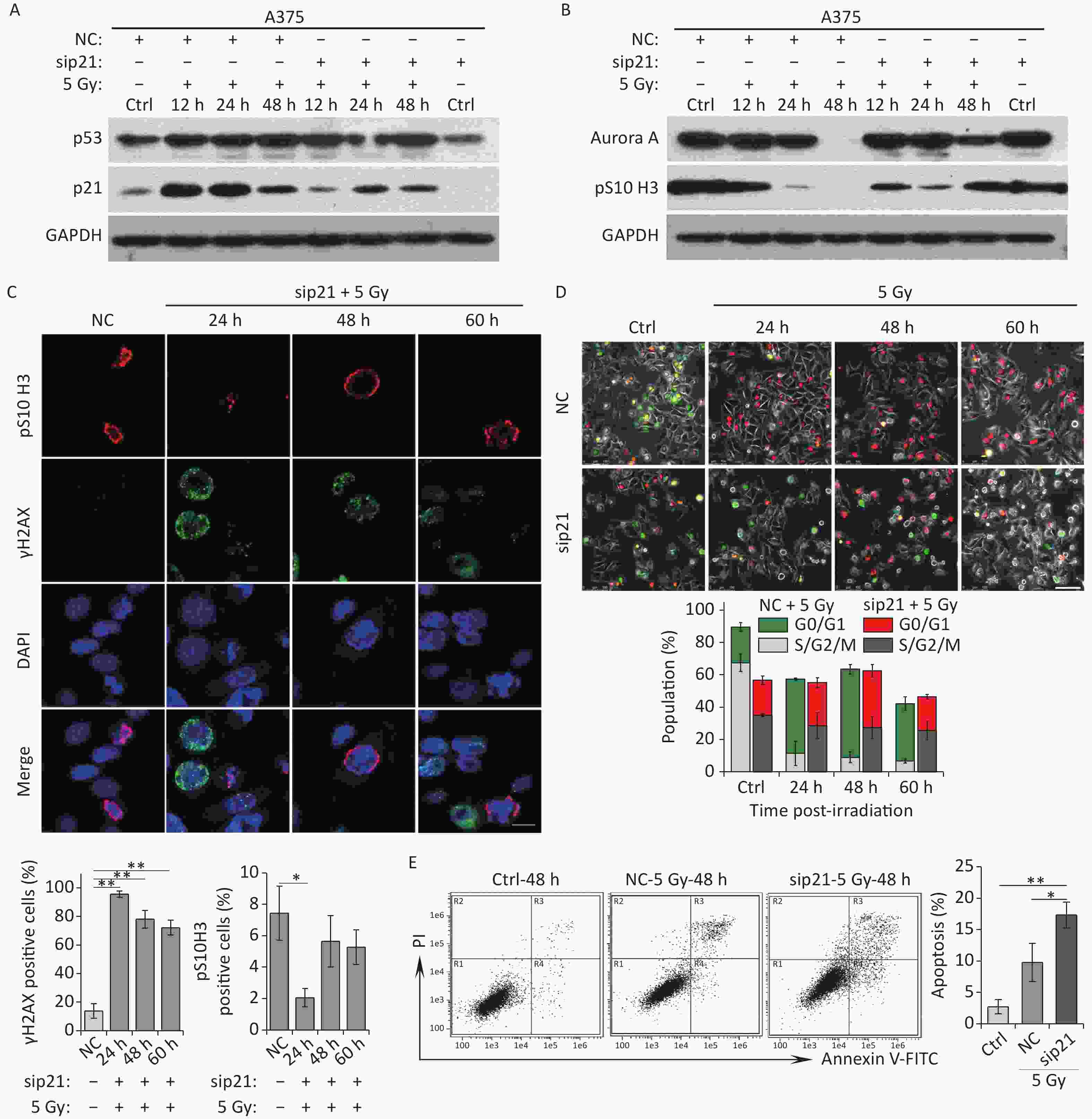
Figure 3. Knockdown of p21 rescues IR-induced Aurora A reduction and senescence induction. (A–B) Western blot analysis of A375 cells transfected with negative control siRNA (NC) or siRNA targeted p21 (sip21). Protein harvested at the indicated time points after X-rays irradiation were subjected to immunoblotting using anti-p53, p21 (A), Aurora A, and pS10 H3 (B) antibodies. (C) A375 cells were transfected with sip21 or directedly transfected with NC siRNA, then these cells were exposed to 5 Gy of X-rays. Representative images of γH2AX foci or pS10 H3 positive nuclei were obtained at the indicated time points post-irradiation (upper panel), and the percentage of γH2AX foci or pS10 H3 positive cells were calculated (lower panel). Scale bar, 10 µm. (D) A375 cells were infected with FUCCI lentiviruses expressing Cdt1-RFP (red) and geminin-GFP (green) and transfected with NC or sip21, then irradiated with 5 Gy of X-rays. Representative micrographs were captured at the indicated time points (upper panel) and the population was calculated (lower panel). Scale bar, 100 µm. (E) Flow cytometric analysis of apoptotic cells in A375 control cells (Ctrl) and cells transfected with sip21 or NC after exposure to 5 Gy of X-rays, at 48 h post-irradiation (left). Quantification of apoptotic cells (right). *P < 0.05, **P < 0.01, and ***P < 0.001, compared to Ctrl.
-
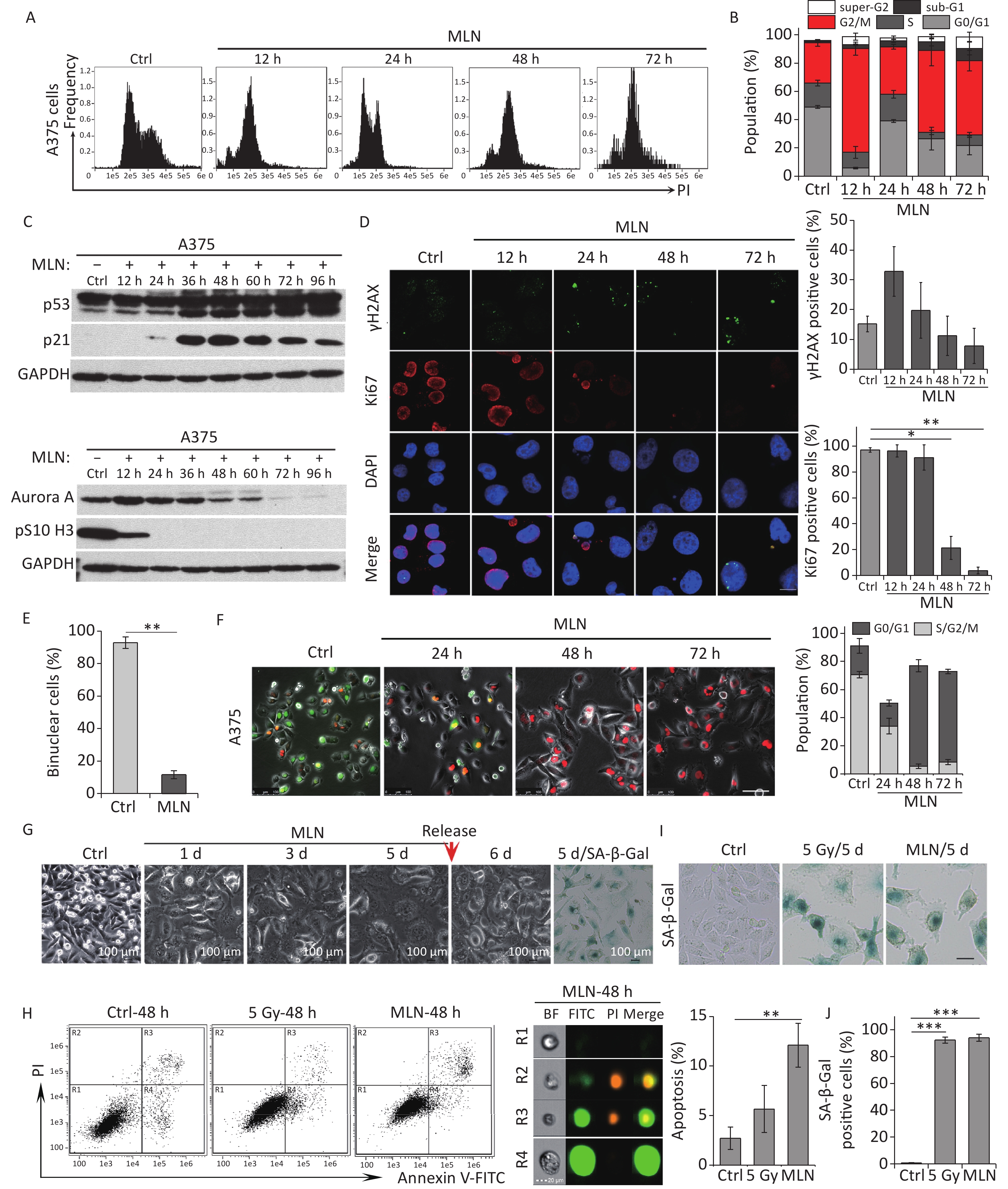
In irradiated cells, p21 upregulation was responsible for Aurora A degradation, mitosis skipping and cellular senescence, while p21-depelation lead to M phase entry and cellular apoptosis. Next, we investigated the role of Aurora A degradation in the induction of senescence in G2-arrested cells. To determine whether the loss of Aurora A kinase activity could cause cellular senescence in tumor cells, A375 cells were treated with Aurora A kinase inhibitor MLN8237 (MLN) at a dose of 0.5 μmol/L and the cell cycle progression of these cells was analyzed by detecting DNA content using flow cytometry. MLN treatment efficiently induced G2/M cell cycle arrest (Figure 4A and 4B). Combined with the finding that the mitotic markers pS10 H3 and Ki67 were lost after MLN treatment (Figure 4C and 4D), these data confirmed that the Aurora A kinase inhibitor induced G2 arrest in tumor cells. A375 cells were simultaneously treated with cytochalasin B and MLN for binuclear cell analysis. Notably, compared to cells treated with cytochalasin B alone, MLN treatment significantly inhibited mitotic entry and binuclear cell formation (Figure 4E). These results indicate that cells that lose Aurora A kinase activity cannot complete the G2-M transition or nuclear division.
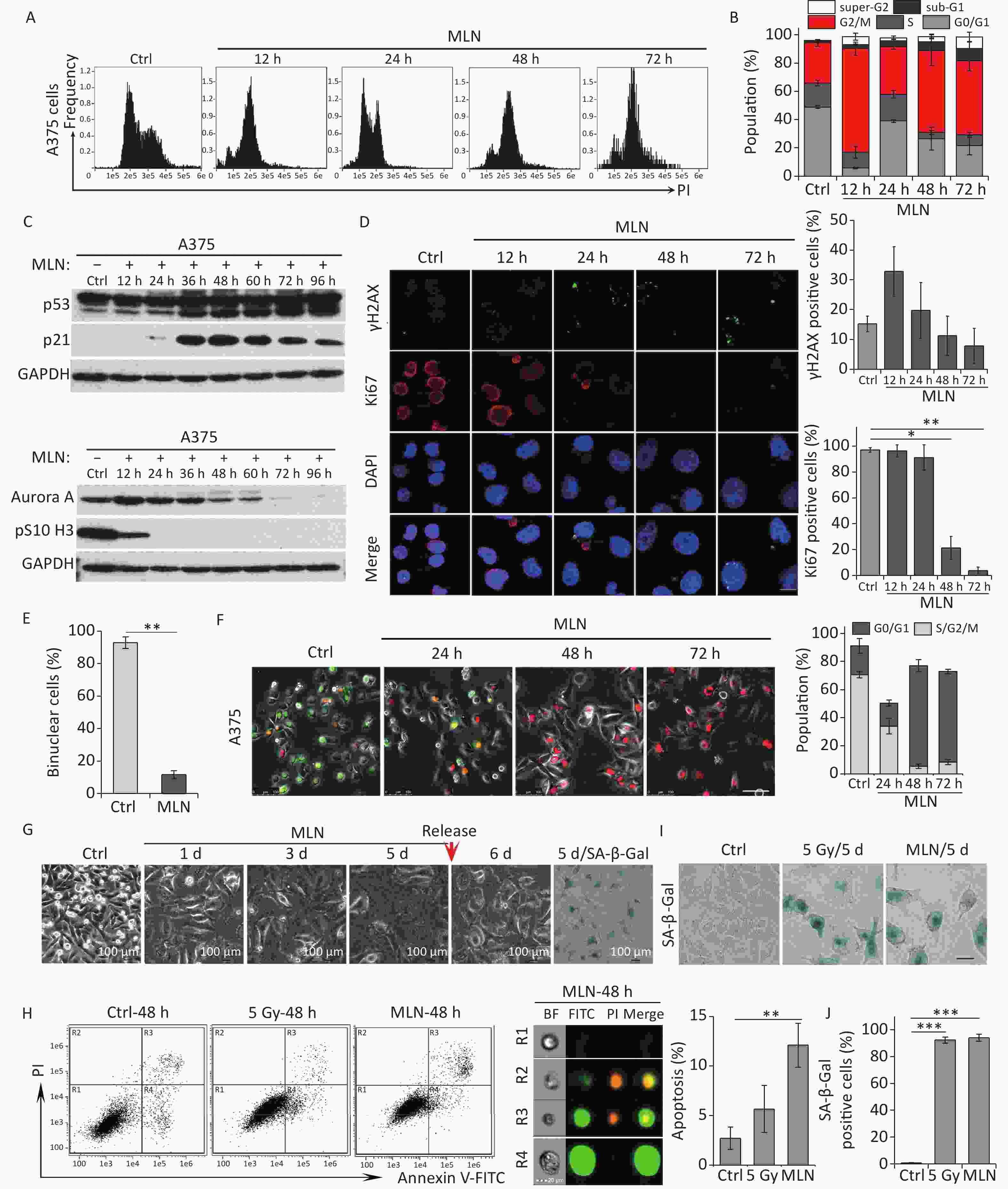
Figure 4. Aurora A kinase inhibition leads to G2 blockade and senescence in A375 cells. (A) Cell cycle distribution of A375 control (Ctrl) cells and cells treated with Aurora A inhibitor MLN (0.5 µmol/L) for 12 h, 24 h, 48 h, or 72 h. (B) Data analysis of cells in (A). (C) Cell lysates of A375 cells treated with MLN for the indicated times were subjected to immunoblotting using anti-p53, p21, Aurora A, and pS10 H3 antibodies. (D) Representative immunostaining images of anti-γH2AX and Ki67-positive nuclei in control (Ctrl) cells or cells treated with MLN, analyzed at the indicated time points (left), and the percentage of γH2AX foci or Ki67-positive cells (right). Scale bar, 10 µm. (E) The percentage of binucleated cells in A375 cells treated with cytochalasin B (Ctrl) or cytochalasin B combined with MLN. (F) A375 cells were infected with FUCCI lentiviruses expressing Cdt1-RFP (red) and geminin-GFP (green). Cells were treated with MLN for the indicated time periods and representative images from the indicated time points were acquired after MLN treatment (left). The graph (right) shows the population analysis data. Scale bar, 100 µm. (G) Representative images of cellular morphology of A375 cells treated with MLN for the indicated time periods and released from MLN by fresh medium washing for 1 d (5 d + 1 d), or SA-β-Gal staining performed 5 d after treatment with MLN (5d/SA-β-Gal). Scale bar, 100 µm. (H) Flow cytometric analysis of apoptotic cells in A375 control cells (Ctrl) and cells exposed to 5 Gy of X-rays or 0.5 μmol/L of MLN (left). Representative images of single cells from different gating regions detected by flow cytometry (middle). Quantification of apoptotic cells (right). (I–J) Representative images of control (Ctrl) cells, 5 Gy-irradiated, or MLN-treated cells stained for SA-β-Gal at 5 d after treatment, and the calculated data (J). Scale bar, 100 µm. *P < 0.05, **P < 0.01, and ***P < 0.001, compared to Ctrl.
Although our DNA content analysis results implied that a large fraction of cells was arrested at the G2 phase after treatment with MLN, we also sought to confirm whether the Aurora A inhibitor induced mitosis skipping after G2 arrest. Therefore, a live-cell imaging assay was performed on A375-FUCCI cells. After MLN treatment for 36 h, the proportion of cells expressing Cdt1 (red) increased without mitotic entry (Figure 4F). In addition, cells treated with MLN for 5 days exhibited typical senescent phenotypes such as an enlarged and flattened morphology (Figure 4G) and robust SA-β-Gal activity (Figure 4G and 4I). However, the percentage of apoptotic cells just slightly increased compared to that in cells treated with IR (Figure 4H). Together, these results suggest that Aurora A activity is crucial for the G2-M transition and that inhibition of kinase activity results in G2 arrest and skipping of mitosis, eventually causing cellular senescence via tetraploidy in G1 phase.
-
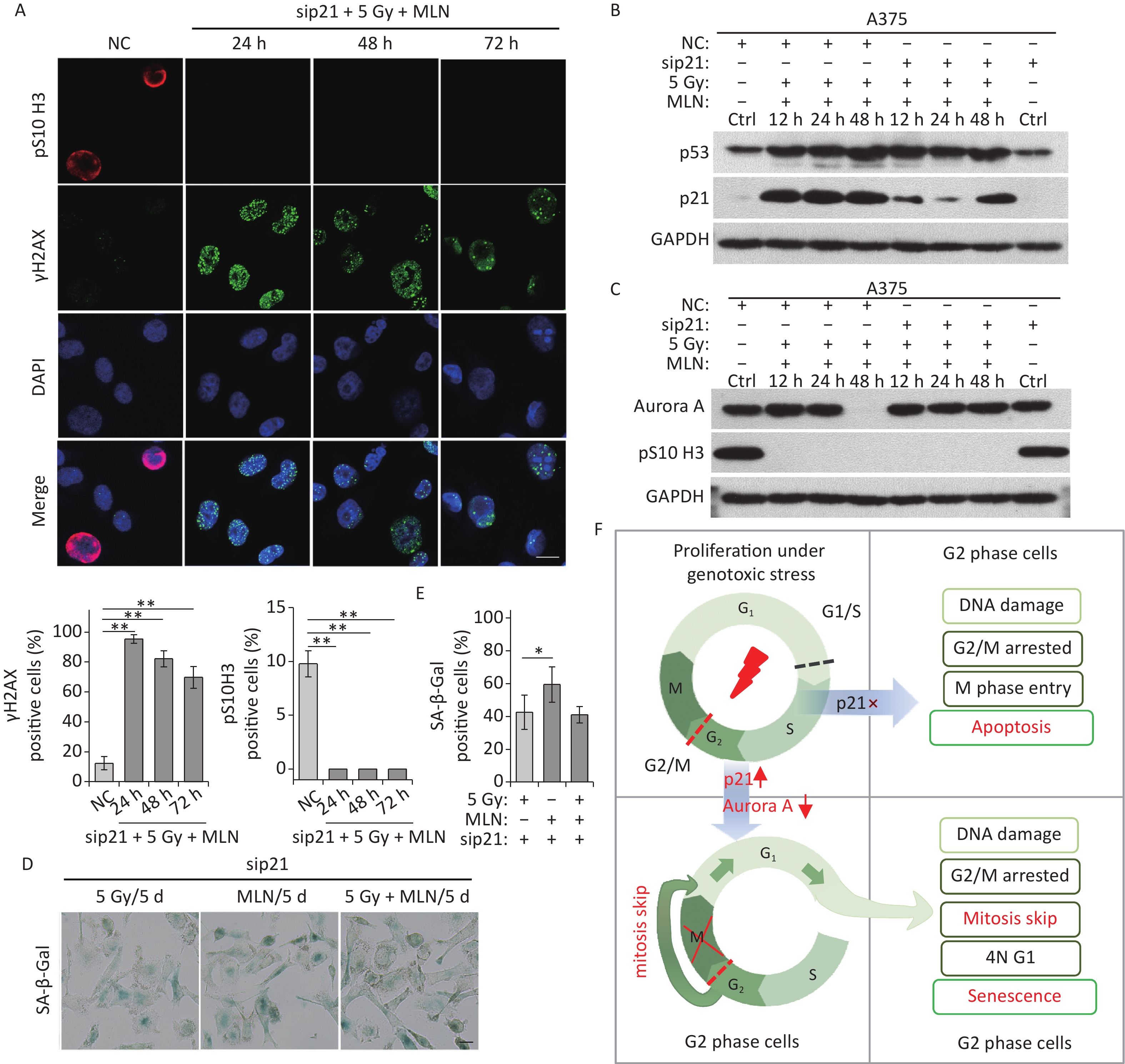
Although the Aurora A kinase inhibitor MLN induced G2 phase arrest, mitosis skipping, and tetraploid G1 senescence in A375 cells, p21 upregulation was also detected in A375 cells treated with MLN for 36 h (Figure 4C and 5A). To determine whether Aurora A inhibition-induced senescence was dependent on p21 upregulation in response to DNA damage, p21 was depleted using siRNA in A375 cells prior to MLN treatment (Figure 5B). The live-cell imaging assay revealed that p21 knockdown did not alter the senescent fate of these cells due to inhibition of Aurora A kinase activity (Figure 5C–5F). Without p21 upregulation and DNA damage (Figure 5B and 5D), although a portion of cells underwent apoptosis (Figure 5G), the majority of the cells treated with MLN underwent G2 arrest and mitosis skipping, followed by tetraploid G1 senescence, suggesting that inhibition of Aurora A kinase activity was sufficient to stimulate the entry of G2-arrested cells into senescence via mitosis skipping and G1 tetraploidy.
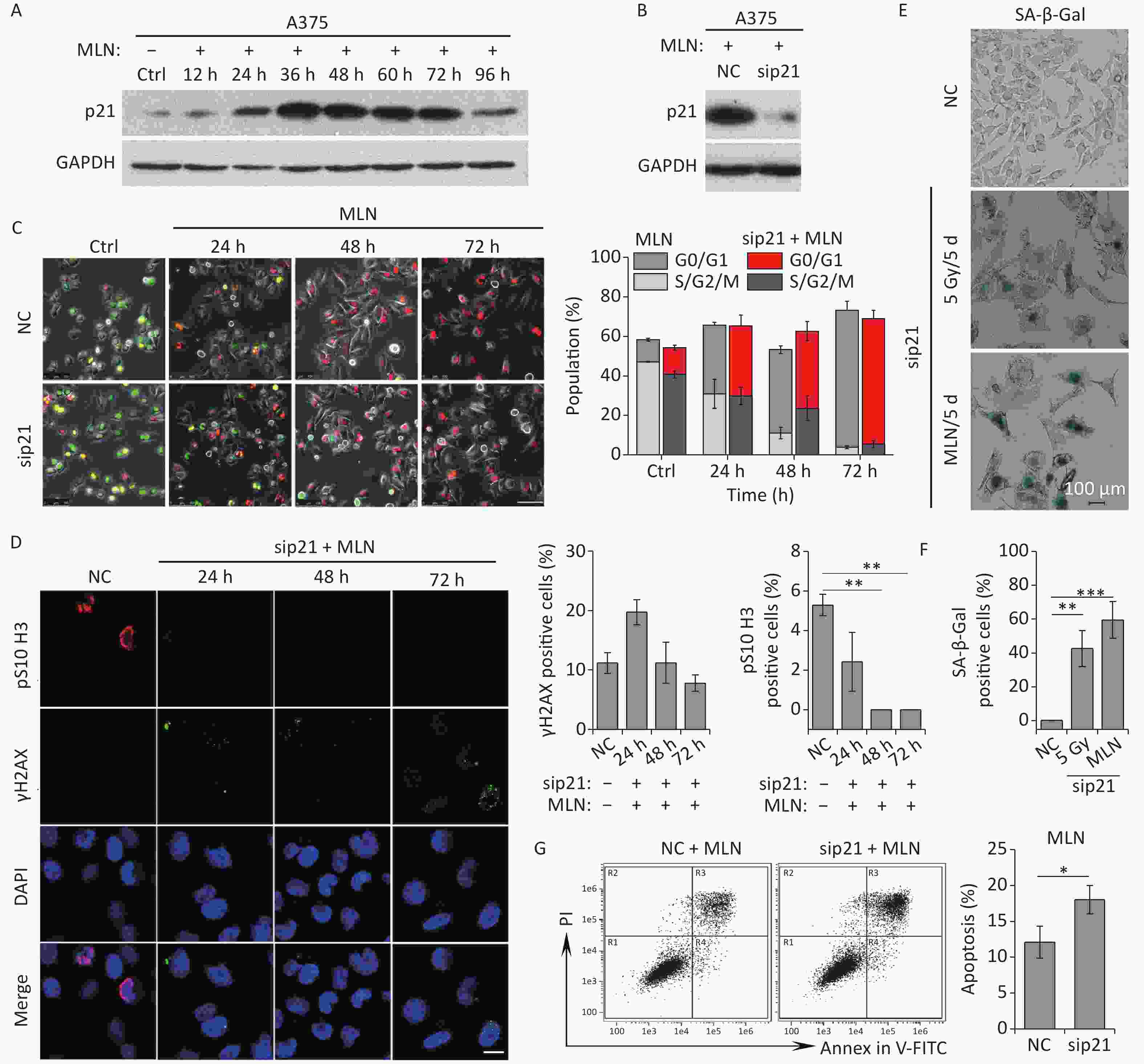
Figure 5. Effect of p21 knockdown on MLN-induced senescence in A375 cells. (A) Cell lysates of A375 cells treated with MLN for the indicated time periods were subjected to immunoblotting using anti-p21 antibodies. GAPDH was used as loading control. (B) Cell lysates of A375 cells transfected with NC or sip21 at 48 h after MLN treatment were subjected to immunoblotting using anti-p21 antibodies. GAPDH was used as loading control. (C) A375 cells were transfected with NC or sip21 and infected with FUCCI lentiviruses expressing Cdt1-RFP (red) and geminin-GFP (green). Cells were treated with MLN for the indicated time periods. The resulting cells were photographed 3 d after treatment. Representative images at the indicated time points are shown (left), and the cell population was analyzed (right). Scale bar, 100 µm. (D) A375 cells were transfected with NC or sip21. Representative immunostaining micrographs of anti-pS10 H3-positive nuclei and γH2AX foci in untreated A375 control (Ctrl) cells or cells treated with MLN for the indicated time periods are shown (left), and the data were measured (right). Scale bar, 10 µm. (E) A375 cells were transfected with NC or sip21. Representative images of NC, 5 Gy-irradiated, and MLN-treated cells stained for SA-β-Gal at 5 d after treatment (left), with the analyzed data shown in (F). Scale bar, 100 µm. (G) Flow cytometric analysis of apoptotic cells in A375 cells transfected with NC or sip21 and treated with 0.5 μmol/L of MLN (left). Quantification of apoptotic cells (right). *P < 0.05, **P < 0.01, and ***P < 0.001.
This was confirmed in p21-depleted A375 cells treated with X-rays and MLN using DNA damage (γH2AX foci) and mitosis entry (pS10 H3 immunofluorescence staining) analyses. The results showed that p21 knockdown did not alter DNA damage repair in A375 cells (Figure 6A), whereas Aurora A kinase inhibition prevented mitotic entry caused by p21-depletion in IR-exposed A375 cells (Figure 6A and 6C). In addition, the expression levels of p53, p21, and Aurora A were examined using immunoblotting. Significantly, the inhibition of Aurora A kinase did not alter p53 expression in response to DNA damage, although the expression of p21 was decreased dramatically by siRNA treatment (Figure 6B). Though the protein level of Aurora A was rescued by IR-induced degradation in p21-depleted cells (48 h time point in Figure 6C), kinase activity was inhibited by MLN (Figure 6C). Ultimately, although p21 was depleted by siRNA transfection, A375 cells still underwent senescence after IR treatment (Figure 6D–6E). Collectively, these results suggest that the induction of cellular senescence caused by Aurora A deficiency is independent of p21 expression.
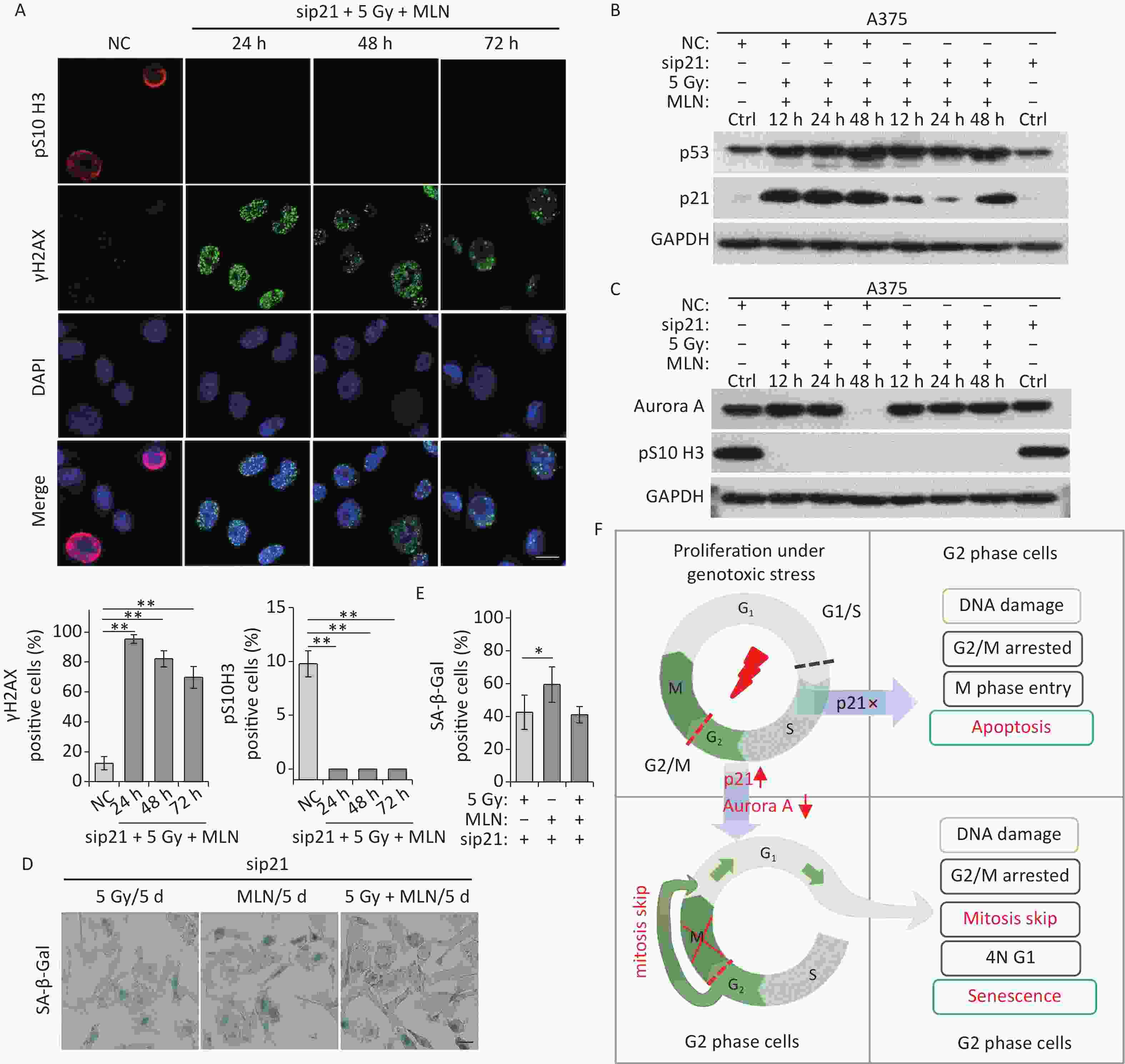
Figure 6. MLN-induced senescence in p21-depleted A375 cells is independent of DNA damage. (A) A375 cells were transfected with sip21 prior to treatment with 5 Gy of X-rays and MLN or only transfected with negative control siRNA (NC). Representative immunostaining images of anti-pS10 H3-positive nuclei and γH2AX foci in untreated cells (NC) or cells treated with MLN combined with X-rays, captured at the indicated time points, are shown (upper panel), and the data were analyzed (lower panel). Scale bar, 10 µm. (B–C) A375 cells were transfected with NC or sip21 prior to treatment with 5 Gy of X-rays and MLN. Cell lysates harvested at the indicated time points were subjected to immunoblotting using the indicated antibodies. (D) A375 cells were transfected with NC or sip21, and then treated with 5 Gy of X-rays, MLN, or 5 Gy of X-rays combined with MLN. Representative images of cells stained for SA-β-Gal at 5 d after treatment, and the calculated data are shown in (E). Scale bar, 100 µm. (F) Schematic of the proposed model describing the fate determination of G2-arrested cells induced by IR. *P < 0.05, **P < 0.01, and ***P < 0.001, compared to Ctrl.
-
In this study, we demonstrated that IR induced non-programmed mitotic failure and accumulation of tetraploid senescent cells in the A375 and 92-1 cell lines. Senescent cell accumulation poses a serious threat to the health of most organisms and facilitates tumor development[30]. However, our analyses show that excessive DNA damage elicited by IR leads to an exit from the cell cycle, resulting in irreversible G1 or G2 arrest following a senescent-like phenotype (Figure 1 and Supplementary Figure S1). Although we confirmed that severe DNA damage caused a large fraction of A375 cells to arrest at G2, live-cell imaging combined with the FUCCI reporter system revealed that these G2-arrested cells were unstable and skipped mitosis before senescence induction, generating G1 cells with a tetraploid nucleus (Figure 1F). Robust evidence suggests that most senescent signals activate the p53/p21 and/or p16/pRb pathways (Figure 2). Considering that mitosis skip is necessary for senescence induction in G2-arrested cells and that p21 is crucial for the maintenance of G2 arrest[15,31], we speculated that p21 is a key regulator of mitosis skip. Indeed, in p21-depleted IR-treatment cells, although they incurred severe DNA damage, a fraction of G2-arrested cells still overrode the G2/M checkpoint and completed the G2-M transition, although these mitotic cells did not complete nuclear division and ultimately underwent apoptosis (Figure 3). Collectively, our results emphasize that the G2-M transition is a crucial checkpoint that determines the apoptotic or senescent fate of cells with severe DNA damage, with p21 levels serving as the key regulator.
p21-mediated inhibition of Cdk1 and Cdk2 in the G2 phase has been proposed to prematurely activate APC/CCdh1 to degrade various APC/C substrates, resulting in long-term growth arrest at G2 in response to DNA damage[32,33]. Our previous studies have confirmed that high-dose X-rays treatment induces p21 upregulation and the decline of G2-M transition regulators, including Cdk1, Plk1, cyclin B1, and Aurora A[13,14,26]. Here, we showed that knockdown of p21 in A375 cells rescued the decline in the G2-M transition induced by IR, especially the decline in Aurora A kinase levels (Figure 3B). In addition, p21 knockdown stimulated mitotic entry, followed by apoptosis (Figure 3D and 3E). Interestingly, although p21 upregulation was accompanied by a decline in Cdk1, Plk1, cyclin B1, and Aurora A in IR-treated cells, only treatment with MLN, a specific inhibitor of Aurora A kinase, induced senescent-like phenotypes in A375 cells independent of p21 expression (Figures 4 and 5). Other kinase inhibitors (AZD5438 for Cdk1/cyclin B1 complexes and BI6727 for Plk1 kinase) did not induce long-term G2 arrest or senescent-like phenotypes (data not shown). These results highlight the core role of Aurora A in IR-induced senescence induction of G2-arrested cells via mitotic skipping. Based on the obtained data, we developed a cellular model to describe the fate determination of G2-arrested cells induced by IR. Genotoxic stress induces DNA damage and triggers G2 phase arrest in proliferating cells. In this process, if p21 is not expressed in damaged cells, the cells enter the M phase and undergo apoptosis with severe, unrepaired DNA damage. Otherwise, DNA damage-induced p21 upregulation can trigger Aurora kinase A decline and thus induce long-term G2 arrest and skipping of mitosis in damaged cells, which ultimately undergo tetraploid G1 phase senescence (Figure 6F).
Aurora A kinase is amplified and overexpressed at the transcript and protein levels in various malignancies but is restricted or absent in most adult tissues. It is considered a promising therapeutic target, and a number of clinical trials focused on Aurora A have been carried out[34-37]. Our results demonstrated that the inhibition of Aurora A kinase induced cellular senescence in tumor cells independent of the p53/p21 pathway. Senescent cells can be long-lived and they accumulate in aged and damaged organs. The elimination of senescent cells is known to increase healthy lifespan and reduce the severity of age-associated diseases[38-42]. For example, pharmacological perturbation of the function of FOXO4, a transcription factor essential for the survival of senescent cells, reverses age-associated defects and tissue dysfunction caused by chemotherapy in a mouse model[43]. Scientists and pharmaceutical companies are keen to identify drugs known as “senolytics” that are able to eliminate senescent cells in the hope of rolling back or at least forestalling the ravages of age[44], and numerous promising senolytics have been described in the literature, including small molecules, antibodies, and peptides[3,44,45]. Further studies are required to determine whether Aurora A kinase inhibitor-induced senescent cells can be eliminated using these senolytics in vitro and in vivo. Therefore, using Aurora A kinase inhibitors combined with senolytics would be a promising therapeutic strategy for improving the outcomes of cancer treatment.
-
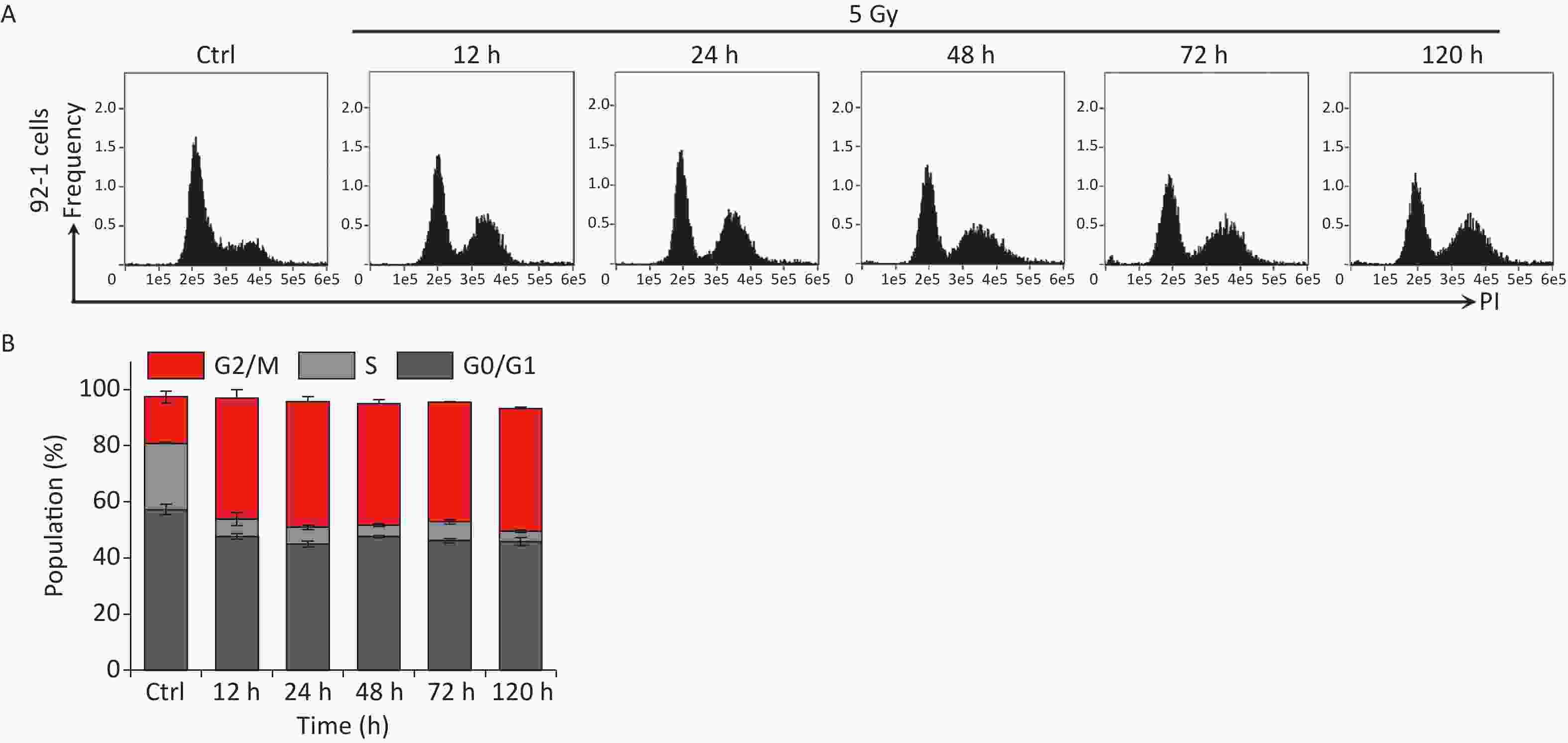
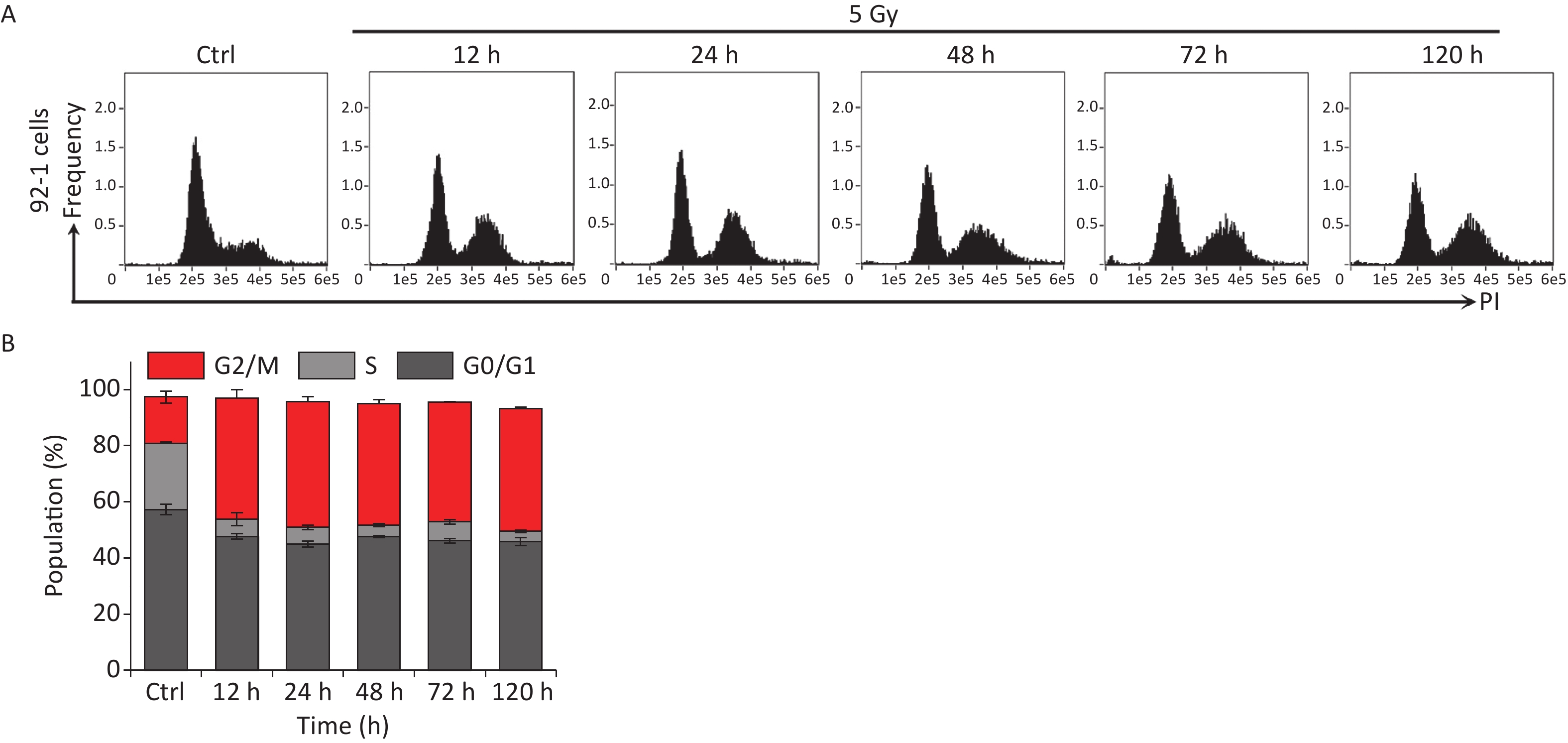
Figure S1. IR induces persistent cell cycle arrest in 92-1 cells. (A) 92-1 cells were exposed to 5 Gy of X-rays following stained with PI, and analyzed by flow cytometry at indicated time points after IR radiation. (B) The cell cycle distribution of cells in (A).
HTML
Cell Culture and Irradiation
Cell Cycle Analysis
Immunofluorescence
RNA Interference
Time-lapse Microscopy
Senescence-associated β-Galactosidase (SA-β-Gal) Staining
Western Blot Analysis
Apoptosis Assay
Statistical Analysis
G2 Cells Skip Mitosis during Senescence Induction in Response to IR
p53/p21 Activation Results in Aurora A Downregulation in Response to IR
Knockdown of p21 Rescued IR-induced Aurora A Reduction and Senescence Induction
Aurora A Kinase Inhibition Triggers Mitosis Skipping and Cellular Senescence
Cellular Senescence Induced by Aurora A Kinase Inhibition is Independent of p21
COMPETING INTERESTS The authors declare no competing financial interests.
&These authors contributed equally to this work.
 22403+Supplementary Materials.pdf
22403+Supplementary Materials.pdf
|

|

 Quick Links
Quick Links
 DownLoad:
DownLoad: