
-
Cervical cancer (CC) is one of the most common gynecological malignancies; approximately 604,127 new cases were reported worldwide in 2020, of which nearly 85% occurred in low- and middle-income countries[1,2]. In China, the mortality rate of CC in 2022 was as high as 55.07%[3]. Cervical squamous cell carcinoma (CSCC) accounts for approximately 70% of all CCs worldwide, which is significantly higher than the rate of cervical adenocarcinoma[4]. Although surgery is an effective treatment for early-stage CC, 20%–30% of CC patients in advanced stages experience recurrence or distant metastasis after chemotherapy administered concurrently with radiation therapy, and the 5-year survival rate is only 5%–15%[5]. A comprehensive description of the genomic and molecular characteristics of 228 primary CCs (including 144 CSCC cases) was reported in The Cancer Genome Atlas (TCGA) research network, although only 18 Asians were included[6]. CSCC remains a disease with high morbidity and mortality in China, and the number of sequencing samples available for evaluating genomic alterations in CSCC is small. Therefore, it is necessary to establish a comprehensive genetic profile of the Chinese population with a larger sample size through next-generation sequencing.
Most CSCCs are caused by high-risk subtypes of human papillomavirus (HPV), and the disease could therefore be prevented through well-established screening and vaccination programs[7]. However, the prevalence and mortality of CSCC have remained relatively high because of low vaccination rates. In addition to increasing the rate of vaccination, the development of new treatments for CSCC is critical. In recent years, immunotherapy has emerged as a potentially effective therapeutic approach. Immune checkpoint inhibitors (ICIs) have been successfully applied to the treatment of different types of cancer, such as gastric, lung, and head and neck cancer, which has significantly prolonged the survival of patients[8-10]. Pembrolizumab was the first Food and Drug Administration (FDA)-approved first-line immunotherapy for CC; however, it showed positive results in a minority of CC patients[11]. In addition to ICIs, several immunotherapeutic approaches for advanced cervical cancer, including HPV therapeutic vaccines and adoptive cellular therapy, are under study, and promising efficacy data are emerging[12,13].
The genetic instability of tumor cells often leads to numerous somatic mutations, and the expression of non-synonymous mutations generates tumor-specific antigens called neoantigens[14]. Neoantigens are presented by human leukocyte antigen (HLA) class I/II molecules and can activate CD8+ and CD4+ T cells, resulting in the induction of immune responses[15]. Because neoantigens are not expressed in normal tissues and are highly immunogenic, they have emerged as novel targets for tumor immunotherapy. Extensive analysis of the genomic variations in CC identified a close association with mutated genes such as PIK3CA, FBXW7, EP300, MLL3, CASP8, and FADD[16]. However, research aimed at identifying CSCC neoantigens through the analysis of genetic mutations is lacking.
In this study, we explored the genomic characteristics of CSCC using whole-exome sequencing (WES) data and identified potential neoantigens by WES and RNA sequencing (RNA-seq). The present findings improve our understanding of genetic alterations in CSCC and identify relevant neoantigens, which may provide effective immunotherapeutic targets for the treatment of CSCC.
-
Written informed consent was obtained from each individual before enrollment in the study. Primary tumor tissues and peripheral blood were collected from patients diagnosed with CSCC at the Obstetrics and Gynecology Department of the Chinese Liberation Army General Hospital between January 1, 2021 and May 1, 2022. Sample collection, HPV typing, and pathological examination were performed according to the guidelines or requirements of the patients. Data on clinical characteristics were collected from medical records.
The study included 60 samples from 30 CSCC patients. Twenty-nine surgically resected tumor tissues (4–6 g per sample) were snap-frozen in liquid nitrogen and stored at –80 °C, and one was paraffin-embedded. Additionally, 30 blood samples (3–5 mL per sample) were collected as controls and stored in anticoagulant tubes containing ethylenediaminetetraacetic acid. The blood and anticoagulant were thoroughly mixed and stored at –20 °C. All slides were diagnosed by two experienced pathologists using hematoxylin and eosin (H&E) staining.
-
Tumor and matched normal DNA were extracted using the TIANamp Genomic DNA Kit (DP304, TIANGEN, Beijing, China) using fresh-frozen tumor tissues and paired blood samples. The QIAamp DNA FFPE Tissue Kit (56404, Qiagen, Hilden, Germany) was used to extract genomic DNA (gDNA) from formalin-fixed paraffin-embedded (FFPE) tissues. To ensure the quality of the gDNA, two methods were employed: (1) Agarose gel electrophoresis to analyze the degree of DNA degradation and contamination; and (2) Qubit® 3.0 Fluorometer (Invitrogen, USA) to quantify the DNA concentration accurately. Finally, DNA samples with a gDNA concentration ≥ 20 ng/µL and a minimum of 0.4 µg gDNA per sample were used for library construction.
WES was performed using 0.4 µg of gDNA from each sample. Library construction and capture experiments were performed using the Agilent SureSelect Human All Exon V6 Kit (Cat. No. 5190–8864, Agilent Technologies, Santa Clara, CA, USA) following the manufacturer’s instructions. During this process, index codes were added to each sample. The gDNA was fragmented into pieces measuring approximately 180–280 bp using a hydrodynamic shearing system (Covaris, Massachusetts, USA). After end repair, phosphorylation, and A-tailing, adapter oligonucleotides were added to create libraries. High-fidelity polymerase was used to amplify DNA fragments with ligated adaptor molecules on both ends to ensure sufficient library volume. The libraries were then hybridized with a solution of biotin-labeled probes, and exons were captured using streptomycin magnetic beads. In preparation for sequencing, PCR was used to add index tags to the captured libraries. After purification and quantification, the index-encoded samples were clustered using the HiSeq PE Cluster Kit V2.5 (Illumina). The DNA libraries were sequenced on the Illumina HiSeq X-TEN platform (San Diego, CA, USA), generating 150 bp paired-end reads after cluster generation.
-
To create the library, a minimum of 1 µg of total RNA was necessary. RNA was extracted from fresh tumor tissues using the Qiagen RNeasy Mini kit (74106, Qiagen, Germany) and RNA-Seq libraries were prepared using the NEBNext® UltraTM RNA Library Prep Kit (E7530L, Illumina, USA). The library was quantified using the Qubit2.0 Fluorometer detection kit (Q32866, Invitrogen, USA) and diluted to 1.5 ng/µL. The Agilent 2100 bioanalyzer (2100, Agilent, USA) was used to detect the insert size of the library and accurately determine the effective concentration (higher than 2 nmol/L) to ensure quality. Once the library passed inspection, it was categorized according to the effective concentration and target off-machine data volume. Finally, Illumina sequencing was performed, which generated 150 bp paired-end reads.
-
Raw data were preprocessed by Fastp (v.0.23.4, https://github.com/OpenGene/fastp) to obtain clean data using the following steps: (1) adapter trimming; (2) removal of reads with > 10% N bases; (3) removal of reads in which > 50% of the length contained low-quality bases (quality threshold < 5); and (4) sliding window trimming, in which the bases with an average quality below the cutoff value (default was 20) in the sliding window (default was 4 bp) were cut. BWA[17], Picard (http://broadinstitute.github.io/picard/), and GATK tools[18] were used for read alignment, variant calling, and identification of single-nucleotide variants (SNVs) and small insertions and deletions (InDels). Default parameters were used in all software programs for identifying mutations in matched normal-tumor samples. To further annotate candidate somatic mutations, we used Funcotator (FUNCtional annOTATOR)[19] and generated mutation annotation format (MAF) files that included position, function, and sequencing data supporting the mutation status.
Somatic copy number alterations (CNAs) were evaluated using the CNVkit[20] pipeline (v.0.9.10). The default log2 threshold was applied to detect copy number gains or losses in target regions. Heatmaps of copy number alterations were obtained by loading the resulting files with segmented copy numbers into Integrative Genomics Viewer (IGV, v.2.15.9)[21] for visualization. The GISTIC (Genomic Identification of Significant Targets in Cancer) 2.0 pipeline (v.2.0.23)[22] was then applied to detect the significantly amplified and deleted regions with somatic CNAs with FDR (false discovery rate) < 0.20. A confidence level of 0.90 was set to determine significance. The GISTIC2.0 output files were visualized by the R package ggplot2.
Synonymous and nonsynonymous somatic SNVs were analyzed using the R package maftools to identify the type of point mutations in each tumor sample[23]. The mutational signature contribution of each tumor sample was estimated using the R package deconstructSigs[24], which accurately reconstructed the mutational profiles of tumor samples by identifying linear combinations of pre-defined features. This tool established the correspondence between the 96 mutation spectrum and the 30 mutational characteristics of the Catalog of Somatic Mutations in Cancer (COSMIC) database. The calculated weights were assigned to the mutational signatures, in which a higher weight indicates a more significant contribution.
Two computational methods, OncodriveCLUSTL[25] and OncodriveFML[26], were used to detect potential driver genes. The OncodriveCLUSTL algorithm uses a sequence-based clustering technique to identify linear clustering bias in the observed somatic mutations. OncodriveFML is a tool that detects genes under positive selection by analyzing the functional impact bias of the observed somatic mutations. Default values were used for all software parameters, and driver genes were identified according to the following criterion: genes with an FDR < 0.25 in both OncodriveCLUSTL and OncodriveFML were considered as driver genes. MutSigCV[27] was used to perform convolution tests to identify significantly mutated genes (SMGs). This software comprehensively analyzes somatic SNVs and InDels to obtain SMGs whose mutation rate is significantly higher than the background mutation rate. Genes with FDR < 0.20 were considered as SMGs.
RNA-seq raw reads that passed the Illumina RTA quality filter were first preprocessed with Trim Galore (v.0.6.10) to remove adapter sequences and for base quality control. Then, STAR software (v.2.7.10b) was used to align the remaining RNA reads to the NCBI human reference genome (GRCh38). Finally, the number of reads aligning to each gene in the mapping results was calculated as Fragments Per Kilobase per Million (FPKM) values using RSEM software (v.1.3.3).
-
HLAscan tool (v.2.1.2)[28] was used to determine HLA types across the patients’ whole-exome sequences by aligning reads to HLA sequences from the international ImMunoGeneTics project/human leukocyte antigen (IMGT/HLA) database. Neoantigen analysis was performed using the NeoPredPipe pipeline[29], which integrates ANNOVAR and netMHCpan to process neoantigens predicted from multi-region Variant Call Format (VCF) files. ANNOVAR correctly annotates variants from VCF files to identify non-synonymous variants, generating peptide sequences based on variant bases. Before executing netMHCpan, HLA haplotypes were cross-referenced with available HLA haplotypes, and epitopes of 8–11 mer length (known to be likely for peptides presented by human MHC class I molecules) were specified to make predictions. NetMHCpan 4.1[30] was used to detect the binding affinity strength of each mutant peptide to sample-specific HLA alleles to identify exome-derived neoantigens. Finally, the results were filtered according to half-maximal inhibitory concentration (IC50) and rank value (IC50 < 500 nmol/L and %Rank < 2%). A neoantigen was considered expressed if a mutated gene Tumor-FPKM ≥ 0.5 when RNA-seq was available.
-
All graphical analyses were performed in the R statistical environment (v. 4.2.2). P-value calculation methods and multiple testing corrections are reported in the text. Because of the limited sample size, all analyses were conducted at a two-sided significance level of 0.2 (exceptional cases stated otherwise). Linear correlations were assessed using Pearson’s correlation coefficient.
-
A total of 60 paired samples from patients who had undergone radical hysterectomy without radiotherapy or chemotherapy were analyzed. The samples included 30 primary tumor tissues and 30 matched peripheral blood samples. Of the 30 patients, 22 were HPV-positive, and 8 were HPV-negative (Supplementary Table S1, available in www.besjournal.com). The patients ranged in age from 24 to 77 years, with an average age of 50.3 years. The clinical stages (International Federation of Gynecology and Obstetrics [FIGO] 2018) of the patients were as follows: 12 in stage I, 12 in stage II, and 6 in stage III. The pathological types of all samples were squamous cell carcinoma with varying levels of differentiation: 6 cases were low, 17 were middle, and 7 were middle-low. Among the patients, 9 had lymph node metastasis, 22 had lymphovascular space involvement, 3 had parametrial invasion, 21 had deep stromal invasion (infiltration depth > 1/2), and 3 had a positive vaginal resection margin. During the follow-up period of 6–19 months (median, 12.5 months), 3 cases (10%) experienced tumor recurrence.
Gene Indels SNVs Tot Muts* Sample affect Sample percent (%) FDR CT‡ Present in CGC Reported in previous research Predicted by OncodriveCLUSTL Predicted by OncodriveFML Predicted by MutSigCV K3CAPI 0 8 8 8 26.67 0.048 0.181 0.836 Yes Yes BICRA 0 3 3 3 10.00 0.181 0.064 1 No No FBXW7 1 4 5 5 16.67 0.181 0.165 1 Yes Yes Note. *Tot Muts denotes the total mutations occurred in certain genes. ‡FDR CT denotes corrected P value. FDR, false discovery rate; SNVs, single-nucleotide variants; InDels, insertions and deletions; CGC, Cancer Gene Census. Table 1. Driver genes identified by whole exome sequencing
-
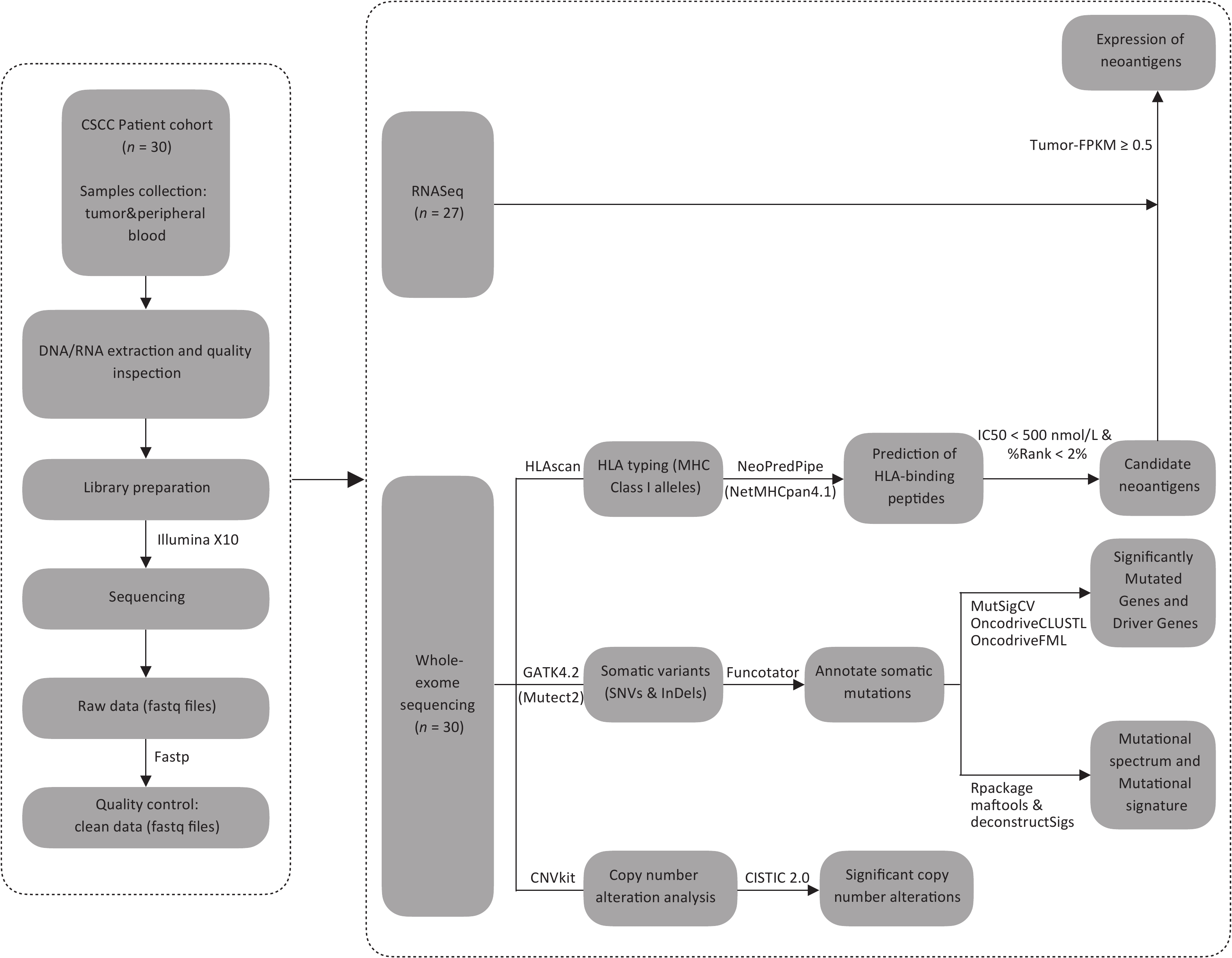
After performing WES on 30 CSCCs and matched control samples (peripheral blood cells) using the Illumina X10 platform (Illumina Inc., San Diego, CA, USA), we assembled a concatenated quality report of the WES data (Supplementary Table S2, available in www.besjournal.com). The control samples had at least 57 Mb of target exons covered with an average depth of 160.76x (ranging from 112.96× to 243.10×), and the tumor samples had at least 155 Mb of target exons covered with an average depth of 400.22× (ranging from 289.22× to 516.15×). More than 93.03% of tumor-targeted regions were effectively covered by at least 30× reads. Figure 1 shows the schematic of the research process.
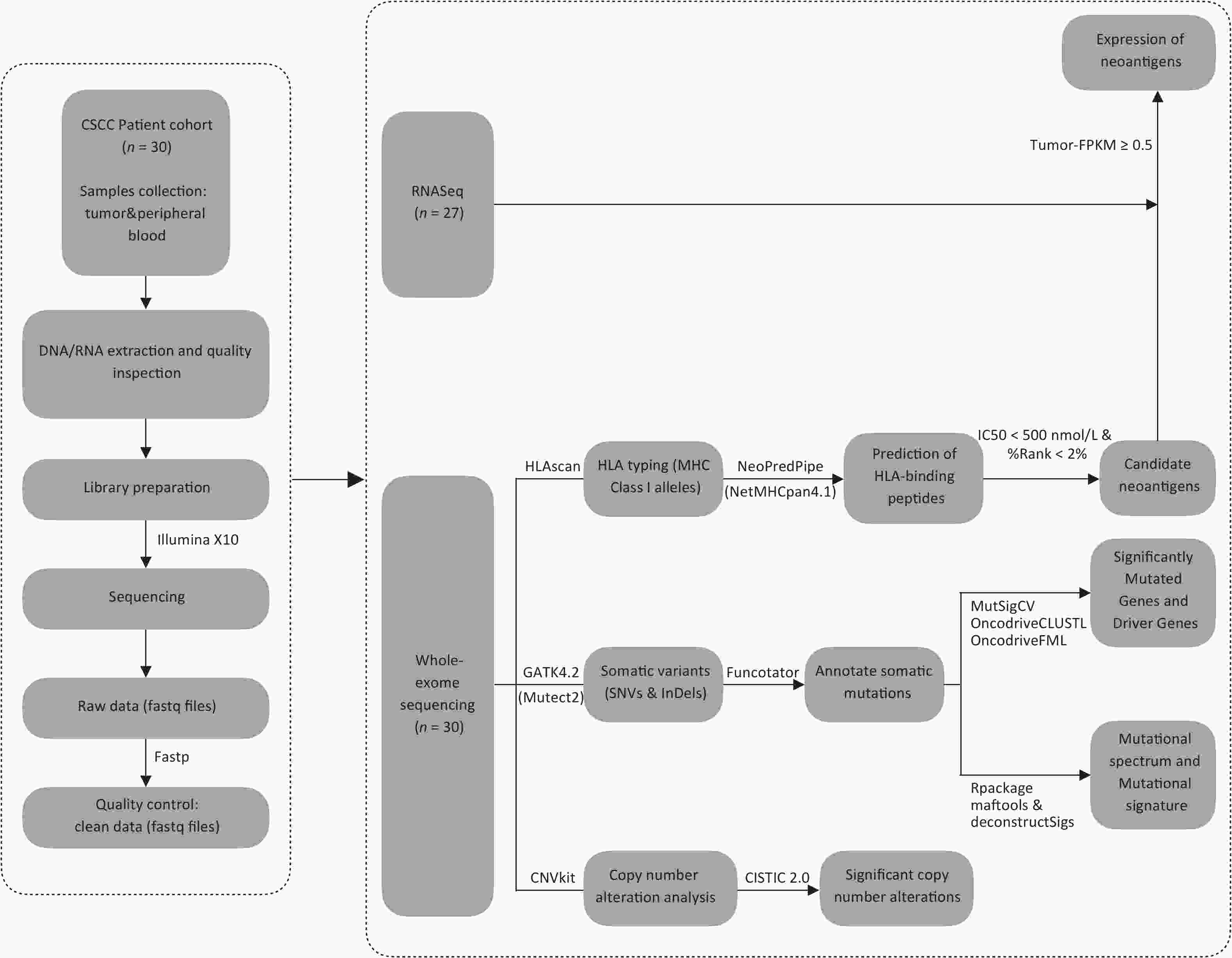
Figure 1. The flowchart. CSCC, cervical squamous cell carcinoma. FPKM, Fragments Per Kilobase per Million. HLA, human leukocyte antigen. IC50, half-maximal inhibitory concentration. SNVs, single-nucleotide variants. InDels, insertions and deletions.
Sample-ID Number of
nonsynomous mutationsNumber of neoantigens HLA-A* HLA-B* HLA-C* Sequencing strategies Stage SCCP01T 66 52 02:07/30:01 - 01:02/12:02 WES/RNA-seq IIIC1 SCCP02T 72 86 24:02/33:03 58:01 01:02/03:02 WES IIA2 SCCP03T 220 258 02:01/03:01 35:08/44:02 05:03 WES/RNA-seq IIA SCCP04T 67 97 03:01/31:01 51:02 12:02/15:02 WES/RNA-seq IIB SCCP05T 96 119 02:01/11:01 51:01/51:02 08:01/15:02 WES/RNA-seq IB2 SCCP06T 182 311 02:01/02:06 51:01 03:03/15:02 WES/RNA-seq IB2 SCCP07T 264 163 03:01 35:01 04:01 WES/RNA-seq IB3 SCCP08T 50 76 11:01/30:01 13:02 03:04/06:02 WES/RNA-seq IB2 SCCP09T 193 333 11:01/24:02 13:01/15:01 03:03/03:04 WES/RNA-seq IIA SCCP10T 98 151 02:01/02:03 13:01/48:01 03:04/08:03 WES/RNA-seq IB2 SCCP11T 102 115 02:07/31:01 40:01 01:02/03:04 WES/RNA-seq IIIA SCCP12T 77 124 02:01/30:01 15:02/44:03 08:01 WES/RNA-seq IB1 SCCP13T 106 128 02:01/02:07 40:01/54:01 01:02/03:04 WES/RNA-seq IB3 SCCP14T 79 109 02:06/11:01 40:01/40:06 01:02/08:01 WES/RNA-seq IIA1 SCCP15T 71 51 01:01/11:01 37:01 06:02/07:02 WES/RNA-seq IIA1 SCCP16T 218 290 02:01/02:06 15:11/35:01 03:03 WES IB2 SCCP17T 188 271 02:06/24:02 15:11/51:01 03:03/14:02 WES/RNA-seq IIIC1 SCCP18T 99 128 02:03/03:01 27:07/40:01 07:02/15:02 WES/RNA-seq IB3 SCCP19T 361 487 01:01/02:06 07:02/51:01 07:02/14:02 WES/RNA-seq IIA1 SCCP20T 72 67 03:01 07:02/37:01 06:02/07:02 WES/RNA-seq IIIC1 SCCP21T 79 121 11:01/24:02 15:02/27:07 08:01/15:02 WES/RNA-seq IB2 SCCP22T 68 53 02:01/03:01 13:02/ 03:03/06:02 WES/RNA-seq IIB SCCP23T 72 90 01:01/11:01 35:03/37:01 06:02/12:03 WES/RNA-seq IIIC1 SCCP24T 83 66 02:01 15:11 03:03/08:01 WES/RNA-seq IIIC1 SCCP25T 378 324 01:01/30:01 13:02/54:01 01:02/06:02 WES/RNA-seq IIA1 SCCP26T 180 118 02:07/33:03 37:01/46:01 01:02/06:02 WES/RNA-seq IIA1 SCCP27T 121 186 11:01/30:01 13:02/14:01 06:02/08:02 WES/RNA-seq IB2 SCCP28T 204 392 11:01/11:12 15:02/35:03 08:01/12:03 WES/RNA-seq IIA2 SCCP29T 167 150 11:01 13:01/13:02 03:04/06:02 WES/RNA-seq IIA SCCP30T 58 44 02:01/32:01 13:01 03:04/12:02 WES IB1 Note. HLA, human leukocyte antigen. *List separator. Table 2. Number of nonsynonymous mutations, neoantigens and HLA class I allotypes of 30 patients
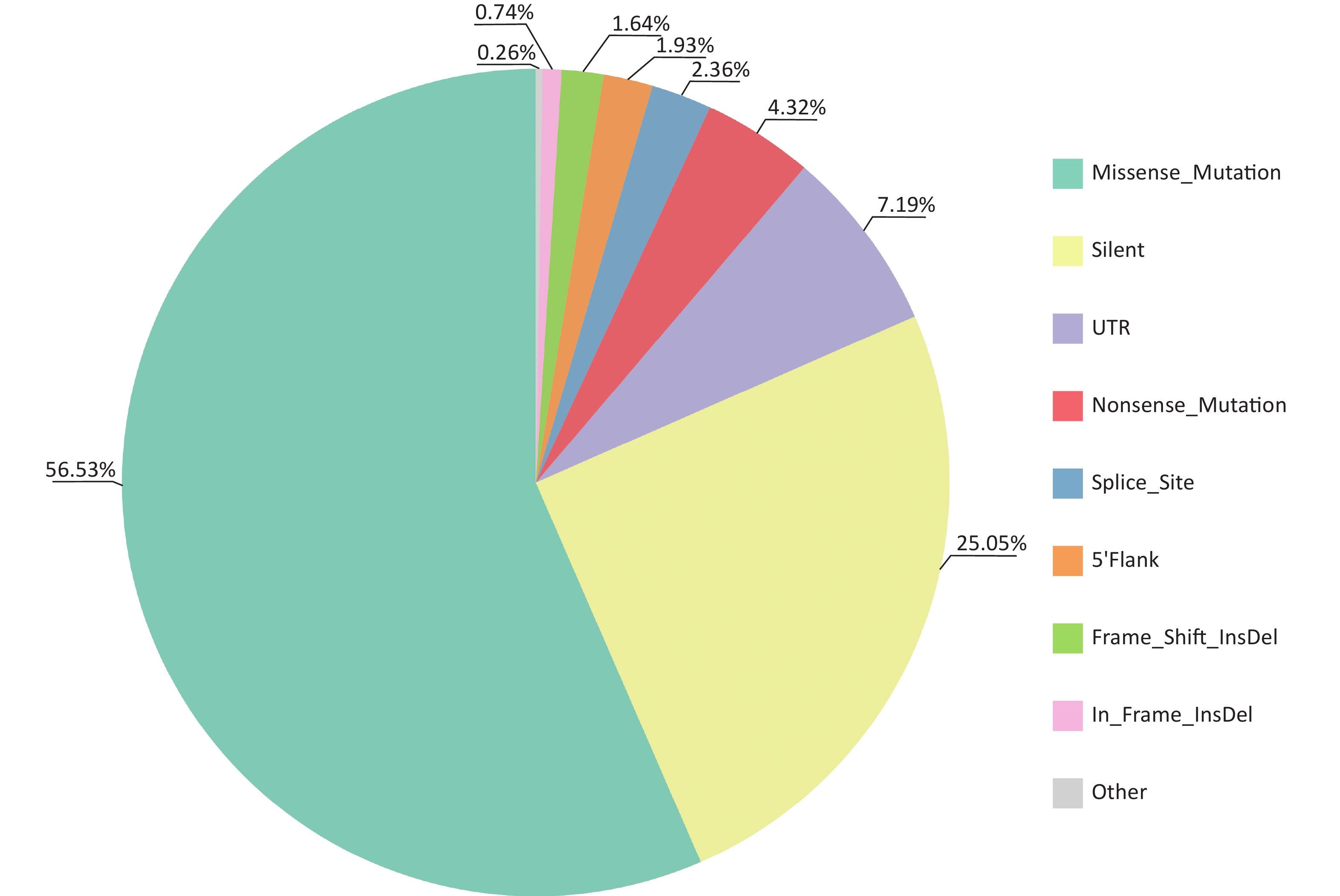
Multiple tools were used to identify somatic mutations (SNVs and Indels) in CSCCs. A total of 6,232 mutations involving 3,216 genes in exons and splice regions were detected, including 3,523 missense, 1,561 synonymous, 269 nonsense, 147 splicing, 46 in-frame InDels, 102 frameshift InDels, 448 UTR mutations, 120 5'Flank mutations, and 16 other types of mutations (
Supplementary Tables S3 –S4 , available in www.besjournal.com). Missense mutations were the most frequent type of somatic mutation in the CDS (coding sequence) regions, accounting for 56.53% of all mutations (Figure 2). Each sample had an average of 345 mutations (range, 152–1,030), indicating high heterogeneity among CSCC samples. Lastly, we calculated the aggregated mutation density from non-silent mutations in the CSCC data, resulting in mean and median mutation densities of 2.73/Mb and 1.97/Mb, respectively.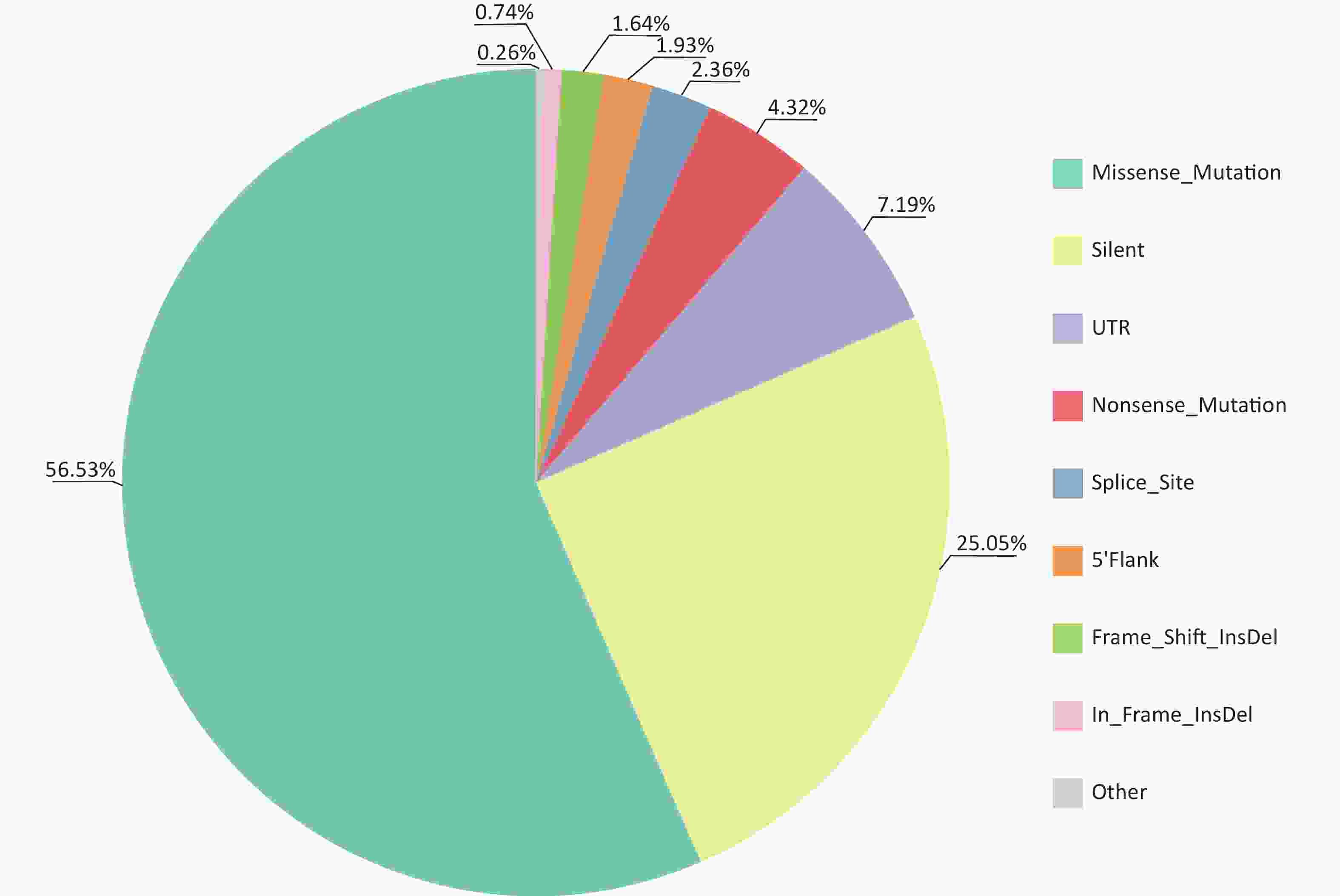
Figure 2. Distribution of somatic mutations identified in CSCCs by WES. CSCC, cervical squamous cell carcinoma; WES, whole-exome sequencing.
-
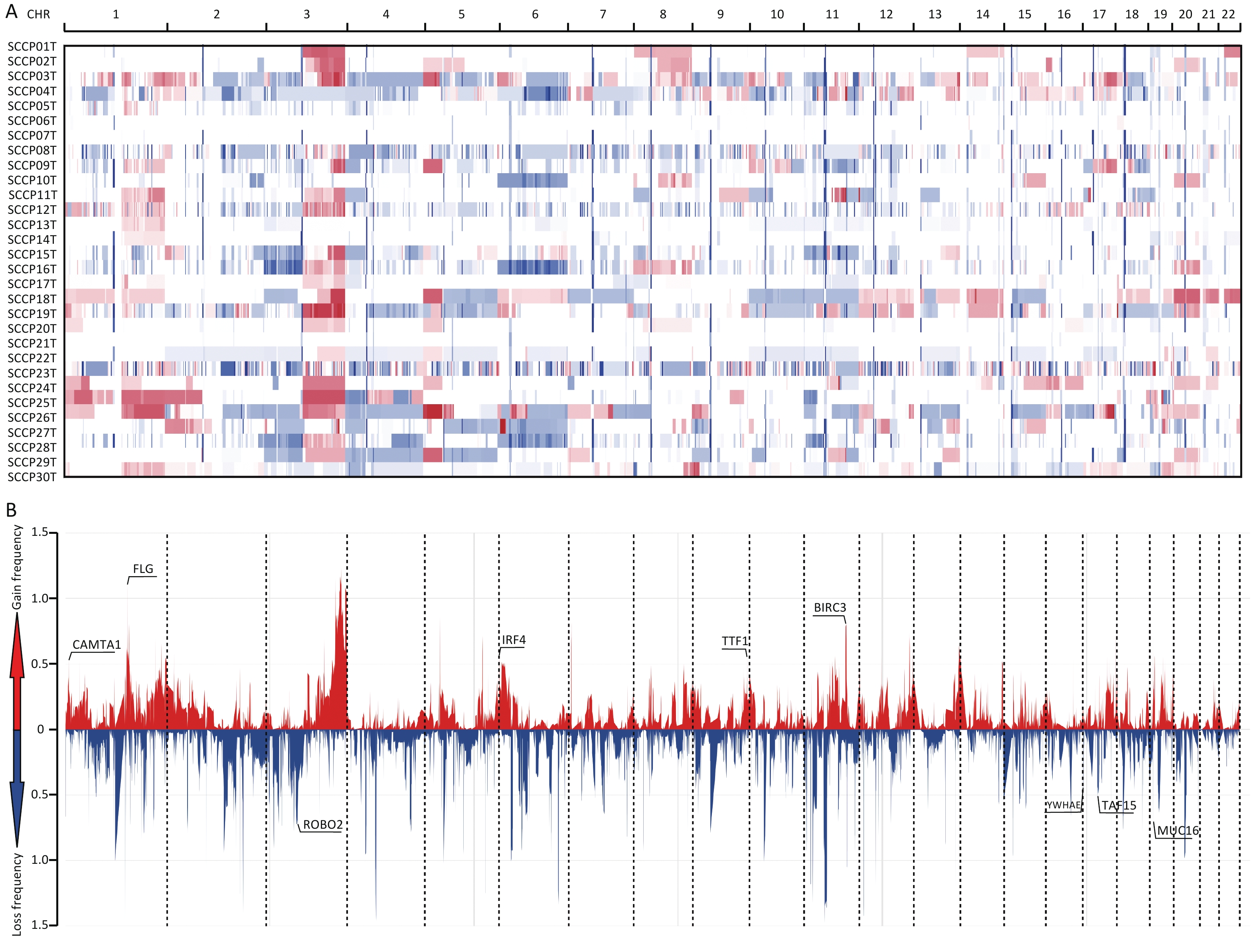
Huang J.[16] et al. compared whole-genome sequencing to WES data for somatic CNA analysis in CC, which demonstrated that WES data is also suitable for CNA analysis. In this study, we used WES data to analyze CNAs in CSCCs and identified 6766 CNAs in 30 samples, with an average of 225.87 CNAs per sample. Significant gains or losses of copy number were detected on many chromosome arms in the CSCC samples. Chromosomes 1p, 1q, 3p, 3q, 5p, 16q, 18p, 19q, 20p, and 20q showed frequent copy number gains, whereas chromosomes 4p, 4q, 11p, 11q, 13p, 15q, and 22p showed frequent copy number losses (Supplementary Table S5, available in www.besjournal.com). The overall CNA results were generally consistent with those of other studies on CC[31-34].
Significantly amplified and deleted regions were identified using the GISTIC2.0 algorithm (with a threshold of FDR < 0.20). We subsequently identified 64 focal events, of which 19 were amplification peaks and 45 were deletion peaks. The analysis revealed that these focal events involved 535 genes, some of which are known oncogenes or tumor suppressor genes, such as FLG (1q21.3), BIRC3 (11q22.3), IRF4 (6p25.3), TTF1 (9q34.13), CAMTA1 (1p36.23), MUC16 (19p13.2), ROBO2 (3p12.3), TAF15 (17q12), and YWHAE (17p13.3) (Supplementary Table S6, available in www.besjournal.com). Among the 64 focal events, significantly amplified cytobands were 3q27.1, 1q21.3, 5p13.2, 7p22.1, and 11q22.3, and significantly deleted regions were 4q13.2, 2q11.2, 7p11.2, 1q21.1, and 11p14.3 (both with the top five lowest q values) (Figure 3). The regions with the highest significance in terms of recurrent copy number amplification and deletion were 3q27.1 and 4q13.2, encompassing multiple genes such as AP2M1, DVL3, PSMD2, EIF2B5, ECE2, ALG3, ABCF3, VWA5B2, CAMK2N2, HTR3C, HTR3E, MIR1224, HTR3E-AS1, UGT2B11, UGT2B28, and LOC105377267. This result indicated that genes in these two regions may be related to CSCC occurrence.
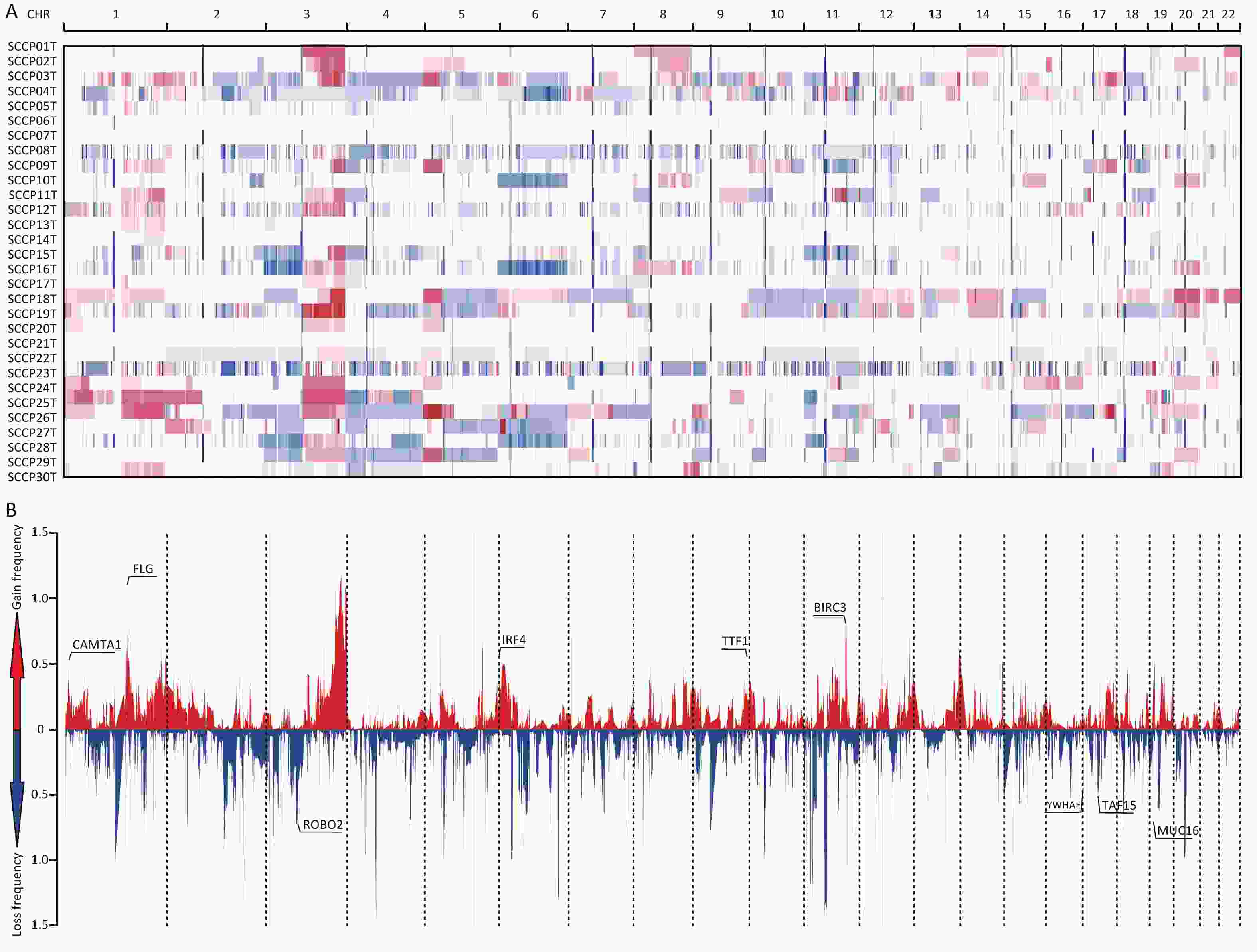
Figure 3. Distribution of copy number variations in cervical squamous cell carcinoma. (A) Clustered heatmap of samples. In the heatmap, blue represents downregulated expression, red represents upregulated expression, and white represents no change in expression. (B) Distribution of CNA in the chromosomes. The y-axis shows the variation scores of GISTIC software; the higher the score is, the higher the significance is. CNA, copy number alterations.
-
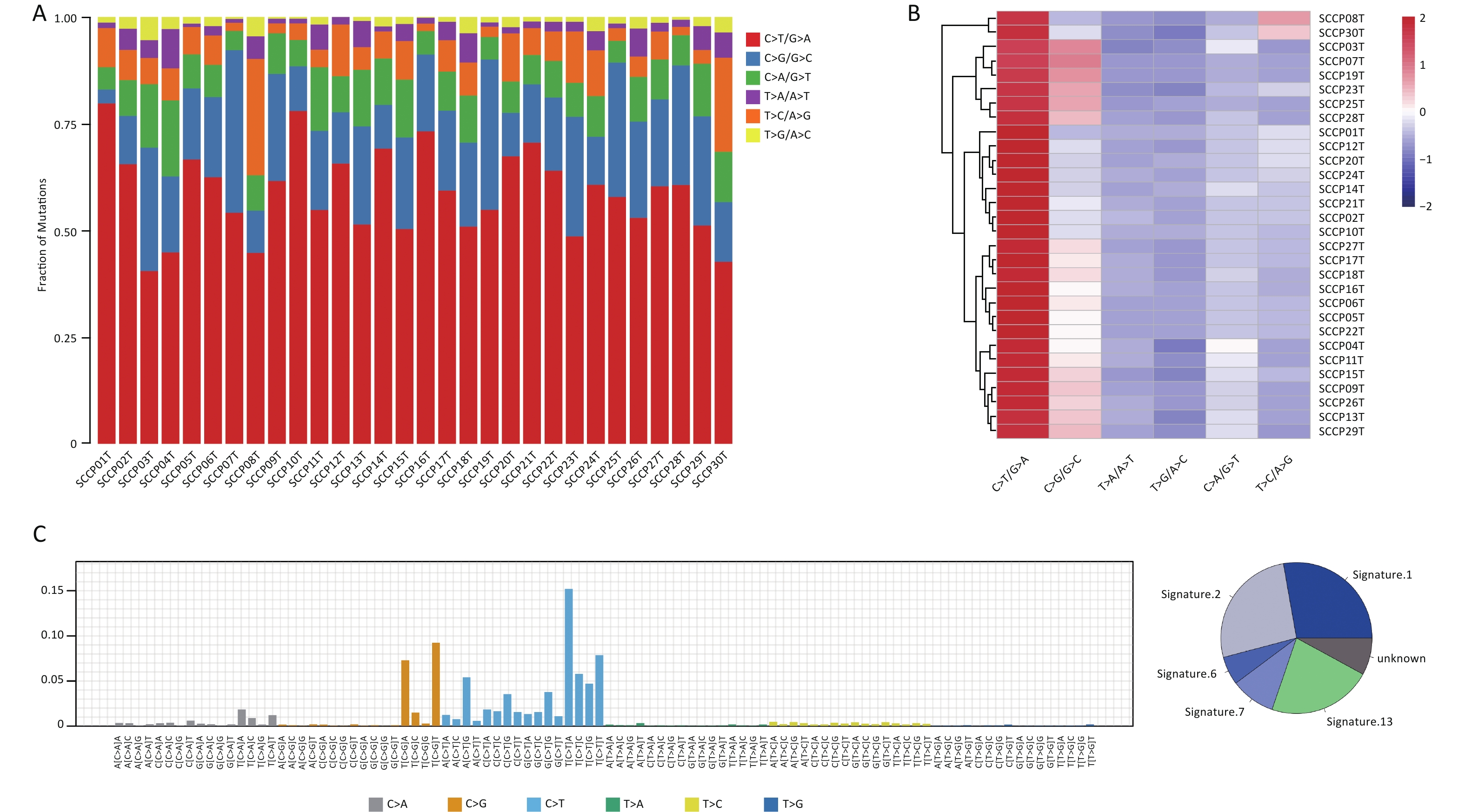
The mutation spectrum displayed the type and number of point mutations in each tumor sample (Figure 4A, B). C>T/G>A transitions (58.38%) and C>G/G>C transversions (23.39%) were the most common point mutations in the present cohort. Previous studies reported a high C>T transition rate (54%, 55.0%, 63.47%) and a high C>G transversion rate (-, 18.9%, 21.85%) in CC samples through mutation spectrum analysis[6,16,35], which is consistent with the present results. The mutational processes leading to tumorigenesis are diverse, and certain mutational signatures can reveal the specificity of the mutational process and the potential etiology of a cancer[36]. To explore this further, we added one base each upstream and downstream from the point mutation site to form a three-base pattern and then counted 96 (6×4×4) mutation combinations. We mapped 96 mutation spectra to the 30 mutational signatures of the COSMIC database[37] and identified three mutational signatures in CSCC samples, namely, signature 2 (26.33%), 6 (6.06%) and signature 7 (9.58%) (Figure 4C).
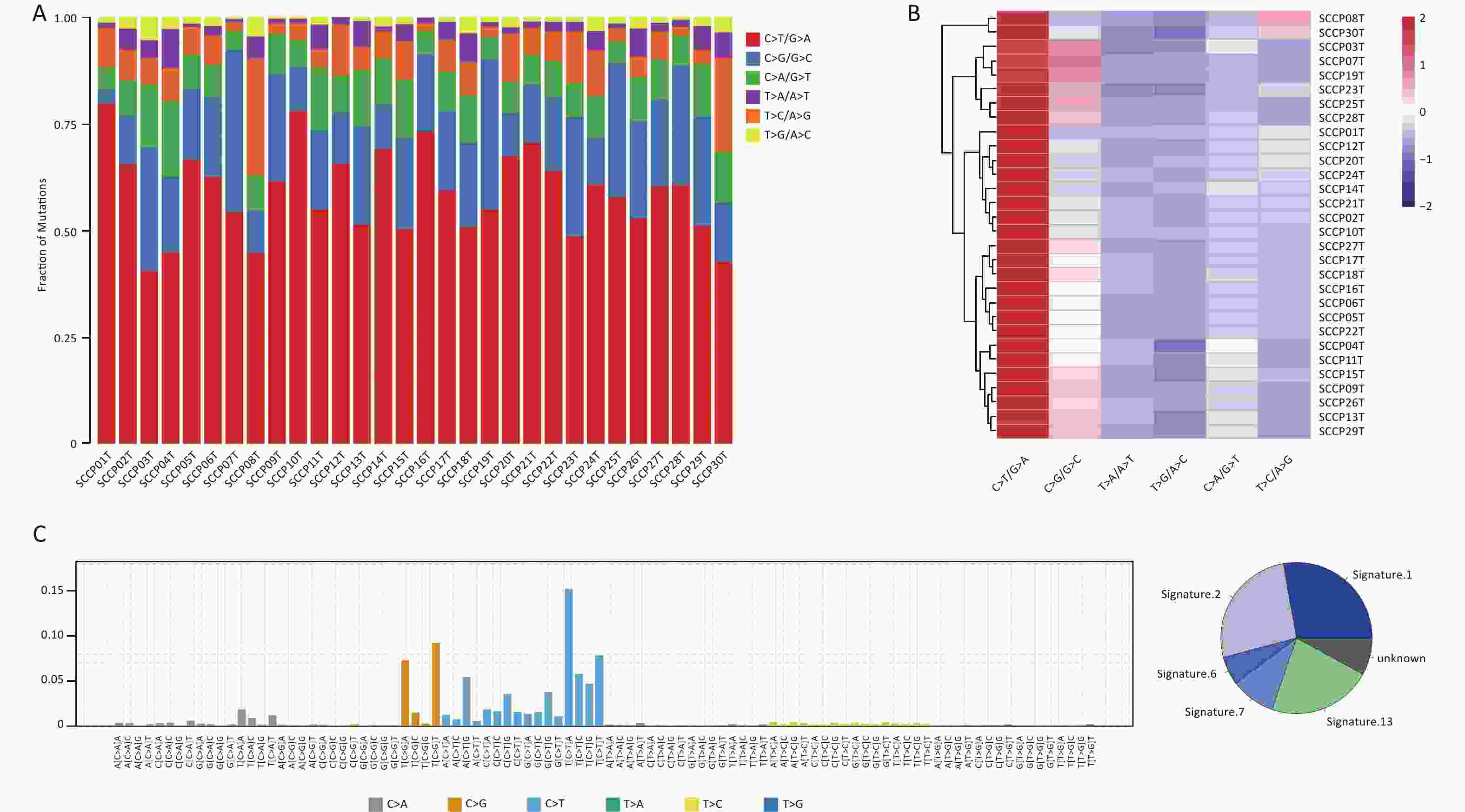
Figure 4. Distribution of single-nucleotide variations in different cervical squamous cell carcinoma samples. (A) Column diagram of various mutations identified by WES. (B) Clustered heatmap of various types mutations in 30 cases by WES. (C) Mutational signatures in this study. Proportion of mutational signatures distribution in each group (on the left). Inset pie chart shows the proportion of mutational signatures in each group (on the right). WES, whole-exome sequencing.
-
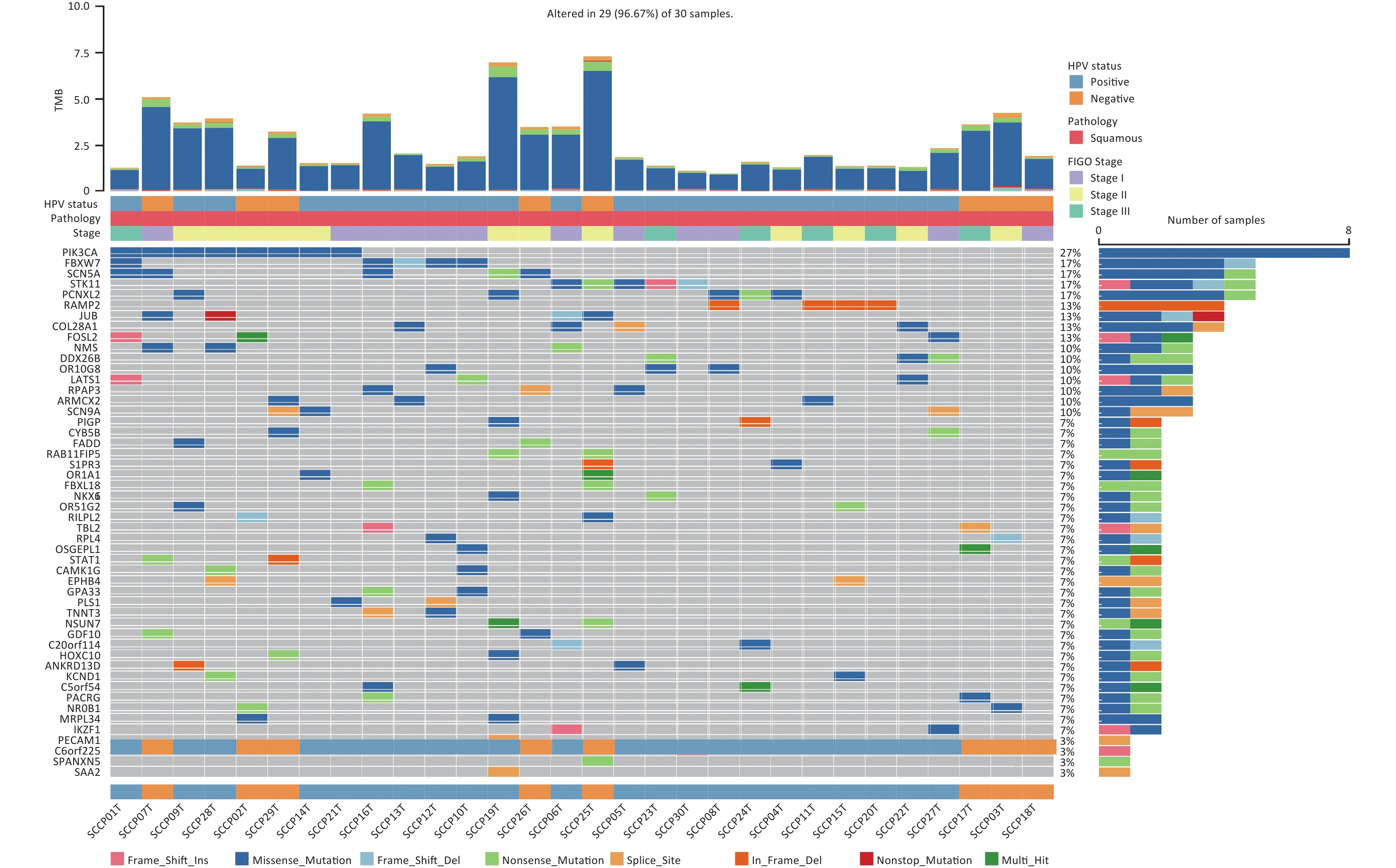
The MutSigCV algorithm was applied to identify SMGs across all tumor samples (FDR < 0.20). Then, the top 50 mutated genes of all tumor samples were selected for visualization. In addition to SMGs, the clinical features of 30 patients, including HPV status, pathology, FIGO stage, and tumor mutation burden (TMB) were determined (Figure 5). Among these genes, RAMP2 (13.33%, 4/30) and FOSL2 (13.33%, 4/30) showed statistically significant levels of recurrent mutations (FDR < 0.20) (Supplementary Table S7, available in www.besjournal.com). They were identified as SMGs in this cohort and confirmed as novel SMGs. Additionally, PIK3CA (26.67%, 8/30), FBXW7 (16.67%, 5/30), SCN5A (16.67%, 5/30), STK11 (16.67%, 5/30), and PCNXL2 (16.67%, 5/30) had high mutation rates but did not have statistically significant recurrent mutations. However, they were enriched in non-silent mutations, indicating that they may play important roles in CSCC. We identified three driver genes according to the filtering criteria (Table 1). Among the predicted driver genes, PIK3CA and FBXW7 were reported in previous studies[6,16], and BICRA (10.00%, 3/30) is novel. The point mutations of PIK3CA were all clustered in the helical domain, including E545K (62.50%, 5/8) and E542K (37.50%, 3/8), whereas no mutations were detected in the kinase domain of the gene (Supplementary Figure S1A, available in www.besjournal.com). Five hotspot mutations were identified in FBXW7, including two R385C (40.00%), one R399G (20.00%), one R425S (20.00%) within the WD40 domain, and one L154fs (20.00%) outside the domain (Supplementary Figure S1B). This mutational characteristic of FBXW7 has not been reported in previous studies on CSCC.
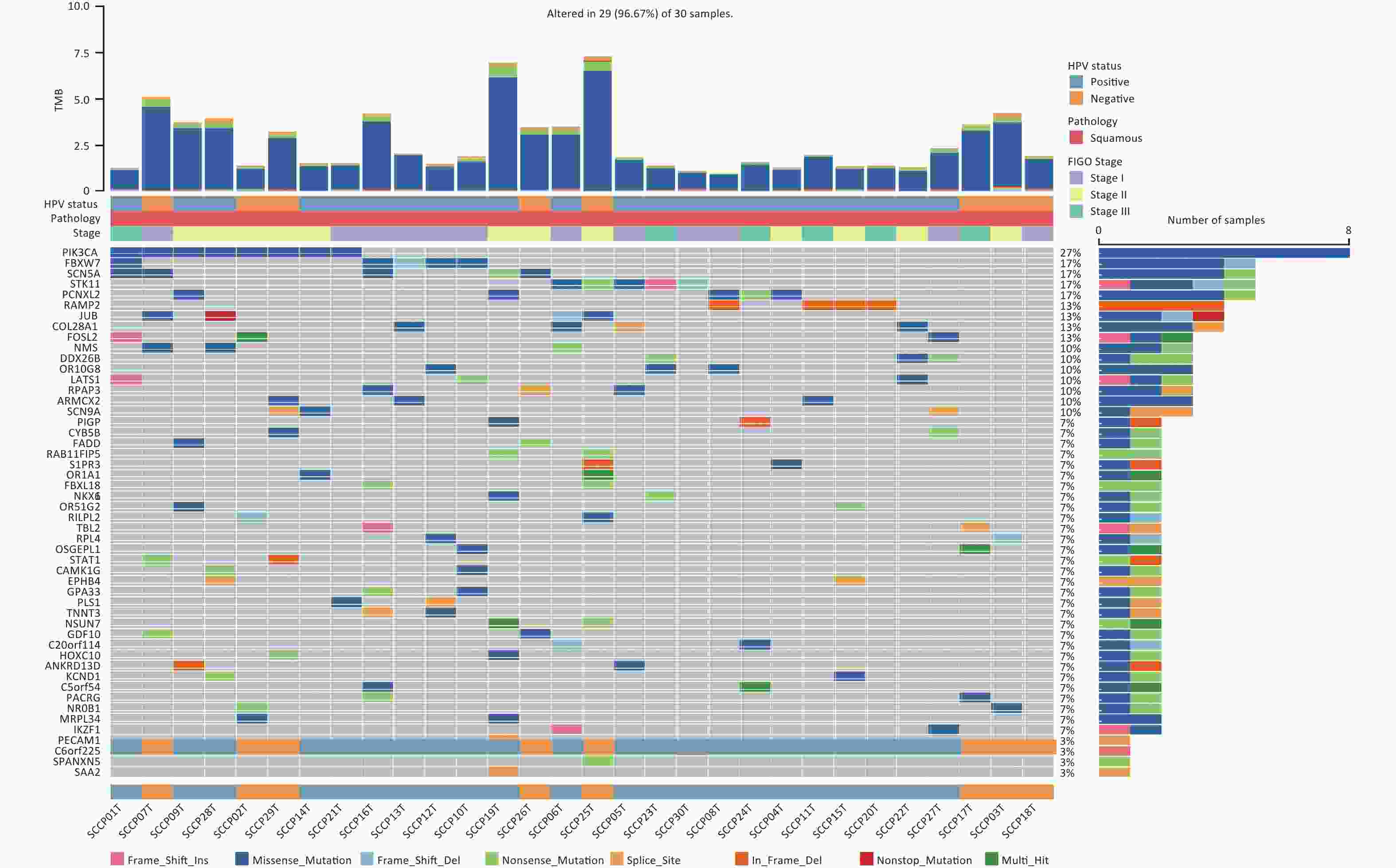
Figure 5. The alteration spectrum of the top 50 significantly mutated genes in 30 cervical squamous cell carcinoma samples. TMB, tumor mutation burden. HPV, human papillomavirus. FIGO, International Federation of Gynecology and Obstetrics.
-
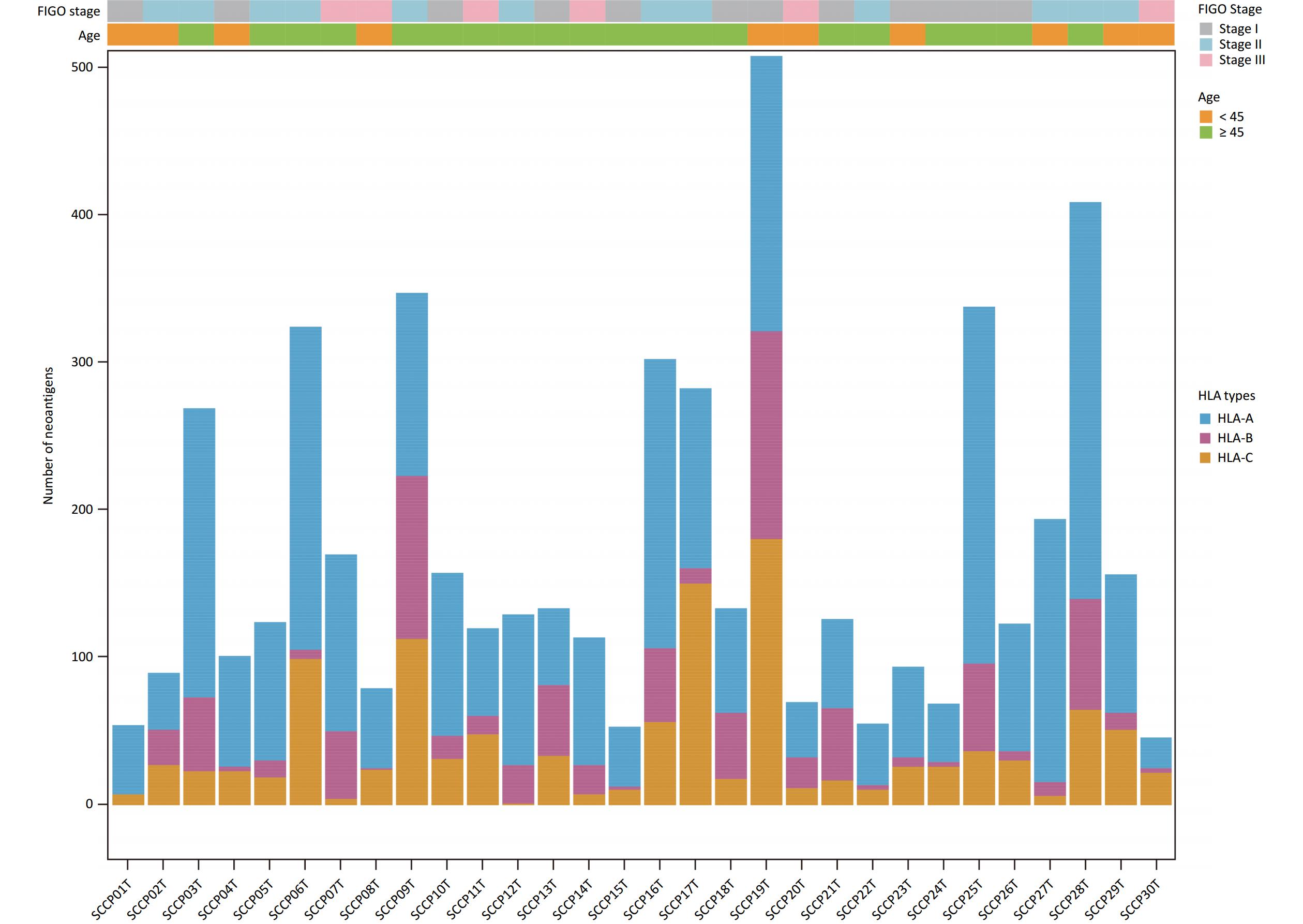
In this study, WES data for CSCCs was used to predict neoantigens and RNA-seq data was used to examine the expression of these neoantigens. The HLAscan tool was used to identify four-digit HLA class I alleles in CSCC samples, which showed that HLA-A*02:01 (33.33%), HLA-B*13:02 (16.67%), and HLA-C*06:02 (30.00%) were the most common alleles at the HLA-A, HLA-B, and HLA-C loci respectively (Table 2).
The NeoPredPipe pipeline incorporating the NetMHCpan 4.1 program was applied to predict neoantigens with filtering criteria of IC50 < 500 nmol/L and %Rank < 2%. Potential neoantigens were detectable in all samples, and 4960 potential neoantigens were identified (
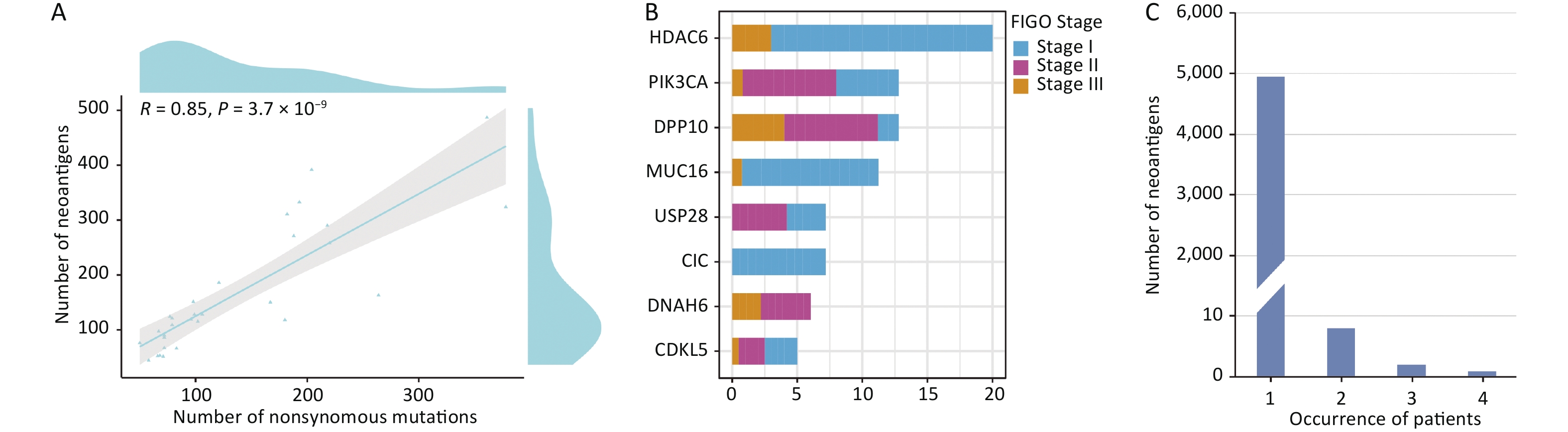
Supplementary Table S8 , available in www.besjournal.com). The average number of non-synonymous mutations in CSCC samples was 136.37 (range, 50–378), with an average of 165.33 neoantigens (range, 44–487) (Table 2). Figure 6 displays the predicted HLA class I neoantigens for each CSCC sample. Pearson correlation analysis showed a strong linear positive correlation between the number of neoantigens and the number of somatic non-synonymous mutations (Pearson correlation coefficient R = 0.85, P = 3.7 × 10−9) (Figure 7A), consistent with previous studies[35,38].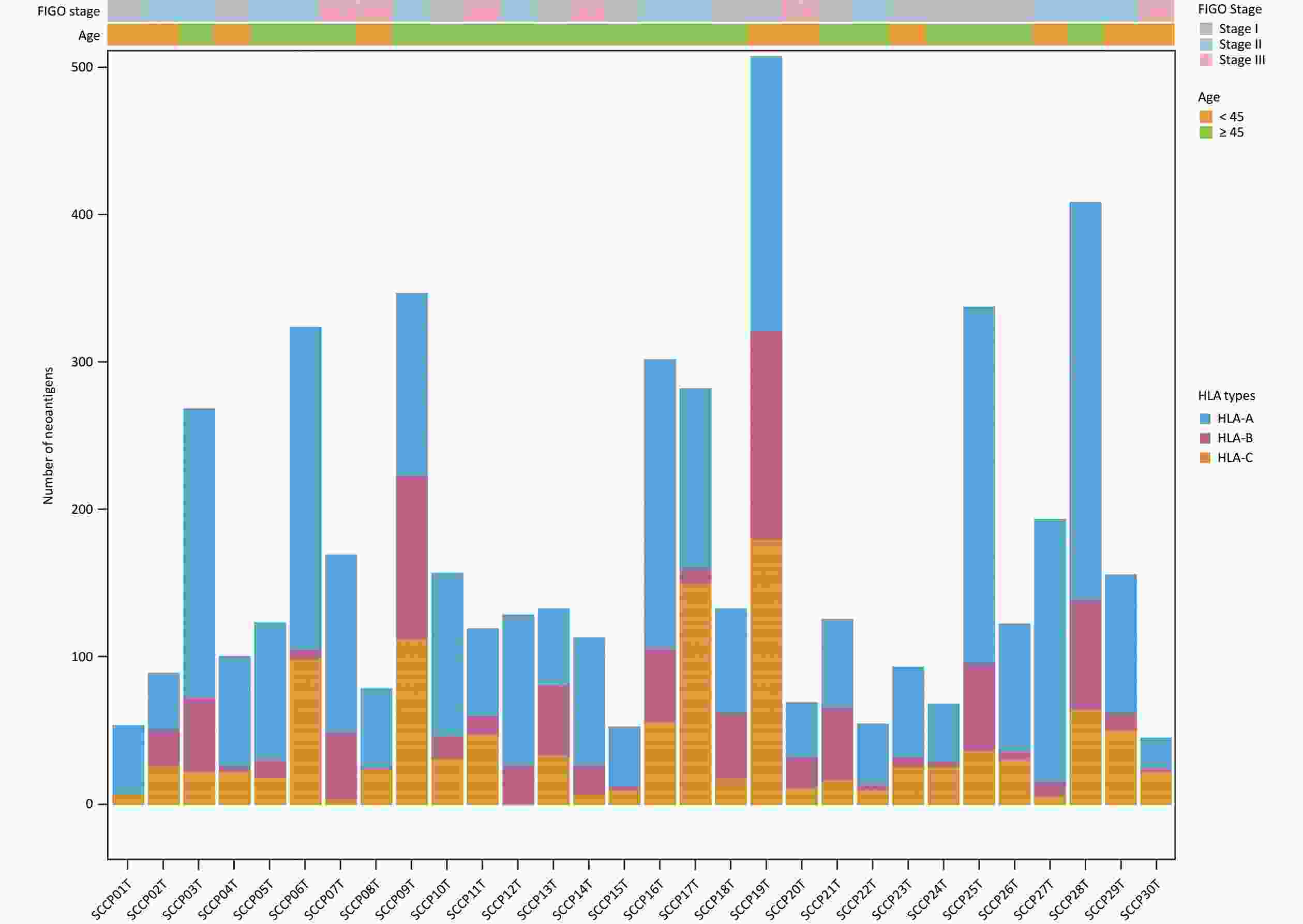
Figure 6. Predicted HLA class I neoantigens in 30 squamous cell carcinoma. Different colors represented different HLA types. HLA, human leukocyte antigen.
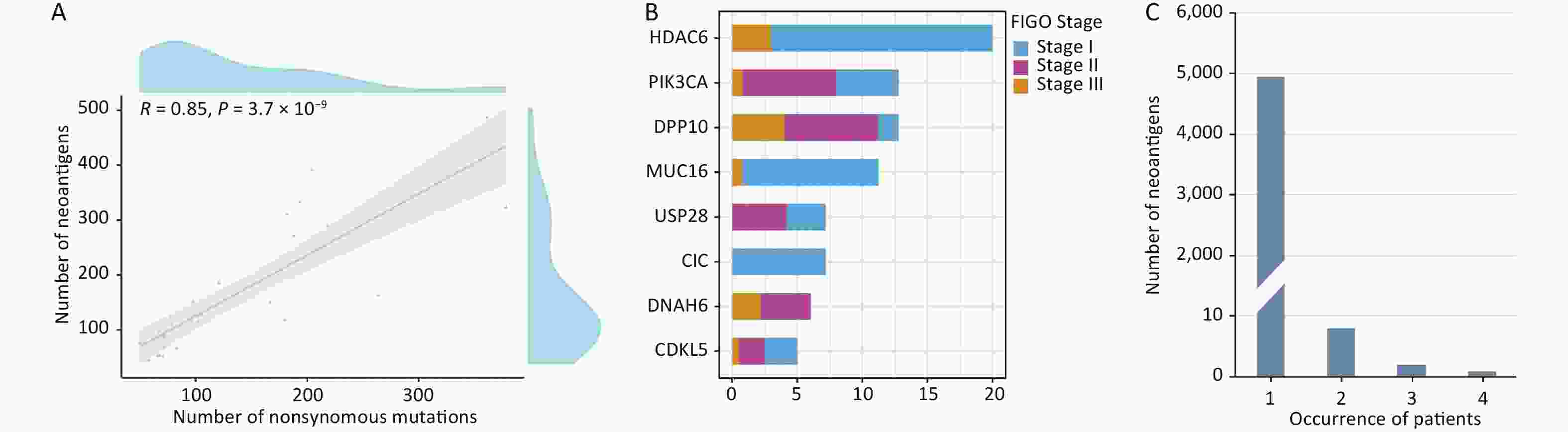
Figure 7. Distribution of potential neoepitopes genes in different stages of CSCC. (A) Correlation between the number of mutations and the number of exome-derived neoepitopes (Pearson’s correlation coefficient, R = 0.85, P < 0.001). (B) Barplots showed the number of potential neoantigen genes in CSCCs. (C) Neoantigen recurrence in our cohort. CSCC, cervical squamous cell carcinoma. FIGO, International Federation of Gynecology and Obstetrics.
The study also identified multiple recurrently mutated antigen genes, which were defined as “potential neoantigen genes”. Figure 7B shows the potential neoantigen genes at different FIGO stages:
HDAC6 (3/30), DPP10 (3/30), PIK3CA (8/30), MUC16 (5/30), CIC (3/30), USP28 (3/30), DNAH6 (3/30), and CDKL5 (3/30) (all genes were found in at least three CSCC samples). Most potential neoantigen genes were present in known oncogenic driver genes such as MUC16, which promotes cervical cancer progression via JAK2/STAT3 phosphorylation-mediated cyclooxygenase-2 expression[39]. Furthermore, PIK3CA is an immune-related gene deposited in the Immunology Database and Analysis Portal (ImmPort)[40].
In the present analysis of neoantigens, each unique immune target was detected in a particular CSCC patient, suggesting that neoantigens are highly heterogeneous. This result is consistent with previous reports that the vast majority of potential neoantigens are patient-specific[41]. Therefore, we explored the heterogeneity of potential neoantigens in CSCC. The results showed that 99.78% (4949/4960) of the neoantigens were present in only one patient, whereas only 0.22% (11/4,960) of the neoantigens were present in at least two patients (Figure 7C). “STRDPLSEITK”, generated by PIK3CA (E545K), was the most commonly shared neoantigen. This result suggests that identifying shared neoantigens for CSCC could be challenging.
RNA-seq data were used to examine the expression of neoantigens, and the results showed that there were 4,559 potential neoantigens in 27 pairs of samples, of which 58.13% (2,650/4,559, 2,650 neoantigens involving 939 genes) were expressed (Supplementary Table S9, available in www.besjournal.com).
-
The Tumor-Specific Neoantigen database (TSNAdb) is a collection of neoantigen information from 7748 samples of 16 different cancers in TCGA database[42]. CTdatabase contains high-throughput and carefully curated data on cancer-testis antigens[43]. The accuracy of the present neoantigen results was verified by comparison with TCGA-CESC (cervical squamous cell carcinoma and endocervical adenocarcinoma) data in TSNAdb and CTA data in CTdatabase. Among the 4,960 potential neoantigens identified in this study, 114 neoantigens involving 27 genes were identified in both databases. In TSNAdb, 46 neoantigens corresponding to eight genes were identified in TCGA-CESC dataset (Table 3), whereas in CTdatabase (CTAs), 68 neoantigens corresponding to 19 genes were identified (
Supplementary Table S10 , available in www.besjournal.com). Six of the 27 neoantigen-related genes were reported as therapeutic targets in the Therapeutic Target Database[44] and the corresponding drugs were also included. Alpelisib and BAY 80-6956, which are related to the PIK3CA target, have been approved by the FDA for the treatment of breast cancer and follicular lymphoma, respectively[45,46], whereas drugs targeting MAPK1, MUC16, MAGEA3, MAGEC2, and MAGEC1 are currently in clinical trials. Additionally, PIK3CA was included in a clinical trial as a potential therapeutic target in the treatment of CC (NCT02957266).Sample Protein Mutation AA HLA types Identity Length (AA) %Rank Affinity (nmol/L) Drugs SCCP14T CADPS R737W HLA-A02:06 YLRDLLEWA 9 0.502 25.53 − SCCP14T CADPS R737W HLA-B40:01 LEWAENGAM 9 0.436 52.67 − SCCP14T CADPS R737W HLA-B40:01 LEWAENGAMI 10 1.173 237.85 − SCCP24T CADPS R737W HLA-A02:01 YLRDLLEWA 9 0.407 24.09 − SCCP13T DENND5B D339H HLA-A02:01 FLHAPVPYL 9 0.007 3.86 − SCCP13T DENND5B D339H HLA-A02:01 SLLHFLHAPV 10 0.526 5.35 − SCCP13T DENND5B D339H HLA-A02:01 FLHAPVPYLM 10 0.469 13.82 − SCCP13T DENND5B D339H HLA-C03:04 FLHAPVPYL 9 0.133 34.66 − SCCP13T DENND5B D339H HLA-A02:01 HFLHAPVPYL 10 1.087 44.79 − SCCP13T DENND5B D339H HLA-B54:01 LPASLLHFLHA 11 0.194 52.16 − SCCP13T DENND5B D339H HLA-A02:07 FLHAPVPYL 9 0.014 256.91 − SCCP13T DENND5B D339H HLA-A02:01 LHFLHAPVPYL 11 1.597 272.86 − SCCP13T DENND5B D339H HLA-C01:02 HAPVPYLMGL 10 0.187 380.05 − SCCP13T DENND5B D339H HLA-C01:02 FLHAPVPYL 9 0.129 391.67 − SCCP13T DENND5B D339H HLA-A02:01 SLLHFLHAP 9 1.536 453.38 − SCCP07T MAPK1 E322K HLA-C04:01 YYDPSDKPI 9 0.017 389.35 BVD-523, ASTX029, HH2710 SCCP01T MAPK1 R135K HLA-A30:01 KGLKYIHSA 9 1.615 482.21 BVD-523, ASTX029, HH2711 SCCP06T MUC16 A4577T HLA-A02:01 SMGDTLASI 9 0.265 30.46 Oregovomab, Abagovomab SCCP06T MUC16 A4577T HLA-A02:06 SMGDTLASI 9 0.523 58.48 Oregovomab, Abagovomab SCCP06T MUC16 A4577T HLA-A02:01 SMGDTLASISI 11 1.565 324.65 Oregovomab, Abagovomab SCCP06T MUC16 A4577T HLA-C15:02 SSMGDTLASI 10 1.661 353.86 Oregovomab, Abagovomab SCCP07T PIK3CA E542K HLA-A03:01 AISTRDPLSK 10 0.14 53.93 Alpelisib, BAY 80-6946 SCCP07T PIK3CA E542K HLA-A03:01 KAISTRDPLSK 11 0.299 469.89 Alpelisib, BAY 80-6947 SCCP09T PIK3CA E542K HLA-A11:01 AISTRDPLSK 10 0.312 99.32 Alpelisib, BAY 80-6948 SCCP09T PIK3CA E542K HLA-A11:01 KAISTRDPLSK 11 0.273 256.36 Alpelisib, BAY 80-6949 SCCP09T PIK3CA E542K HLA-A11:01 ISTRDPLSK 9 0.629 333.21 Alpelisib, BAY 80-6950 SCCP21T PIK3CA E542K HLA-A11:01 AISTRDPLSK 10 0.312 99.32 Alpelisib, BAY 80-6951 SCCP21T PIK3CA E542K HLA-A11:01 KAISTRDPLSK 11 0.273 256.36 Alpelisib, BAY 80-6952 SCCP21T PIK3CA E542K HLA-A11:01 ISTRDPLSK 9 0.629 333.21 Alpelisib, BAY 80-6953 SCCP21T PIK3CA E542K HLA-C15:02 STRDPLSKI 9 0.135 453.03 Alpelisib, BAY 80-6954 SCCP01T PIK3CA E545K HLA-A30:01 STRDPLSEITK 11 0.01 38.96 Alpelisib, BAY 80-6955 SCCP02T PIK3CA E545K HLA-B58:01 ITKQEKDFLW 10 0.049 13.03 Alpelisib, BAY 80-6956 SCCP02T PIK3CA E545K HLA-B58:01 EITKQEKDFLW 11 1.03 391.32 Alpelisib, BAY 80-6957 SCCP14T PIK3CA E545K HLA-A11:01 STRDPLSEITK 11 0.053 91.72 Alpelisib, BAY 80-6958 SCCP28T PIK3CA E545K HLA-A11:12 STRDPLSEITK 11 0.053 91.72 Alpelisib, BAY 80-6959 SCCP28T PIK3CA E545K HLA-A11:01 STRDPLSEITK 11 0.053 91.72 Alpelisib, BAY 80-6960 SCCP29T PIK3CA E545K HLA-A11:01 STRDPLSEITK 11 0.053 91.72 Alpelisib, BAY 80-6961 SCCP03T SLC26A3 V340I HLA-C05:03 VGDCFDIAM 9 0.841 391.96 − SCCP19T VCPIP1 D434N HLA-A02:06 GIHPSLVANV 10 0.749 171.9 − SCCP19T VCPIP1 D434N HLA-A01:01 VANVHQYFY 9 0.173 244.29 − SCCP19T VCPIP1 D434N HLA-C14:02 LVANVHQYF 9 1.247 361.85 − SCCP19T VCPIP1 D434N HLA-A01:01 LVANVHQYFY 10 0.643 448.15 − SCCP28T ZBED4 E664K HLA-B15:02 KMIALDLQPY 10 1.429 89.83 − SCCP28T ZBED4 E664K HLA-C12:03 IAKMIALDL 9 0.66 132.87 − SCCP28T ZBED4 E664K HLA-A11:12 VAKKITSLIAK 11 1.798 386.39 − SCCP28T ZBED4 E664K HLA-A11:01 VAKKITSLIAK 11 1.798 386.39 − Note. AA, amino acids. HLA, human leukocyte antigen. Table 3. A list of candidate neoantigens validated by TSNAdb database
-
There is limited data on genomic alteration profiles and neoantigens of CSCC in Chinese patients. In this study, we used WES to analyze the somatic mutational landscape in a cohort of patients with CSCC (30 samples). Analysis of somatic non-synonymous mutations was used to identify potential neoantigens that can serve as new targets for CSCC immunotherapy. RNA-seq data was used to examine the expression of candidate neoantigens.
A total of 6,232 somatic mutations were identified in 30 CSCC samples, with an average of 207.73 mutations per sample, which is slightly lower than the average of 225.65 mutations per sample in TCGA database for CC[6]. Analysis of nonsilent mutations showed a mean mutation burden of 2.73/Mb, which was slightly higher than TCGA mutational burden of 2.53/Mb (excluding hypermutated tumors)[6]. Chung[34] et al. reported that cervical adenocarcinoma has a lower mutational burden than CSCC, which may explain the difference in mutational burden between this study and TCGA database. CNA analysis in CSCC identified 6,766 CNAs (225.87 per sample), which is considerably higher than the number reported in TCGA database for CC[6]. GISTIC2.0 analysis revealed 19 amplification peaks and 45 deletion peaks. Furthermore, 15 (50%) patients had high-level copy number amplification at 3q27.1, which was consistent with findings in lung squamous cell carcinoma and esophageal squamous cell carcinoma[47,48]. ALG3, which is one of the genes covered by this region, helps tumor cells generate high mannose N-linked glycans. Aberrant expression of high-mannose N-linked glycans is associated with cancer progression[49,50,51]. ALG3 is significantly overexpressed in radioresistant breast cancer tissues and promotes radioresistance and cancer stemness by inducing the glycosylation of TGF-β receptor II (TGFBR2)[50]. Although ALG3 is an effective therapeutic target in breast cancer patients with high ALG3 levels[50], whether it promotes the development of CSCC remains to be determined. HTR3C, another gene in the 3q27.1 region, is a biomarker for predicting lung cancer prognosis[52]. A risk model that includes HTR3E together with 13 other central immune-related genes (CBLC, TNF, PSMC4, TRAV30, PDIA3, FGF8, PDGFRA, ESRRA, SBDS, CRHR1, LTA, NR2F1, TNFRSF18) was used to predict the prognosis of endometrial carcinoma[53]. MIR1224 acts as a tumor suppressor in the occurrence and development of cancers and can be used as a tumor biomarker for early diagnosis and prognosis prediction[54]. UGT2B28 (located at 4q13.2) is a predictor of progression in prostate cancer and can be therapeutically targeted by using a combination of AR/EGFR inhibitors[55].
Analysis of CSCC samples identified three mutational signatures corresponding to signatures 2, 6, and 7 in the COSMIC database (Figure 4C). These are slightly different from those found in the CESC dataset in TCGA database, specifically signature 2 (72.5%) and 6 (13.2%). The APOBEC family of proteins specifically catalyze the conversion of cytosine in the genome to uracil, which is related to base excision repair and DNA replication mechanisms[56]. It can be activated by HPV virus infection and is involved in the immune response[56]. APOBEC, which is closely related to cervical carcinogenesis, is the source of signature 2 and signature 13 in human cancers[6,57]. These studies suggest that APOBEC causes CSCC mutations under the control of HPV. Additionally, signature 6 is a novel early warning biomarker for CC associated with the deficiency of DNA mismatch repair.
This study identified three genes (PIK3CA, FBXW7, and BICRA) predicted to act as driver genes that could potentially promote tumor formation and development. BICRA, a component of the SWI/SNF chromatin remodeling complex, was identified as a novel driver gene of CSCC. However, we did not find published evidence supporting that this gene is associated with cancer risk or development. We observed eight missense mutations in PIK3CA (26.67%), which is consistent with results reported previously (26% in Cancer Genome Atlas Research Network[6], 16.7% in Huang et al.[16]). The results further indicated no significant difference in the PIK3CA mutation rate between cervical adenocarcinoma and CSCC. We identified two SMGs that may be associated with CSCC: RAMP2 and FOSL2. RAMP2 downregulation may promote distant metastasis of cancers and is associated with a low survival rate in oral squamous cell carcinoma[58,59], whereas FOSL2 is closely related to the occurrence of ovarian cancer, lung cancer, and breast cancer[60,61,62]. The results indicate that these two genes may play crucial roles in the occurrence and development of tumors. However, further studies are needed to validate these findings and to develop these factors as potential biomarkers for CSCC.
Antigen presentation plays a crucial role in the human immune response to cancer. Immunotherapies for cancer are often based on targeting antigens presented by major histocompatibility complex/HLA molecules[63]. Significant advances have been made in immunotherapy strategies for the treatment of solid tumors (such as breast cancer, prostate cancer, and non-small cell lung cancer). However, the currently available approaches are not sufficient to cure CC. Neoantigens are optimal targets for immunotherapy and are up-and-coming therapeutic options. We identified 4960 neoantigens in this study and found that the number of neoantigens was positively correlated with the number of somatic non-synonymous mutations, whereas it showed no obvious correlation with clinical stage. HDAC6 and DPP10 are two potential neoantigen genes that are detected in the early stage of CSCC. HDAC6 is a unique HDAC family member that regulates the Ras/MAPK/ERK, PI3K/Akt, and Wnt signaling pathways, which are associated with cellular proliferation and are activated in most tumors[64]. Liu et al. suggested that DPP10 inhibits colon cancer stem cell proliferation by regulating microRNAs such as miR-127-3p[65]. A previous study demonstrated that DPP10 methylation levels are significantly correlated with cervical neoplasia progression[66]. We found that almost every neoantigen was present in one sample, further highlighting the difficulty in ubiquitous neoantigen identification in CSCC. We used RNA-seq data to examine the expression of neoantigens and found that five potential neoantigen genes (including PIK3CA, MUC16, USP28, CIC, and CDKL5) were expressed. Further extended clinical studies are needed to determine whether these genes are of value for CSCC immunotherapy. The validation with two available neoantigen-related public databases (TSNAdb and CTdatabase) identified 114 neoantigens involving 27 genes that may serve as candidate targets for neoantigen vaccines.
-
In this study, the comprehensive genomic characteristics of CSCC were determined using WES data. WES and RNA-seq data were used to narrow the scope of neoantigens for individualized immunotherapy in CSCC. Further experimental verification is needed to obtain effective neoantigens.
Identifying Comprehensive Genomic Alterations and Potential Neoantigens for Cervical Cancer Immunotherapy in a Cohort of Chinese Squamous Cell Carcinoma of the Cervix
doi: 10.3967/bes2024.064
- Received Date: 2023-09-15
- Accepted Date: 2024-02-23
-
Key words:
- Cervical squamous cell carcinoma /
- Genome alteration /
- Neoantigens /
- Immunotherapy
Abstract:
| Citation: | Meng Wu, Jialu Zhou, Zhe Zhang, Yuanguang Meng. Identifying Comprehensive Genomic Alterations and Potential Neoantigens for Cervical Cancer Immunotherapy in a Cohort of Chinese Squamous Cell Carcinoma of the Cervix[J]. Biomedical and Environmental Sciences, 2024, 37(6): 565-580. doi: 10.3967/bes2024.064

|

 Quick Links
Quick Links
 DownLoad:
DownLoad: