
HTML
-
Viruses are a cause of serious health problems in humans, both in developed and developing countries; in particular, some pathogenic viruses have a potential for rapid and global spread with high morbidity and mortality. The last 30 years of the 21st century have witnessed some outbreaks of significant public health issues caused by viruses, such as Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) in 2003 [1-3], swine-origin H1N1 influenza virus in 2009 [4-5], avian-origin influenza A H7N9 virus in 2013 [6-8], Middle East Respiratory Syndrome (MERS-CoV) in 2012 [9-12], and Ebola virus from 2014 to 2015 [13-14]. Therefore, accurate identification and analysis of the causative agent of viral infectious disease is extremely urgent and crucial for active control and prevention of virus spread.
In recent years, next-generation sequencing (NGS) has been an emerging method for the identification of viral pathogens. It does not require a priori knowledge of potential infectious agents and is a more powerful tool than traditional viral diagnostic methods, including electron microscopy, cell culture, antigen detection, nucleic acid detection, cytology, histology, and serology [15]. Multiple viruses can be detected simultaneously, and novel or highly divergent viruses can be discovered and genetically characterized using NGS [16-24]. However, on account of the low virus abundance and high background noise in clinical specimens, the clinical application of NGS in virus detection is still limited. Moreover, other limitations, such as the relatively high cost of library preparation and sequencing, complicated and tedious procedures, as well as tremendous data processing and analysis, also hinder the widespread use of NGS in clinical practice.
At present, there are many ways to increase the signal-to-noise ratio (ratio of viral genome to host genome), such as ultracentrifugation, filtration, nuclease treatment, sequence-independent amplification (like random PCR), or a combination of these [25]. Particularly, a combination method of centrifugation, filtration, nuclease treatment, and random-PCR (rPCR) showed the greatest increase in the proportion of viral sequences [26]. In addition, rPCR is a widely used and efficient method in which reverse transcription is performed by using oligonucleotides made up of random hexamers tagged with a known sequence for the enrichment of low-abundance viral nucleic acids [27-30]. Recently, a substracted primer set named non-ribosomal hexanucleotides (hexanucleotides) was designed to further reduce noise for reverse transcription, and its viral enrichment ability was verified by cDNA representational difference analysis (RDA) and compared with that of random hexamers [31]. NGS-based evaluation of the viral enrichment abilities of both random hexamers and hexanucleotides is yet unreported. Directly pooling samples at the very beginning (without barcodes) or pooling individual libraries (with barcoded adaptors) for NGS may reduce sequencing costs, but the subsequent verification tests of the interested sample from pooled samples or the individual library construction of each pooled sample are rather laborious and time consuming. In addition, fragmented DNA or RNA libraries are always recommended to acquire much information from the original sample; however, the shear time is difficult to control because DNA or RNA originates from samples with different virus abundance and this treatment may increase the occurrence of artefactual recombination [32]. Moreover, the processing and analysis of tremendous NGS data is always complex, time consuming, and highly dependent on excellent profe ssional bioinformaticians, which is impractical in many laboratories.
To address these limitations, in the present study, we describe a simplified, rapid, and relatively cost-effective NGS approach in which the barcoded random hexamer or non-ribosomal hexanucleotide primers are used in the reverse transcription step for viral enrichment followed by sample pooling of each enriched product in one tube as a single sample for fragmentation-free library construction and sequencing, and the high-throughput sequencing data are rapidly analyzed by an in-house pipeline. We demonstrate that multiple viruses can be accurately identified from cell culture materials or clinical specimens and verify that the viral enrichment abilities of both primer sets are nearly comparative in model experiments (sequencing three representative viruses). Furthermore, direct testing of clinical specimens associated with hand, foot, and mouth disease (HFMD) using this improved hexanucleotide primer-based NGS approach provides further detailed genotypes of viral pathogens and identifies other potential viruses or differentiated misdiagnosis events appearing in the clinic.
-
Influenza B virus (Flu B), Coxsackievirus A16 (CVA16), and Epstein-Barr virus (EBV) were chosen as representatives of segmental RNA virus, non-segmental RNA virus, and DNA virus, respectively. Cell culture supernatants of Flu B and EBV were kindly provided by the Key Laboratory for Medical Virology of the China CDC. The CVA16-positive sample and 20 clinical specimens were collected from pediatric patients clinically diagnosed with severe or mild HFMD in the Hebei province of China in 2015. All samples were stored at -80 ℃ before use and their information is listed in Table 1. In this study, patients diagnosed with HFMD who had more serious complications, including meningitis, encephalitis, cardiorespiratory failure, acute flaccid paralysis, or death, were classified as severe HFMD. Patients diagnosed with HFMD, but without any serious complications, were classified as mild HFMD.
Specimens Specimen Type Clinical Diagnosis Laboratory Diagnosis (Real-time RT-PCR) CT Value Clinical specimen 1 Stool Mild HFMD HEV 32.9 Clinical specimen 2 Stool Mild HFMD HEV 30.6 Clinical specimen 3 Stool Mild HFMD HEV 34.0 Clinical specimen 4 Rectal swabs Mild HFMD HEV 32.0 Clinical specimen 5 Rectal swabs Mild HFMD HEV 34.0 Clinical specimen 6 Stool Mild HFMD HEV 30.2 Clinical specimen 7 Throat swabs Mild HFMD HEV 29.8 Clinical specimen 8 Throat swabs Mild HFMD HEV 33.0 Clinical specimen 9 Throat swabs Mild HFMD ﹣ 36.2 Clinical specimen 10 Throat swabs Mild HFMD HEV 32.0 Clinical specimen 11 Stool Severe HFMD HEV 27.9 Clinical specimen 12 Stool Severe HFMD HEV 28.1 Clinical specimen 13 Stool Severe HFMD ﹣ 35.4 Clinical specimen 14 Stool Severe HFMD HEV 26.2 Clinical specimen 15 Stool Severe HFMD HEV 34.2 Clinical specimen 16 Stool Severe HFMD HEV 32.8 Clinical specimen 17 Stool Severe HFMD ﹣ 35.2 Clinical specimen 18 Rectal swabs Severe HFMD HEV 34.1 Clinical specimen 19 Throat swabs Severe HFMD EV 71 31.0 Clinical specimen 20 Stool Severe HFMD ﹣ 35.7 Table 1. Identification of Specified Viral Pathogens in Different Samples Using Real-time RT-PCR
This project was approved by the Institutional Review Boards of the Centre of Disease Control and Prevention of China and the Ethical Review Committee of the Institute for Viral Disease Control and Prevention, Center for Disease Control and Prevention of Hebei. Individual written informed consent was obtained from the parents or guardians of all participants. All methods were carried out in accordance with the approved guidelines, including any relevant details.
-
Clinical specimens were thawed at 37 ℃ followed by preprocessing to reduce background noise as in a previously published protocol [33]. Briefly, the viral transport medium was vortexed thoroughly and centrifuged at 1500 ×g for 10 min to remove debris. Then, the supernatant was filtered through a 0.22 μm filter (Millipore) and dispersed into aliquots frozen at -80 ℃. An aliquot was treated with a cocktail of nucleases, including 10 units TURBO™, DNase (Ambion), 25 units Benzonase®nuclease (Sigma), 5 units DNase I (Takara), 2 μg RNase A (Takara), 5 units RNase T1 (Thermo Scientific), 4 units RNase One (Promega), 4 units micrococcal nuclease (New England Biolabs), and 15 units mung bean nuclease (Promega), to degrade any extracellular nucleic acids at 37 ℃ for 90 min. Then, viral nucleic acids were extracted from the preprocessed samples using the QIAamp Viral RNA Mini Kit (Qiagen) per the manufacturer's instructions.
-
For detection of Flu B, CVA16, or EBV, real-time RT-PCR was performed in a 50 μL reaction solution containing 5 μL RNA or DNA, 0.5 μL of each forward and reverse primers (Flu B; F, 5'-TCCTCAACT CACTCTTCGAGCG-3', and R, 5'-CGGTGCTCTTGACCA AATTGG-3' [34], CVA16, F, 5'-GGGAATTTCTTTAGC CGTGC-3', and R, 5'-CCCATCAARTCAATGTCCC-3' [35], EBV; F, 5'-AACATTGGCAGCAGGTAAGC-3', and R, 5'-ACTTACCAAGTGTCCATAGGAGC-3' [36])and other reagents of GoTaq®qPCR Master Mix (Promega). Thermal cycling parameters of reverse transcription were 42 ℃ for 15 min; denaturation at 95 ℃ for 10 min; and 40 cycles of 95 ℃ for 10 s, 60 ℃ for 30 s, and 72 ℃ for 30 s on an ABI 7900HT Fast Real-Time PCR System (Applied Biosystems).
For detection of clinical specimens from patients diagnosed with severe or mild HFMD, real-time RT-PCR was conducted using primer pairs and corresponding probes of HEV, EV71, and CVA16 per a previously published protocol [35]. Samples were considered positive only if threshold cycle (CT) values were < 35.
-
Enrichment of purified viral nucleic acids was conducted by using a modified RT-PCR method. Briefly, 8 μL viral RNA was mixed with 0.5 mmol/L dNTPs and 50 pmol random hexamers or hexanucleotides tagged with known barcode sequences (A, AATCACATAGGCGTCCGCTG; B, GGCA GGACCTCTGATACAGG; C, GGCACTCGACTCGTAACA GG; D, GTCTCTGTCACGACGGTCAG; E, GGATAGTCGG CGTGCACCAA; F, AACACCTCTGGAACACGCCC; G, GGT CCAGGCACAATCCAGTC; H, ACGGTGTGTTACCGACGT CC; I, TGCGAACCGTTACGGGTGGA; J, GGACAAGCAC AGCATAGCCA), which were not only used for viral nucleic acid enrichment but also for pooling and demultiplexing each sequencing viral library [37-38]. Then the mixture was heated at 70 ℃ for 5 min and immediately placed on ice. First-strand cDNA synthesis was performed per the SuperScript®III Reverse Transcriptase kit (invitrogen) manual. Thereafter, the second strand of cDNA was synthesized using 3'-5' exo- Klenow Fragment (New England Biolabs) followed by amplification of double-stranded cDNA using the barcode oligonucleotide and KAPA HiFi Hot Start Ready Mix PCR kit (KAPABiosystems) for enrichment of viral contents. The PCR program included 3 min of initial denaturation at 95 ℃; 25 cycles of 98 ℃ for 20 s, 60 ℃ for 30 s, and 72 ℃ for 30 s; and a final extension at 72 ℃ for 3 min.
-
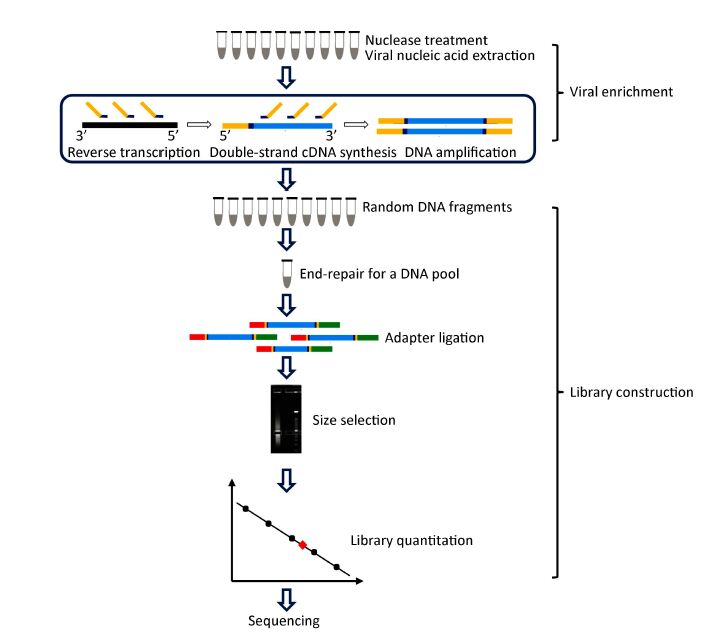
The enriched products were purified separately by using the MinElute Reaction Cleanup Kit (Qiagen) per the manufacturer's specifications. Groups of 10 purified DNA samples were pooled at equal mass as one mixture for library construction (Figure 1). Briefly, a total of 500 ng (50 ng of each sample) of the mixture used as the input was placed in an end-repair reaction (Ion Plus Fragment Library kit, Life Technologies) and purified using Agencourt Ampure Beads (Beckman Coulter). Then, Ion Torrent sequencing adaptors were ligated on both ends of the pooled DNA followed by purification with an appropriate volume of Agencourt Ampure Beads to remove adapter dimers. The 290-330 bp fractions were recycled from the pooled DNA on E-Gel®SizeSelect™, 2% Agarose Gel (Invitrogen) and subjected to Agilent®2100 Bioanalyzer analysis using an Agilent®high-sensitivity DNA kit (Agilent Technologies). The qualified library quantified by qPCR using Ion Library Quantification Kit (Life Technology) was subsequently subjected to emulsion-PCR with Ion PGM™, Template Hi-Q Kit (Life Technology) and ES enrichment with Ion PGM™, Enrichment Beads (Life Technology). Then, the recovered template-positive Ion Sphere™, Particles were loaded onto a 316v2 chip and sequenced on an Ion Torrent PGM™, sequencer using Ion Torrent Hi-Q sequencing kit (Life Technologies).
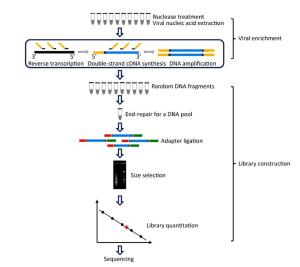
Figure 1. A schematic overview of viral enrichment and library construction in the improved NGS approach. Viral nucleic acid enrichment was initially performed using barcoded oligonucleotide primers (non-ribosomal hexanucleotides or random hexamers) in the reverse transcription step and each enriched product was pooled together in one tube as a single sample for subsequent fragmentation-free library construction and sequencing.
-
The raw sequencing data from each sequencing run were demultiplexed using vendor software (PGM) according to the barcode sequence to generate individual BAM files of each sample. Then, these files containing raw sequencing reads were directly processed and analyzed by in-house virus identification pipeline (VIP) in a Bio-Linux operating environment. VIP, recently developed in our laboratory [39], is a one-touch computational pipeline for virus identification and discovery from metagenomic NGS data and contributes to timely virus diagnosis (-10 min) in the performance of unbiased NGS data. The program run is accomplished in minutes and generates VIP reports, including virus populations, coverage statistics of certain gene or complete genome of viral pathogens, corresponding average depth of sequencing, and coverage files of read distributions on the reference viral genome.
Clinical Specimens and Ethical Approval
Sample Preprocessing and Viral Nucleic Acid Purification
Real-time RT-PCR
Viral Nucleic Acid Enrichment
Library Construction and Sequencing
NGS Data Processing and Analysis
-
Three representative virus-positive (Flu B, CVA16, and EBV) samples and 20 clinical specimens diagnosed with severe or mild HFMD were detected by real-time RT-PCR. The results showed that all the cell culture materials and CVA16 sample were confirmed positive. As shown in Table 2, the CT values of EBV and CVA16-positive sample were 30.6 and 30, respectively. Regarding Flu B-positive samples, two samples (Flu B-1 and Flu B-2) contained slightly high viral loads (CT values were 21 and 25, respectively), whereas the third one (Flu B-3) contained low viral loads (CT value was 29).
Samples Sample Type CT Valuesa Random Hexamer Primers Non-ribosomal Hexanucleotide Primers Total Readsa Mean Read Length (bp)a Virus-Related Reads (%)a Mapping Reads of Gene or Complete Genomea Coverage (%)a Average Depth of Sequencing (×) a Total Readsb Mean Read Length (bp)b Virus-Related Reads (%)b Mapping Reads of Gene or Complete Genomeb Coverage (%)b Average Depth of Sequencing (×)b EBV Cell supernatant 30.6 174,795 178 30.3 membrane protein, 34,277 100 2771.57 123,955 104 19.8 membrane protein, 2,928 98.05 195.67 CVA16 throat swabs 30 423,172 157 13.1 genome 35,624 85.61 85.68 187,994 175 16.6 genome 700 93.53 11.36 FluB-1 Cell supernatant 21 564,046 161 9.9 segment 1 15,366 100 132.65 183,436 153 11.3 segment 1 3,680 99.28 32.42 segment 2 16,697 100 166.63 segment 2 3,817 99.33 28.95 segment 3 15,344 99.78 124.24 segment 3 3,481 99.56 31.19 segment 4 16,047 99.94 182.15 segment 4 5,593 99.94 136.46 segment 5 19,428 100 164.94 segment 5 4,609 99.45 39.72 segment 6 17,698 100 371.31 segment 6 4,642 100 137.03 segment 7 17,812 100 290.81 segment 7 4,008 99.49 68.96 segment 8 14,667 99.90 227.82 segment 8 3,272 99.90 62.02 FluB-2 Cell supernatant 25 283,155 180 10.9 segment 1 6,218 99.95 48.88 44,509 175 12.3 segment 1 860 99.07 7.63 segment 2 7,142 100 60.53 segment 2 1,154 99.12 8.98 segment 3 6,487 99.82 63.50 segment 3 963 99.34 8.69 segment 4 6,772 99.94 78.52 segment 4 904 99.09 8.00 segment 5 8,209 99.94 114.48 segment 5 979 99.51 11.07 segment 6 7,405 100 103.00 segment 6 1,865 98.65 92.37 segment 7 8,244 98.65 202.54 segment 7 1,057 97.98 13.33 segment 8 6,430 99.90 126.79 segment 8 916 98.17 19.80 FluB-3 Cell supernatant 29 3,593 149 13.9 segment 1 60 35.52 0.48 6,777 148 14.7 segment 1 97 47.59 0.65 segment 2 60 53.25 0.98 segment 2 85 43.90 0.61 segment 3 55 48.02 0.88 segment 3 124 58.30 1.14 segment 4 57 57.22 1.05 segment 4 136 73.85 1.60 segment 5 56 36.11 0.45 segment 5 143 80.20 2.42 segment 6 72 60.75 1.67 segment 6 138 81.56 2.87 segment 7 52 38.73 0.65 segment 7 102 60.92 1.20 segment 8 64 44.84 0.85 segment 8 101 77.48 1.64 Note. aData obtained from viral enrichment by random hexamer primers in reverse transcription polymerase chain reaction. bData obtained from viral enrichment by non-ribosomal hexanucleotide primers in reverse transcription polymerase chain reaction. Table 2. NGS Data of Three Representative Viruses Respectively Enriched by Random Hexamer Primers or Non-ribosomal Hexanucleotide Primers in a Model Experiment
Among 20 HFMD clinical specimens, 16 samples were found to be positive with CT values < 35, of which 1 sample was confirmed positive for enterovirus 71 (EV 71) and 15 samples were found to be positive for human enterovirus (HEV). Notably, the remaining four samples tested negative, of which three originated from severe HFMD cases and one originated from a mild HFMD case (Table 1).
-
To evaluate the feasibility of the improved NGS approach, three of the representative viruses-positive samples (Flu B, CVA16, and EBV) described above were initially selected for sequencing following the modified procedure (Figure 1) in a model experiment. Meanwhile, the viral enrichment abilities of both hexanucleotide primers and the traditional random hexamer primers were compared in parallel based on NGS data. Viral nucleic acids of representative viruses were separately primed by barcoded hexamer or hexanucleotide primers in the first- and second-strand cDNA synthesis reaction followed by amplification of double-stranded cDNA using an oligonucleotide barcode. Thereafter, amplified products were pooled together as a single sample for fragmentation-free library construction and sequencing.
The raw sequencing data were assigned back to the original samples individually according to the barcode sequence. The total reads of individual samples after a single run ranged from 3, 593 to 564, 046, and the mean read length varied from 104 bp to 180 bp (Table 2). The proportion of virus-related reads among the total reads was further calculated via an in-house VIP. The results showed that the ratios of virus-related reads captured by hexanucleotide primers were higher than those captured by hexamer primers in RNA samples, whereas the ratio of virus-related reads captured by hexanucleotide primers was slightly lower than those captured by hexamer primers in EBV samples.
Furthermore, as shown in Table 2, the Flu B-related sequence reads enriched by hexanucleotides from samples with high viral loads (Flu B-1 or Flu B-2) revealed similar sequence coverage of Flu B segments 1-8 (range from 97.98% to 100%) compared with the results obtained by random hexamers (98.65%-100%). However, the reads enriched by hexanucleotides from samples with low viral loads (Flu B-3 or CVA16) achieved higher sequence coverage than the results obtained by random hexamers, as seen in segments 1-8 of Flu B (range from 43.90% to 81.56% vs. 35.52% to 60.75%) and the complete CVA16 genome (93.53% vs. 85.61%). Moreover, the EBV-related sequence reads enriched by both hexanucleotides and random hexamers covered the nearly complete sequence of the membrane protein gene of EBV (98.05% vs. 100%). These results indicate that the improved NGS method can accurately identify multiple viral pathogens from cell culture materials or clinical specimens and the hexanucleotide primer-based NGS is probably more suitable for clinical viral identification in view of the sequencing coverage of both RNA and DNA viruses.
-
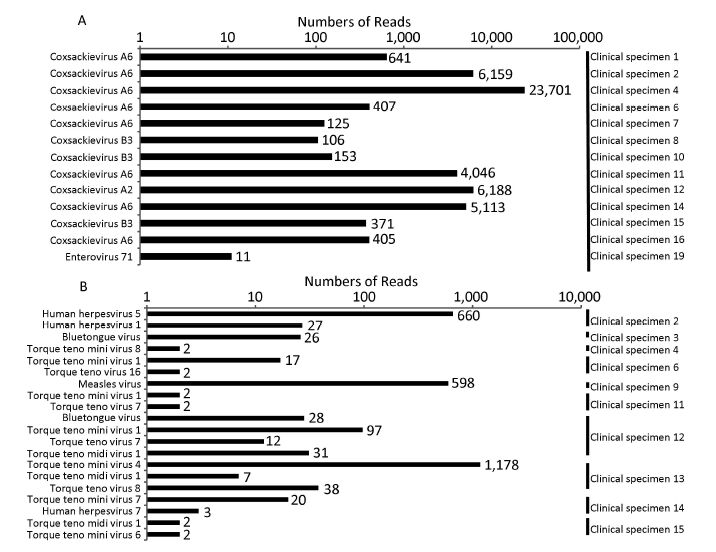
To further confirm that the hexanucleotide primer-based NGS method is viable for clinical applications, all the clinical specimens from pediatric patients clinically diagnosed with mild or severe HFMD were subjected to sequencing following the hexanucleotide primer-based NGS procedure. Viral nucleic acids from each sample (20 samples in total) were enriched by using barcoded hexanucleotide primers in a reverse transcription, double-strand cDNA synthesis, and amplification reaction. After accomplishing library construction, two sequencing libraries (each containing 10 samples) were loaded onto two 316v2 chips (Life Technology) and separately sequenced on an Ion torrent PGM. The raw data from two independent sequencing runs were assigned back to the original samples according to the different barcode sequences. The total number of reads from each sample ranged from 4, 355 to 379, 687 and the mean read length varied from 83 bp to 179 bp (Table 3). All reads originating from each sample were used directly as input for the virus identification via VIP. The results showed that many reads mapped to HFMD-related viral pathogens (Figure 2A), of which eight samples were identified as Coxsackievirus A6, three samples were identified as Coxsackievirus B3, one sample was identified as Coxsackievirus A2, and another one was identified as EV 71. Additionally, the remaining seven clinical specimens were identified as HEV-negatives, of which four samples were also found to be negative by real-time RT-PCR.
Specimens HFMD-associated Viruses Total Reads Mean Read Length (bp) Coverage (%) Average Depth of Sequencing (×) Clinical specimen 1 Coxsackievirus A6 44, 541 176 73.89 2.26 Clinical specimen 2 Coxsackievirus A6 273, 579 138 91.44 12.02 Clinical specimen 3 ﹣ 56, 236 156 ﹣ ﹣ Clinical specimen 4 Coxsackievirus A6 379, 687 151 87.34 45.48 Clinical specimen 5 ﹣ 8, 731 160 ﹣ ﹣ Clinical specimen 6 Coxsackievirus A6 12, 873 145 39.18 1.43 Clinical specimen 7 Coxsackievirus A6 6, 416 163 19.23 0.33 Clinical specimen 8 Coxsackievirus B3 10, 551 114 18.63 0.23 Clinical specimen 9 ﹣ 15, 487 162 ﹣ ﹣ Clinical specimen 10 Coxsackievirus B3 35, 019 96 21.03 0.31 Clinical specimen 11 Coxsackievirus A6 181, 450 168 81.87 10.26 Clinical specimen 12 Coxsackievirus A2 298, 943 164 58.13 15.96 Clinical specimen 13 ﹣ 17, 672 151 ﹣ ﹣ Clinical specimen 14 Coxsackievirus A6 108, 432 179 96.11 13.93 Clinical specimen 15 Coxsackievirus B3 33, 690 153 42.91 0.80 Clinical specimen 16 Coxsackievirus A6 17, 959 134 34.24 1.05 Clinical specimen 17 ﹣ 9, 242 129 ﹣ ﹣ Clinical specimen 18 ﹣ 4, 572 115 ﹣ ﹣ Clinical specimen 19 EV 71 4, 355 83 2.07 0.02 Clinical specimen 20 ﹣ 4, 557 109 ﹣ ﹣ Table 3. FMD-related Viral Pathogens Identified from Clinical Specimens Clinically Diagnosed with Mild or Severe HFMD Using the Improved Hexanucleotide Primer-Based NGS Approach
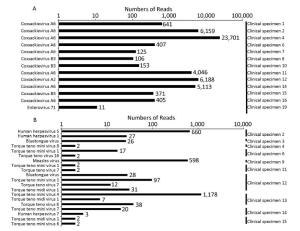
Figure 2. Schematic summary of the number of reads related to detailed virus subtypes. Reads obtained from 13 clinical specimens were related to causative agents of HFMD (A) and reads obtained from 10 clinical specimens were unrelated to causative agents of HFMD (B).
-
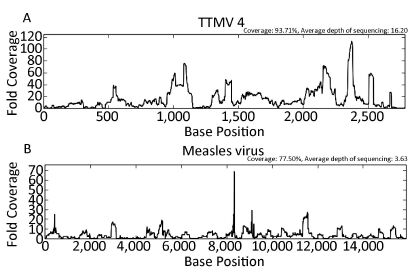
Further analysis showed that several HFMD-unrelated viral pathogens were simultaneously identified in the clinical samples. As shown in Figure 2B, reads of seven samples were assigned to members of the Anelloviridae family, which contains 11 genera [40]. Two of these samples (sample 4 and sample 14) were found to have torque teno mini virus (TTMV)-related sequences, two of these samples (sample 6 and sample 11) were found to have both TTMV- and torque teno virus (TTV)- related sequences, a single sample (sample 15) was found to be positive for both TTMV and torque teno like midi virus (TTMDV), and another two samples (sample 12 and sample 13) were found to be positive for TTMV, TTV, and TTMDV. Notably, clinical specimen 13 contained significant numbers of reads with 93.71% of coverage on reference complete genome of TTMV 4 (Figure 3A). This sample was subsequently confirmed positive by nest RT-PCR using universal primer pairs [41] (data not shown).
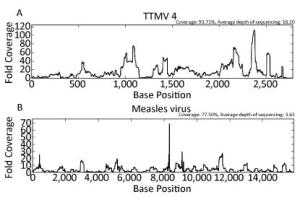
Figure 3. Read distribution in reference viral genomes. All raw reads originating from clinical specimens 14 or 10 were directly used as input for virus detection via in-house VIP. Some reads originating from clinical specimen 14 were assigned to TTMV 4 and covered 93.71% of the reference viral complete genome (GenBank accession number NC_014090) (A). Some reads originating from clinical specimen 10 were assigned to Measles virus and covered 77.50% of the reference viral complete genome (GenBank accession number NC_001498) (B).
Interestingly, reads of several samples were assigned to bluetongue virus and human herpesvirus, of which clinical specimen 2 and clinical specimen 14 were found to have sequences related to three human herpesvirus types. Notablely, clinical specimen 3 and clinical specimen 12 tested positive for bluetongue virus with 71.69% of coverage of the PSMCH1 gene or 76.20% of coverage of the 5'-UTR of the genome, in which the latter was simultaneously co-detected with the presence of TTV, TTMV, TTMDV, and Coxsackievirus A2. Most probably, this severe patient (clinical specimen 12) was infected by multiple viral pathogens.
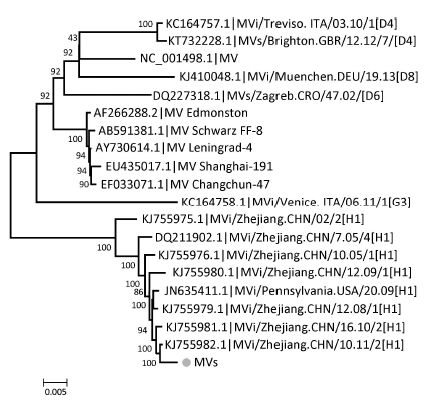
Furthermore, it is worth mentioning that clinical specimen 9 was found to be positive for measles virus with 77.50% coverage of the reference complete genome (Figure 3B). The interesting part is that the sample was diagnosed as mild HFMD in the clinic, but HFMD-associated viral pathogens tested negative by real-time RT-PCR (Table 1). This sample was subsequently verified by real-time RT-PCR using measles virus-specific primer pairs (MF, 5'-TCAGT AGAGCGGTTGGACCC-3'; MR, 5'-GGCCCGGTTTCTCTG TAGCT-3') [42] with a CT value of 23.9. The complete genome sequence of the measles virus has since been assembled combining NGS data and Sanger sequencing data in our laboratory (data not shown). Meanwhile, the complete genome sequence of the measles virus was used for phylogenetic analysis conducted by the neighbour-joining method using MEGA 5.0 software [43] with a bootstrap value of 1, 000. As shown in Figure 4, this measles virus was clustered to a branch of the phylogenetic tree together with measles virus strain genotype H1.
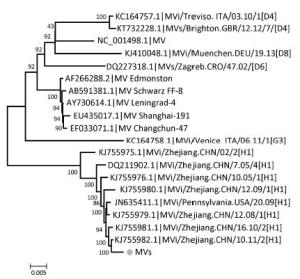
Figure 4. Phylogenetic analysis of measles virus based on complete genomes. One measles virus (grey circle) detected from clinical specimen 10 and 20 representative measles virus strains were analyzed using the Neighbor-Joining method with 1, 000 bootstrap replicates in the MEGA 5.0 program; numbers at the nodes represent bootstrap support. MV, measles virus; MVi, measles virus sequence from isolates; MVs, measles virus sequence from clinical specimens.
Molecular Detection of Samples Using Real-time RT-PCR
Comparison of Barcoded Primer-based NGS for Viral Detection
Hexanucleotide Primer-based NGS for Identification of HFMD-associated Viral Pathogens in Clinical Specimens
Identification of HFMD-unrelated Viral Pathogens in Clinical Specimens
-
NGS is becoming a robust tool to identify and characterize viral pathogens in samples. However,several challenges impede the widespread adoption of NGS in clinical applications. In this study, we described an improved NGS approach based on the barcoded oligonucleotide primers and demonstrated the viability of the method for rapid and direct identification of multiple viral pathogens in HMFD-associated clinical samples.
There are some advantages of the improved NGS approach. Firstly, the procedure of library construction was simplified without the shearing step. In brief, as shown in common whole genome sequencing (WGS) [30, 44], Virus Discovery based on cDNA Amplified Fragment Length Polymorphism (VIDISCA)-combined NGS [45] or virocap-combined NGS [16, 46], these NGS protocols for library construction are usually tedious, laborious, and time consuming because every sample must be processed individually in the entire workflow. In contrast, in the improved NGS protocol, every 10 enriched products from 10 independent samples are pooled together in one tube and the mixture is regarded as a single sample for downstream processing, including end-repair, adapter ligation, size selection, library quantity, and sequencing (Figure 1). Importantly, the improved NGS data obtained from a fragmentation-free library may conserve the natural content of the original samples and reveal factual viral pathogen information. Some artefactual recombination may be introduced into the sheared library regardless of the enzyme digestion or sonication system used for library fragmentation [32]. Moreover, artifactual recombination in NGS generates sequence chimeras, which can affect NGS data analysis by computational pipelines to estimate nucleotide polymorphism frequency or species population diversity and generate incorrect de novo genome assembly [47-48]. Secondly, the improved NGS approach is greatly time saving, labor saving, and cost effective. Compared with the conventional protocol (samples were processed individually throughout the NGS procedure, construction of the fragmented sequencing library was always preferred, tremendous NGS data were analyzed depending on professional bioinformaticians using expensive hardware configurations or commercial software), and the improved method spent 1/10 of the time and cost 1/10 of the capital to process a sequencing library originating from 10 independent samples. Furthermore, NGS data analysis by using in-house VIP saves further time and cost in that VIP needs minimum hardware configurations and can run on a common desktop personal computer with a 3.4 GHz Intel Core i7-4770 and 16 GB of RAM (running Ubuntu 14.04 LTS), and it can be easily manipulated by someone without a background in bioinformatics. Notably, in a practical application, VIP was utilized to analyze NGS data of one unknown sample from a severe Dengue outbreak in Guangzhou, demonstrating that VIP has contributed to timely virus diagnosis (-10 min), allowing detailed virus information to be obtained in acutely ill patients, and has the potential to perform in unbiased NGS-based clinical studies that demand a short turnaround time [39]. In general, the improved approach took approximately 20 h for viral pathogens analysis of 10 samples, comprising 1.5 h for RNA extraction; 4.5 h for viral enrichment; 3 h for library construction; 8.75 h for emulsion PCR, ES, and sequencing; and 2.25 h for data analysis. Therefore, the improved NGS approach combined with in-house VIP is suitable for rapid identification of viral pathogens in clinical application, especially in emerging viral outbreaks when many samples await detection.
Initially, as model experiments, the improved NGS approach was adopted to compare the viral enrichment abilities of random hexamers and non-ribosomal hexanucleotides through reverse transcription followed by PCR and sequencing using different samples containing known viruses with different viral loads. The data from one EBV sample, Flu B samples (three different viral loads), and one CVA16 sample showed that the hexanucleotides are comparable to the traditional random hexamers in the sequence performance of EBV and Flu B with high viral loads (CT values 21 or 25), because both reads of EBV and Flu B covered the nearly complete sequence of EBV membrane protein and Flu B segments 1-8 (Table 2). On the contrary, random hexamers are slightly inferior to hexanucleotides in the coverage of the complete CVA16 genome and Flu B segments 1-8 in samples with low viral loads (CT values 30 and 29 for CVA16 and FluB, respectively). These results indicate that sequence-independent amplification by barcoded oligonucleotide primers combined with the improved NGS approach can work efficiently to directly detect viral pathogens in cell culture materials and clinical specimens. Moreover, for RNA viral samples, the ratios of virus-related reads originating from hexanucleotide-enriched samples are higher than those originating from random hexamer-enriched samples (Table 2), which is consistent with the results of Endoh et al. using the RDA method [31], suggesting that NGS-based sequence-independent amplification with hexanucleotides can further reduce noise compared with that of random hexamers. Remarkably, our improved NGS-based sequence-independent amplification with hexanucleotides is more suitable for detection of viral pathogens, not only in cell culture materials but also in clinical specimens with low viral loads, than the previously reported RDA-based sequence-independent amplification with hexanucleotides, which might be restricted to infected cells that contain many copies of a virus. This is the first report to simultaneously compare the viral enrichment abilities of random hexamers and hexanucleotides on an improved NGS platform.
Furthermore, we used sequence-independent amplification with hexanucleotides followed by the improved NGS approach and real-time RT-PCR to simultaneously detect 20 clinical specimens from pediatric patients clinically diagnosed with mild or severe HFMD in the present study. The total positivity rate of HFMD-associated viral pathogens identified by NGS was 13/20, whereas 16/20 samples were detected by real-time RT-PCR, including 3 samples missed by NGS. Notablely, the viral pathogens detected from these HFMD clinical specimens were detailed typed as Coxsackievirus A6, Coxsackievirus A2, Coxsackievirus B3, and EV 71 via the improved NGS method, whereas all these samples (except EV 71) were classified as HEV without typing information by real-time RT-PCR. Thus, the improved NGS method may play an important role in accurate identification, because it can provide detailed subtype or serotype information on viral pathogens. Meanwhile, these results showed a correlation between the amount of viral RNA from real-time RT-PCR and sequencing reads from the NGS approach. This was particularly strong for Coxsackievirus A6-infected samples where the decreased CT values correlated with increased sequencing reads. This suggests that sequencing reads might be a suitable indicator of viral loads in tested samples. Besides, the prominent advantage of NGS approaches including the improved one mentioned here is that they can provide comprehensive information on samples. The NGS data from twenty clinical specimens showed that these samples contain bacteria, bacteriophage, host information (data not shown), and other viral pathogens unrecognized by specific real-time RT-PCR apart from HFMD-associated viruses. For instance, viral co-infection was detected by NGS, and though few viral sequences were captured, there were enough for identification, such as TTV, TTMV, TTMDV, bluetongue virus, and human herpesvirus in HFMD-positive samples. Interestingly, TTV, TTMV, and TTMDV were also widely found in ot her diseases, especially viral hepatitis [49], asthma [50], and severe lower respiratory tract infections [51]. Here, TTV or TTMV single or dual infection were found in two samples of mild HFMD and TTV, TTMV, or TTMDV single or dual or triple infection were found in five samples of severe HFMD. Up to now, there is little evidence of an association between the infection of TTV, TTMV, or TTMDV and HFMD, we speculate that TTV, TTMV, or TTMDV might be responsible for causing severe HFMD or exacerbating disease progression. Additionally, clinical specimen 9, which was initially diagnosed as mild HFMD in clinic and later tested HEV negative via real-time RT-PCR, was found as measles virus positive using NGS, even though the reads assigned to measles virus appeared low (598) and covered only 77.50% of the complete reference genome. We suspected that the case was probably misdiagnosed by the clinician. One of the clinical characteristics in measles patients is a maculopapular rash [52], which is similar to the vesicular rash caused by HFMD. Thus, it is difficult to differentiate measles and HFMD at early onset. Moreover, this measles case was successfully validated via real-time RT-PCR and was further confirmed by rechecking the medical records (clinically diagnosed as measles a month later). Therefore, NGS analysis in this study demonstrated that misdiagnoses of HFMD do occur in the clinic.
Nevertheless, similar to other previous studies [53-55], the shortcoming of the improved NGS approach is its low sensitivity with respect to agent-specific real-time RT-PCR assays. Here, the improved NGS approach is less sensitive than real-time RT-PCR assays, because a sample with a CT of 33 can be detected whereas one with a CT of 34 is missed, in agreement with Thorburn et al., who state that the NGS method may have a cutoff in the region of CT 32. Furthermore, the average depth of sequencing of clinical samples with low viral loads ranged from 0.02 × to 45.48 ×, far less than that of samples with high viral contents, which ranged from 48.88 × to 126.79 ×. As Lecuit et al. [56] showed, the sensitivity of a deep-sequencing approach for detecting viruses is dependent on sequencing depth. Probably, the relative low sensitivity of the improved NGS approach is attributable to the low sequencing depth of the ultra-low viral contents of clinical specimens. To address this problem, adopting higher throughput methods will probably increase the viral detection rate, such as using a 318v2 chip to replace the 316v2 chip or selecting a greater capacity sequencing platform, such as the Illumina MiSeq or Ion S5.
In conclusion, the improved NGS approach is a simplified, rapid and relatively cost-effective method. It could be used for simultaneous identification of multiple viral pathogens in samples including cell culture material and clinical specimens and provide detailed typing information on specific viral pathogens, as well as differentiation of misdiagnosis events appearing in the clinic.
-
XJM, KN, and CHW conceived of the idea and designed the study; CHW conducted all the experiments; SXQ collected and provided all the HFMD clinical specimens from pediatric patients. SFZ, XNL, and HYZ provided all the primers and probes for HFMD-associated viral pathogen detection and preserved all the HFMD clinical specimens. CHW, XJM, KN, YZ, and JW analyzed the NGS data. CHW wrote the manuscript and prepared all the tables and figures; XJM reviewed the manuscript; all authors approved the manuscript.
-
The authors declare that they have no competing financial interests.
Beijing Municipal Science & Technology Commission Project D151100002115003
This work was supported by the China Mega-Project for Infectious Disease 2016ZX10004-101, 2016ZX10004-215
and Guangzhou Municipal Science & Technology Commission Project 2015B2150820

 Quick Links
Quick Links
 DownLoad:
DownLoad: