




-
Viral encephalitis is one of the most common central nervous system (CNS) infections, and its clinical symptoms include fever, headache, convulsions, and impaired consciousness. Cured patients may develop lasting neurological sequelae, resulting in a huge disease burden on the individuals, their families, and society. Various types of viruses, including herpes, flaviviruses, and enteroviruses, can cause viral encephalitis. According to epidemiological surveys, viral encephalitis occurs in all age groups, with a prevalence of 3.5–7.4/100,000 and a high rate of disability and death[1]. In China, viral encephalitis had an incidence rate of 15.27/100,000, prevalence rate of 74.34/100,000, and death rate of 0.24/100,000 in 2019[2], making it one of the most important infectious diseases threatening national health.
Viral encephalitis is frequently caused by herpes simplex virus-1 (HSV-1), herpes simplex virus-2 (HSV-2), varicella zoster virus (VZV), Epstein-Barr virus (EBV), and human cytomegalovirus (HCMV). Among these, HSV-2 is more likely to cause meningitis[3], and HCMV-induced encephalitis mostly affects immunocompromised patients. Although other herpesviruses can cause encephalitis, only a few cases have been reported.
The main insect-borne pathogens that cause viral encephalitis are the Japanese encephalitis virus (JEV) and the West Nile virus (WNV), both of which belong to the family Flaviviridae and the genus Flavivirus. Japanese encephalitis (JE) is the most common type of viral encephalitis and has the highest disease burden worldwide. In recent years, the incidence of adult JE has been high in the northern region of China, and the population in this area has become the main population affected by JE. In 2013, 407 cases of JE were reported in Shandong Province, of which 73% were over 15 years of age[4]. In 2017, an outbreak of JE occurred in Pingliang City, Gansu Province, with 78 adult cases and 7 deaths[5]. WNV is currently the most widespread flavivirus in the world, and in 2018, cases of WNV infection comprised 94% (2,647) of the arboviral disease cases recorded in the United States. Surveillance revealed a WNV epidemic in Kashgar, Xinjiang, China, from 2013 to 2016[6]. Encephalitis/meningitis caused by other arboviruses has been rarely reported in China.
Enteroviruses (EVs) are a major cause of viral encephalitis/meningitis in the pediatric population in China[7]. After 2008, many outbreaks of hand-foot-mouth disease occurred in our country, and central nervous system damage is the main clinical manifestation of severe hand-foot-mouth disease, in which enterovirus A71 (EV-A71) is the main pathogen[8]; Coxsackievirus (CV) types 6, 10, and 16 (group A) and types 3, 4, and 5 (group B) also cause viral encephalitis[9,10] as do other respiratory viruses, including mumps, measles, and rubella viruses.
Laboratory diagnosis of viral encephalitis pathogens is challenging, and the cause is successfully identified in only approximately 50% of patients with viral encephalitis due to the wide variety of causative pathogens, differences in their viral characteristics, and diversity of pathogenic mechanisms[11]. In recent years, high-throughput sequencing (HTS) technology, also known as next-generation sequencing (NGS), has been increasingly used for infectious disease surveillance[11]. Two different NGS strategies are used for the molecular diagnosis of infectious diseases: Metagenome NGS (including macrotranscriptome sequencing) and targeted amplification sequencing (the sequencing strategy used in this study). Theoretically, macrogenome sequencing is useful for capturing the whole-genome sequences of unknown pathogens with great sequencing depth and massive data output. However, it generates a large amount of host or background genomic data and is expensive[12,13]. By contrast, targeted amplification sequencing is more efficient, accurate, and less costly. Multiplex detection methods for the pathogens that cause viral encephalitis have not been well studied. In this study, we established a platform for the detection of viral encephalitis pathogens using amplicon sequencing.
-
In this study, we screened various viral encephalitis pathogens based on their domestic and international incidences and epidemiological trends. Ultimately, 12 species were selected: HSV-1, HSV-2, VZV, EBV, and HCMV in the family Herpesviridae; JEV and WNV in the family Flaviviridae; EV71, CA6, CA10, and CA16 in the genus Enterovirus; and mumps virus in the family Paramyxoviridae.
-
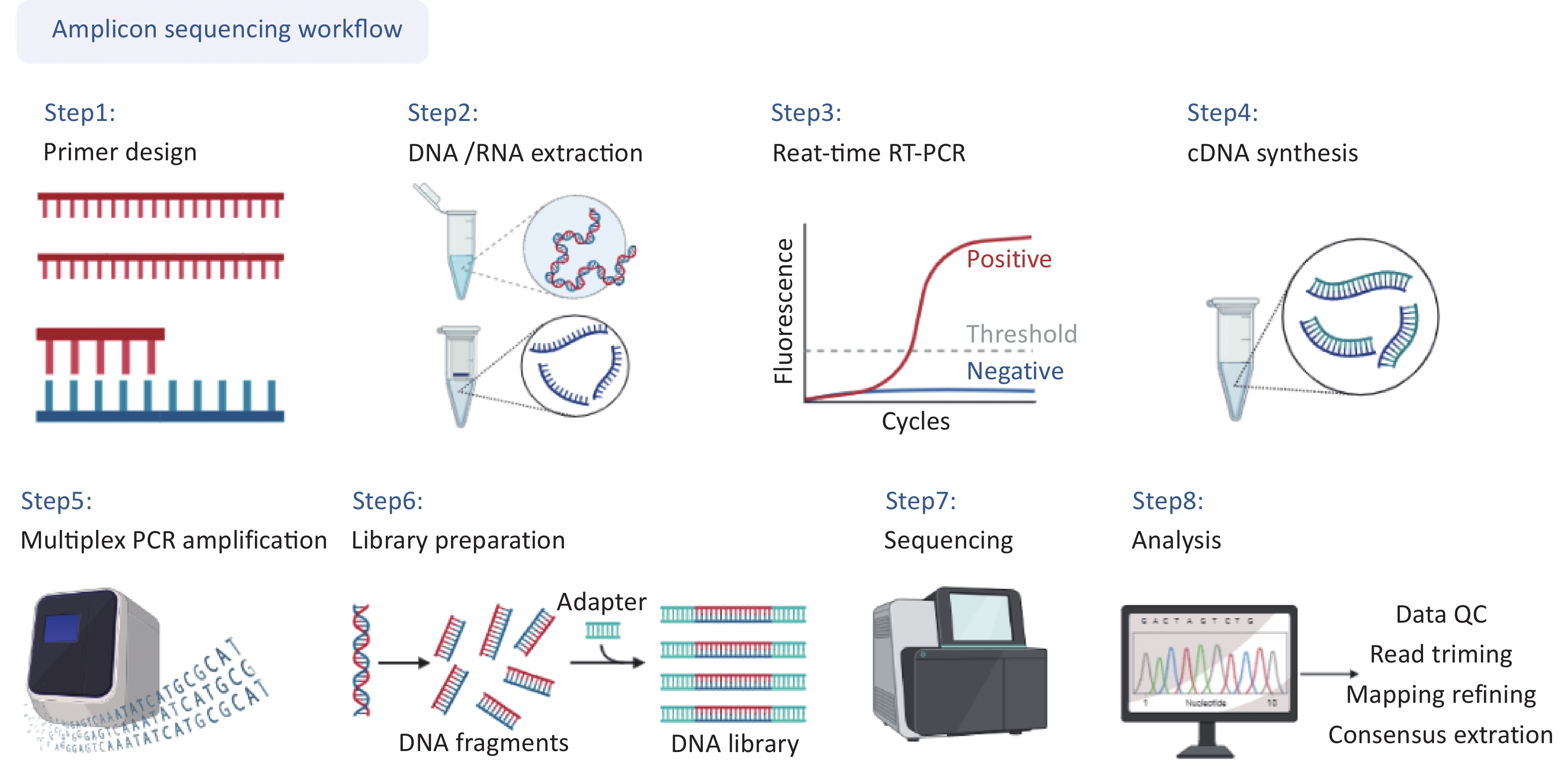
The complete workflow of this study is shown in detail in Figure 1, including (i) primer design, (ii) DNA/RNA extraction, (iii) real-time reverse transcription-polymerase chain reaction(RT-PCR) for concentration estimation, (iv) cDNA synthesis, (v) multiplex PCR amplification, (vi) library preparation, (vii) sequencing, (viii) data analysis.
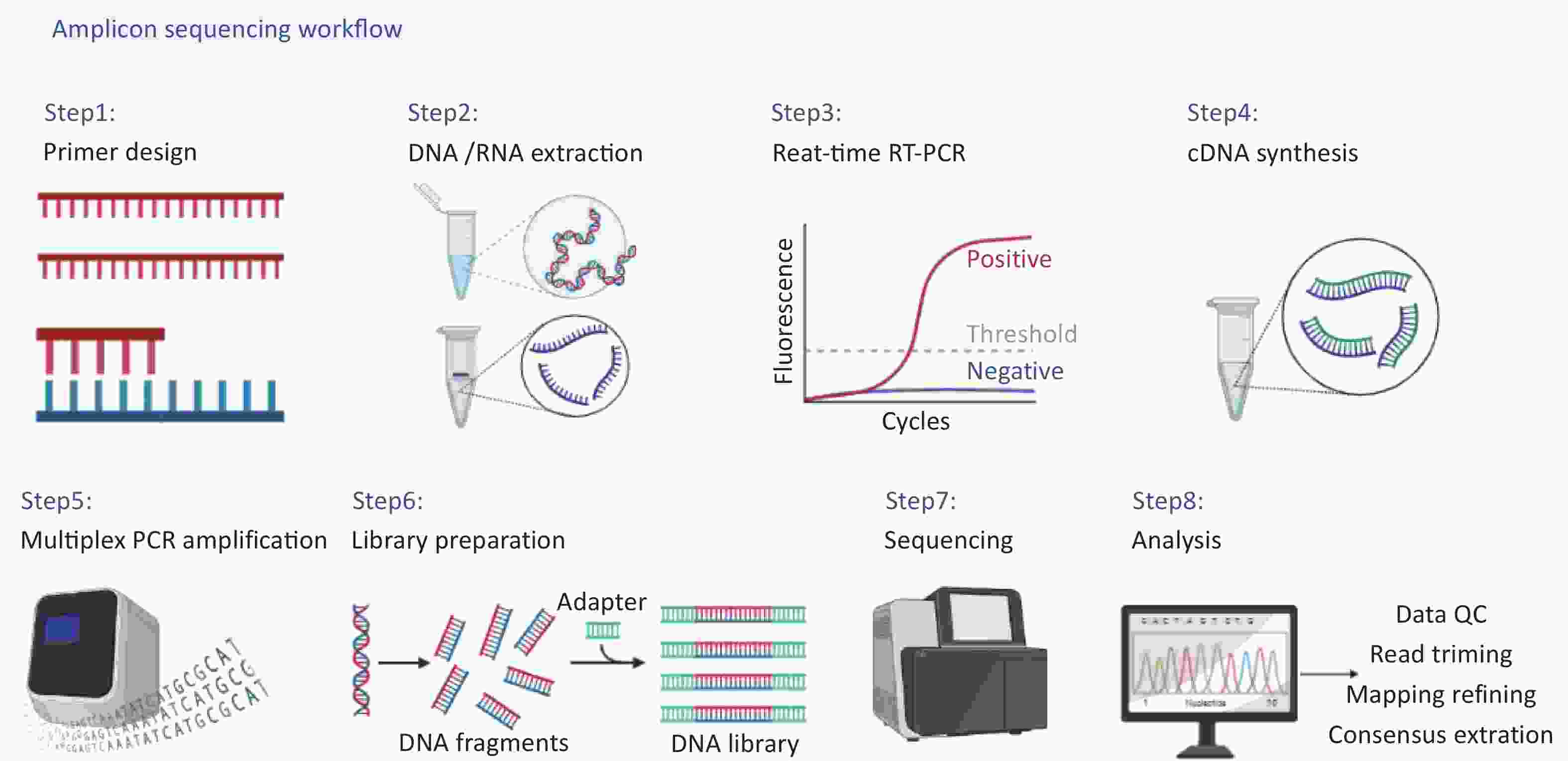
Figure 1. Amplicon sequencing workflow.
-
Primers for the selected pathogens were designed and validated against their target genes (Table 1), and the specificity of the primers was verified using the National Center for Biotechnology Information (NCBI) BLAST. Primers were synthesized by Shanghai Bioengineering Technology Service.
Primer
namePrimer sequence (5'–3') Amplicon size (bp) Target gene Ref HSV-1-F
HSV-1-RGGTCAGCTCGTGATTCTGCA
CGCATCAAGACCACCTCCTC106 UL27 − HSV-2-F
HSV-2-RCTAGTTGTCGCGGTGGGACT
TAGTACACAGTGATCGGGAT212 US5 [14] EBV-F
EBV-RCTCTAACTCCAACGAGGGCAG
GAGGGCCTCCATCATTTCC
320
LMP1
−HCMV-F
HCMV-RAATGATATCCGTACTGGGTCCCATT
TGATGATGGGGATGTTCAGCAT
350
UL83
−VZV-F
VZV-RGGGTTTTGTATGAGCGTTGG
CCCCCGAGGTTCGTAATATC447 ORF22
[15]JEV-F
JEV-RCGTTCTTCAAGTTTACAGCATTAGC
CCYRTGTTYCTGCCAAGCATCCAMCC674 E [16] WNV-F
WNV-RACCAACTACTGTGGAGTC
TTCCATCTTCACTCTACACT445 E [17] EVs-F
EVs-RTTCTGTTTCCCCGGACTGAG
TAAAAGGAAACACGGACACCCA400 VP3 − Mumps-F
Mumps-RAATATCAAGTAGTGTCGATGA
AGGTGCAAAGGTGGCATTGTC316 SH [18] Note. − is a self-designed primer; HSV-1, herpes simplex virus-1; HSV-2, herpes simplex virus-2; EBV, epstein-barr virus; HCMV, human cytomegalovirus; VZV, varicella zoster virus; JEV, Japanese encephalitis virus; WNV, west nile virus; EVs, Enteroviruses. Table 1. Summary of primers for various pathogens
In the initial primer pool, we included a total of 16 pairs of primers, but after optimization, the primer pool of this platform finally conducted verification and later experiments against nine pairs of primers for 12 viruses, including HSV1, HSV2, EBV, HCMV, VZV, JEV, WNV, EV71, CA6, CA10, CA16, and mumps virus, with five pairs referring to relevant literature and four pairs designed in this study. We performed specificity verification on NCBI and PCR verification using the virus strain’s nucleic acid for the primers in the references. The primary reference sequences of the pathogens were downloaded from the NCBI database for the primers designed in this study, and multiple reference sequences for the same virus were compared online by muscle (https://www.ebi.ac.uk/Tools/msa/muscle/). Primers were designed using Geneious Prime software. According to the principles of primer design, primers with excessive mismatches or low specificity should be modified, eliminated, or redesigned manually.
-
The nucleic acids of HSV-1, HSV-2, HCMV, and EBV were derived from the aqueous humor of clinical patients with anterior uveitis; nucleic acids for EV71, CA6, CA10, CA16, and mumps virus were obtained from pharyngeal swabs of patients who were clinically positive for these viruses; and for VZV, nucleic acids were extracted from vaccines. The other two viruses (JEV and WNV) were obtained from strains maintained in Institute of Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention. RNA was extracted from RNA virus strains using the Qiagen Viral RNA mini kit (Qiagen, #52906), and DNA was extracted from DNA virus strains using the QIAamp DNA Mini Kit (Qiagen, #51304), according to the manufacturer’s instructions.
-
Prior to cDNA synthesis, RT-PCR was performed to estimate the concentration of the target template. Real-time RT-PCR was performed using a commercial kits. The reaction system was configured, and the reaction procedure was determined according to the manufacturer’s instructions.
DNA was extracted directly from viruses. For RNA viruses, RNA was extracted, reverse-transcribed into cDNA, and the DNA was amplified. PCR was used to confirm the specificity of primers for DNA viruses, and RT-PCR was used to confirm the specificity of primers for RNA viruses. The amplification products were analyzed using gel electrophoresis to assess amplification specificity, and the fragment sizes of the target bands were compared with the electrophoresis map of the PCR products detected via gel electrophoresis to determine whether they were consistent with the predicted primer-amplified fragment sizes. The PCR products were sent to Shanghai Bioengineering Engineering Service for sequencing. The sequencing results were verified using NCBI BLAST to ensure accuracy.
-
Nucleic acid samples with Ct values < 20 were subjected to 10–100-fold gradient dilutions prior to cDNA synthesis. SuperScript IV VILO Master Mix (Invitrogen, #11756500) was used for cDNA synthesis, according to the manufacturer’s instructions.
-
The primers in Table 1 were diluted to 10 μmol/L and mixed into one tube in equal volumes, and Q5 Hot Start High-Fidelity DNA Polymerase (NEB, #M0493L) was selected for multiplex PCR amplification. The cDNA template has a volume of 5 μL, and amplification was performed in a 50 μL reaction with primer concentration set to 0.02 μmol/L per primer. The reaction procedure for multiplex PCR was as follows: hot start at 98 °C for 30 s, followed by 30 cycles of 98 °C for 10 s, 52 °C for 20 s, 72 °C for 40 s,and 72 °C extension for 3 min.
-
Amplification products were purified using AMPure XP magnetic beads (Beckman Coulter, # A63881) at a sample-to-bead ratio of 1:1. Purified DNA samples were quantified using a Qubit 4.0 fluorometer (Invitrogen; #Q33238) with Qubit double-stranded DNA (dsDNA) high-sensitivity (HS) assay kits (Invitrogen; #Q32851). Depending on the quantification results, all DNA samples were diluted to 0.2 to 0.3 μg/μL prior to library preparation. After diluting each sample to 0.2 μg/μL, equal volumes were mixed into one tube (e.g., 5 μL for each sample), and Qubit quantification was then used to confirm that the mixed sample concentration was indeed 0.2 μg/μL.
-
DNA libraries were constructed using the Nextera XT DNA Library Preparation Kit (#FC-131-1024; Illumina) according to manufacturer’s instructions. After enzymatic fragmentation, library amplification was performed on the fragmented samples using combined dual indices (Nextera DNA Indexes, Illumina, #FC-121-1011). Library quantification was performed using a Qubit 4.0 fluorometer (Invitrogen, #cQ33238) and Qubit dsDNA HS Assay Kit (Invitrogen, # Q32851). Absolute quantification of libraries was performed using the Library Quantification Kit For Illumina. Libraries were scaled and diluted to 1.8 pmol/L and paired-ended 150-cycle high-throughput sequencing was performed on an Illumina Miniseq sequencing platform using the MiniSeq Mid Output Kit (300-cycles) (Illumina, #FS-420-1004).
-
For NGS downstream data, the CLC Genomics Workbench (Qiagen, #832021) was used for data quality control (QC), read trimming, mapping refining, and consensus extraction. NC_001806(Hsv-1), NC_001798(Hsv-2), NC_001348(VZV), NC_009942(WNV), NC_007605(EBV), NC_006273(HCMV), JE_U47032(JEV), NC_009942(WNV), KX_064282(CA6), KP_289398(CA10), JQ_354992(CA16), AY_4653561(EV71), and NC_002200(Mumps virus) were used as the reference sequences.
-
The primers presented in Table 1 were used for the PCR of each pathogen to determine whether they amplified the target fragment. All PCR products were sequenced (Sanger sequencing) to obtain the sequences of the target fragments, and BLAST showed that all sequences matched the reference sequences.
-
Multiple PCR amplifications for different pathogens were performed after mixing the optimized primers (Table 1), and the cDNA library of the pathogens targeted in this study was then sequenced. Target sequences of the pathogens were extracted from the sequencing data at coverage depths greater than 20×. The amplicon lengths were consistent with the predicted sizes of the amplified fragments. Preliminary verification was performed using NCBI BLAST, and all sequences matched their corresponding reference sequences. The experimental data are shown in Figure 2. Panel A shows the results of the pre-experiments (primers were not optimized, and sample concentrations were not adjusted), along with the coverage and sequencing depth for targeted sequence amplification of HSV-1, HSV-2, HCMV, EBV, VZV, JEV, WNV, EV71, CA6, CA10, CA16, and mumps virus. Notably, the primer pool contained two pairs of EBV-specific primers, but amplification using one pair of primers generated few sequences, indicating that the amplification efficiency of this primer pair was poor.

Figure 2. Coverage and sequencing depth map of pathogen target sequences. Coverage and sequencing depth of pathogen target sequences result: A shows the results of the pre-experiments, showing the coverage and sequencing depth of HSV-1, HSV-2, HCMV, EBV, VZV, JEV, WNV, EV71, CA6, CA10, CA16 and Mumps target sequences; B shows the sequencing results of samples at 10-fold dilution, showing the coverage and sequencing depth of VZV, JEV, WNV, EV71, CA6, CA10, CA16 and Mumps target sequences; C shows the sequencing results of the sample at 100-fold dilution, showing the coverage and sequencing depth of HCMV, EBV, VZV, JEV, WNV, EV71, CA6, CA10, CA16 and Mumps target sequences; D shows the sequencing results of samples at 1000-fold dilution, showing the coverage and sequencing depth of HSV-1, HSV-2, HCMV, EBV, VZV, JEV, WNV, EV71,CA6, CA10, CA16 and Mumps target sequences.
Not all nucleic acid templates were derived from clinical samples; the JEV and WNV templates were extracted from virulent strains, and the VZV template nucleic acid was extracted from the vaccine. We generated serial dilutions of these nucleic acid samples (10-, 100-, and 1000-fold), and panel B shows the sequencing results for the 10-fold diluted samples (Ct values: 25.2–29.1), with the coverage and sequencing depths of the VZV, JEV, WNV, EV71, CA6, CA10, CA16, and mumps virus target sequences. Owing to the concentration limitations of some clinical samples, there were only one concentration of HSV-1 and HSV-2 and two concentrations of HCMV and EBV. Therefore, only eight pathogens are shown in panel B. Panel C of Figure 2 shows the sequencing results of the 100-fold diluted samples (Ct values: 29.1–32.7), with the coverage and sequencing depth of the target sequences for 10 pathogens (HCMV, EBV, VZV, JEV, WNV, EV71, CA6, CA10, CA16, and mumps virus). Panel D of Figure 2 shows the sequencing results for the 1000-fold diluted samples (Ct values: 33.1–35.5), with the coverage and sequencing depth of the target sequences for 12 pathogens (HSV-1, HSV-2, HCMV, EBV, VZV, JEV, WNV, EV71, CA6, CA10, CA16, and mumps virus). Amplicons were obtained from all the pathogen samples after gradient dilution. Notably, the number of reads for HCMV was the lowest, mainly due to the amplification efficiency of the primers.
We compiled the experimental data, which included the CT values for each pathogen, number of reads, and reference sequence of the spliced target sequence (Table 2). In the pre-experiments, we found that the sequencing depth was not balanced among pathogens; CA10 had the highest total reads (2,563,545 reads), whereas HSV-2 had the fewest reads (6,308 reads), which may be related to the number of viral copies in the template or the amplification efficiency of the primers. The HSV-1, HSV-2, HCMV, and EBV samples were derived from the aqueous humor of patients with anterior uveitis; the EV71, CA6, CA10, CA16, and mumps virus samples were obtained from pharyngeal swabs of patients who were clinically positive for these viruses, and the remaining samples were nucleic acids recovered from viral strains, which may explain why the sequencing depth was not balanced among the pathogens. BLAST was used to compare the amplification sequences with the Sanger sequencing results, and the findings showed remarkable uniformity for all pathogens.
Virus Pre-experiments 10-fold 100-fold 1,000-fold Ref Ct-value Reads Ct-value Reads Ct-value Reads Ct-value Reads HSV1 27.9 8,678 — — — — 33.9 21,932 NC_001806 HSV2 28.2 6,308 — — — — 33.6 14,562 NC_001798 HCMV 30.1 36,752 — — 30.8 546 34.0 0 NC_006273 EBV 29.6 21,307 — — 29.1 46,482 33.1 23,902 NC_007605 VZV 24.6 774,363 27.0 961,166 29.4 986,698 34.0 2,069,900 NC_001348 JEV 25.8 642,001 27.4 907,263 30.4 102,001 33.5 18,209 JE_U47032 WNV 25.3 1,073,388 27.2 790,729 31.0 430,332 34.7 884,879 NC_009942 EV71 24.1 2,225,726 26.9 2,951,553 31.6 2,178,524 33.7 3,449,881 AY_4653561 CA6 26.9 2,013,312 28.1 2,973,422 32.7 2,195,624 35.5 3,538,523 KX_064282 CA10 21.1 2,401,006 29.1 2,976,273 31.2 2,213,093 33.5 3,597,826 KP_289398 CA16 24.4 2,563,545 28.8 2,959,416 31.4 2,189,642 34.6 3,513,948 JQ_354992 Mumps 23.1 903,858 25.2 928,429 30.5 479,978 33.2 119,500 NC_002200 Note. — Indicates no data. HSV-1, herpes simplex virus-1; HSV-2, herpes simplex virus-2; EBV,epstein-barr virus; HCMV, human cytomegalovirus; VZV, varicella zoster virus; JEV, Japanese encephalitis virus;WNV, west nile virus; EVs, Enteroviruses Table 2. Summary of sequencing results for all pathogens
-
The results showed that the target sequences of various pathogens were obtained at a coverage depth level greater than 20× and the sequence lengths were consistent with the sizes of the predicted amplicons. The sequences were verified using the NCBI BLAST, and all results were consistent with the results of Sanger sequencing. Thus, amplicon-based high-throughput sequencing technology is feasible for the pathogenic detection of viral encephalitis as a supplementary detection method and is also a useful tool for high throughput screening of clinical samples.
With the continuous development of sequencing technologies, HTS has brought new perspectives to the study of CNS infections and has recently been incorporated into guidelines for the management of encephalitis[19]. HTS can be used to identify viral pathogens in patients with encephalitis, meningoencephalitis, or meningitis of unknown origin, and the results can be used to provide effective treatment for immunocompromised patients[20]. Metagenomic next-generation sequencing (mNGS), a type of HTS, is important for the laboratory diagnosis of CNS infections, particularly for the identification of emerging pathogens[21-23]. However, the application of NGS for viral encephalitis has been hampered by the limitations of low viral load in the cerebrospinal fluid (CSF), high number of mNGS background sequences, and high cost. Many studies have actively addressed these shortcomings. For example, researchers have investigated a metagenomic sequencing protocol that includes viral enrichment to reduce the problem of low viral load in the CSF[21]. In some studies, DNA amplification kits were used to overcome insufficient viral nucleic acid levels. However, only few studies have applied deep sequencing for multipathogen detection in viral encephalitis. Multipathogen sequencing is key for both the rapid identification of the causative agent in infections and source investigation during disease outbreaks, which requires tools that are efficient, versatile, convenient, and with minimal complexity.
In this study, we established a technological platform for the detection of viral encephalitis pathogen detection based on amplicon sequencing by combining the benefits of deep sequencing and multipathogen detection. Although multiple PCR detection methods for 18 pathogens in the CSF of children with viral encephalitis have been established and validated for multiple pathogen detection[24], the impact of low viral load in the CSF and the time-consuming and laborious nature of screening large volumes of samples cannot be avoided. The sequencing cost per sample on Illumina and Nanopore platforms increases with the number of samples, although the turnaround time decreases[25]. The platform established in this study can be used to detect a larger number of samples than can be detected using mNGS, thereby reducing sequencing costs. We believe that the application of NGS for the diagnosis of viral nervous system infections should not be limited to mNGS. Compared with mNGS, the advantages of the method developed in this study are its simplicity, standardization, high sensitivity, and cost-effectiveness. This strategy can also be used for the detection of viral encephalitis using second-generation sequencing. We designed or selected one primer pair for each of the 12 pathogens and validated them using nucleic acid standards. The obtained sequences were compared with those in the NCBI Genome Database to validate the nine primer pairs. For the primers designed in this study, the principles of primer design were strictly followed to avoid mismatches, primer dimers, and hairpin structures. Replacing one or more bases at key positions in a hairpin structure can significantly reduce the hairpin Tm, thus ensuring the efficiency of the primer. All primers were checked to ensure that their annealing temperature (55–70 °C), GC content (40%–60%), and hairpin Tm (< 50 °C) were within the appropriate ranges.
When preparing a library for high-throughput sequencing, in multiplex PCR, more than two pairs of primers are added to the same PCR system, and each pair of primers attaches to the corresponding complementary template, if present, allowing the amplification of multiple target DNA fragments in the same reaction system. Multiplex PCR is an efficient, cost-effective, and scalable method for the detection of multiple pathogens by amplifying multiple target fragments. The design of a multiplex PCR assay is much more complex than that of an ordinary PCR assay and is not simply a matter of mixing multiple pairs of specific primers into a single reaction system. The composition of the reaction system and reaction conditions must be repeatedly adjusted based on experimental results to accommodate the simultaneous amplification of multiple fragments. The target region, along with the size of the amplified fragment, template-primer binding kinetics, and optimal cycling conditions, must be carefully considered for amplification. When designing primers for multiplex PCR, one should pay particular attention to the non-complementarity between primers, especially at the 3' end, to prevent the formation of primer dimers, in addition to adhering to the general rules of PCR primer design. There should also be no greater complementarity between any two primers and any other amplification fragments or templates and no greater homology between amplification fragments. Possible primer interactions can be addressed by dividing the interacting primers into two tubes for amplification or replacement.
The detection technology developed in this study is applicable to laboratory-developed tests (LDTs) and has very good development prospects for clinical diagnosis and mass screening of patients with viral encephalitis. LDTs are created by individual laboratories or health systems with requisite experience in designing, validating, and overseeing high-complexity assays. Shanghai, China, has identified a number of public medical institutions as pilot units for high-quality test development and has encouraged hospitals to conduct their own in vitro diagnostic reagent development pilots. The method developed in this study could detect sensitive and specific pathogens in multiple samples, detect co-infected samples, and distinguish the 12 pathogens included in this study with specificity by establishing a technical platform based on amplicon sequencing for the detection of viral encephalitis pathogens. The early and accurate detection of pathogens enables rapid and effective disease control, prevention, and specific clinical treatment. The establishment of this platform provides a viable solution to the current lack of genetic testing technology for early on-site diagnosis, prevention, and control of viral encephalitis. This study framework is also appropriate for the nanopore long-read sequencing technique, which offers a powerful tool for the diagnosis, investigation, and monitoring of infectious diseases[26]. It is also anticipated that other researchers will incorporate other pathogens that cause encephalitis/meningitis syndrome, such as bacteria and parasites, into this platform and perform methodological exploration and establishment of other amplicon sequencing platforms.
HTML
Pathogen Identification
Summary of the Entire Workflow
Primer Design
Virus Nucleic Acid Extraction
PCR and RT-PCR Validation of Primer Specificity
cDNA Synthesis
Multiplex PCR Amplification
Amplicon Purification, Quantification, and Normalization
Library Preparation and NGS Sequencing
Data Analysis
Sanger Sequencing Results
High-throughput Sequencing Results
CONFLICTS OF INTEREST The authors have no conflicts of interest to declare.
&These authors contributed equally to this work.

 Quick Links
Quick Links
 DownLoad:
DownLoad:
