Objective The aim of this study was to describe changes in waist circumference (WC) and prevalence of abdominal obesity over a period of 10 years among Chinese adults in different socio-economic status (SES).Methods Data derived from the China Nutrition and Health Surveillance during 2002 and 2010-2012. We calculated the mean WC and the prevalence of abdominal obesity by gender, place of residence, SES indicators (education, income, and marital status), and body mass index (BMI) categoriesand used pooled t-tests to assess the differences between the two time periods.Results 26.0% of men and 25.3% of women had abdominal obesity in 2010-2012. The age-adjusted mean WC increased by 2.7 cm among men and 2.1 cm among women; the age-adjusted prevalence of abdominal obesity increased by 7.7% among men and 5.3% among women. The rising trends were observed in all subgroups except for a negative growth in high-income women. People living in rural areas with low education and income and with a BMI of 18.5 to 23.9 kg/m2 had a greater absolute and relative increase in WC. People living in rural areas with low income had a greater relative increase in abdominal obesity.Conclusion The mean WC and prevalence of abdominal obesity among Chinese adults have increased during the past 10 years. Gender differences were noted using various SES indicators.
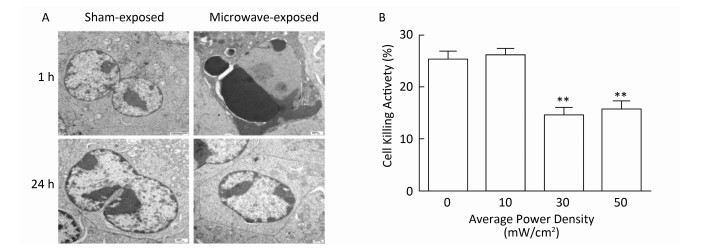
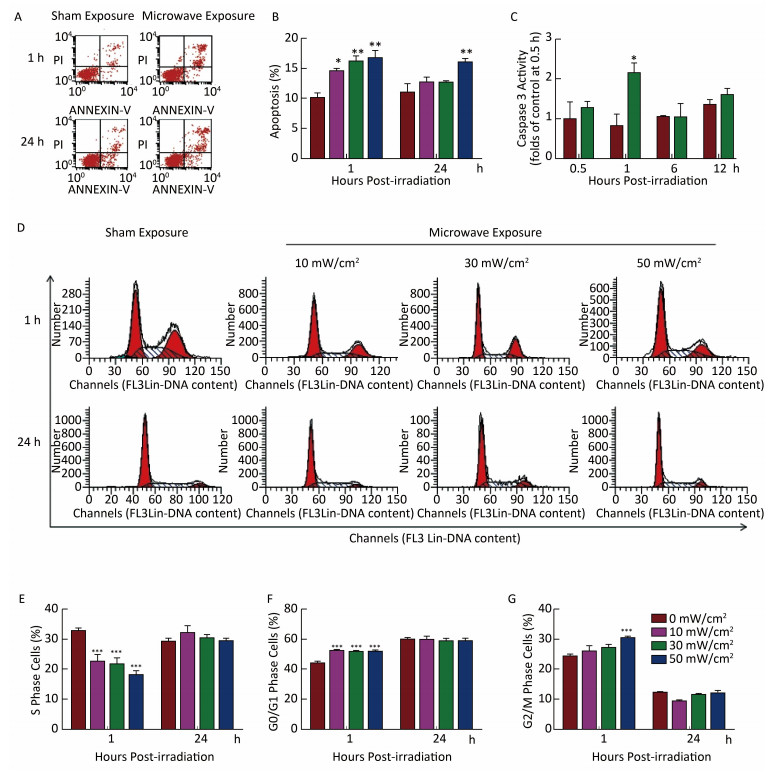
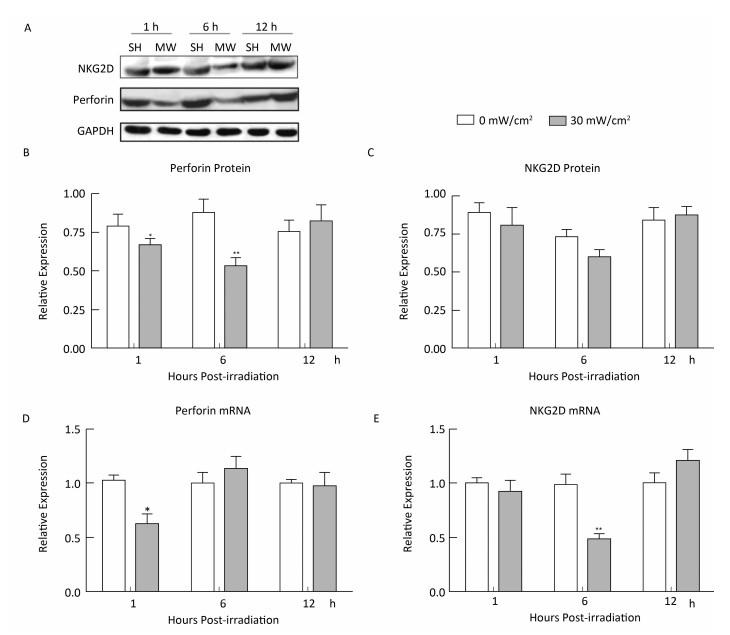
Objective To investigate microwave-induced morphological and functional injury of natural killer (NK) cells and uncover their mechanisms.Methods NK-92 cells were exposed to 10, 30, and 50 mW/cm2 microwaves for 5 min. Ultrastructural changes, cellular apoptosis and cell cycle regulation were detected at 1 h and 24 h after exposure. Cytotoxic activity was assayed at 1 h after exposure, while perforin and NKG2D expression were detected at 1 h, 6 h, and 12 h after exposure. To clarify the mechanisms, phosphorylated ERK (p-ERK) was detected at 1 h after exposure. Moreover, microwave-induced cellular apoptosis and cell cycle regulation were analyzed after blockade of ERK signaling by using U0126.Results Microwave-induced morphological and ultrastructural injury, dose-dependent apoptosis (P < 0.001) and cell cycle arrest (P < 0.001) were detected at 1 h after microwave exposure. Moreover, significant apoptosis was still detected at 24 h after 50 mW/cm2 microwave exposure (P < 0.01). In the 30 mW/cm2 microwave exposure model, microwaves impaired the cytotoxic activity of NK-92 cells at 1 h and down regulated perforin protein both at 1 h and 6 h after exposure (P < 0.05). Furthermore, p-ERK was down regulated at 1 h after exposure (P < 0.05), while ERK blockade significantly promoted microwave-induced apoptosis (P < 0.05) and downregulation of perforin (P < 0.01).Conclusion Microwave dose-dependently induced morphological and functional injury in NK-92 cells, possibly through ERK-mediated regulation of apoptosis and perforin expression.
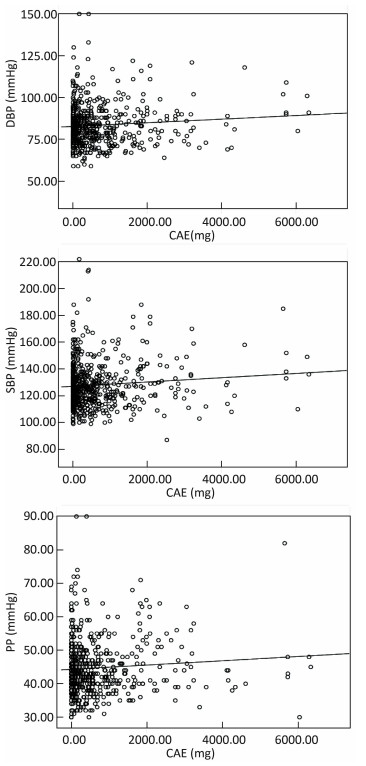
Objective The effects of arsenic exposure from drinking water, arsenic metabolism, and arsenic methylation on blood pressure (BP) were observed in this study.Methods The BP and arsenic species of 560 participants were determined. Logistic regression analysis was applied to estimate the odds ratios of BP associated with arsenic metabolites and arsenic methylation capability.Results BP was positively associated with cumulative arsenic exposure (CAE). Subjects with abnormal diastolic blood pressure (DBP), systolic blood pressure (SBP), and pulse pressure (PP) usually had higher urinary iAs (inorganic arsenic), MMA (monomethylated arsenic), DMA (dimethylated arsenic), and TAs (total arsenic) than subjects with normal DBP, SBP, and PP. The iAs%, MMA%, and DMA% differed slightly between subjects with abnormal BP and those with normal BP. The PMI and SMI were slightly higher in subjects with abnormal PP than in those with normal PP.Conclusion Our findings suggest that higher CAE may elevate BP. Males may have a higher risk of abnormal DBP, whereas females have a higher risk of abnormal SBP and PP. Higher urinary iAs may increase the risk of abnormal BP. Lower PMI may elevate the BP. However, higher SMI may increase the DBP and SBP, and lower SMI may elevate the PP.
Objective Mutations in 23S rRNA gene are known to be associated with macrolide resistance in Mycoplasma pneumoniae (M. pneumoniae). However, these mutations alone do not fully explain the high resistance rates in Asia. The aim of this study was to investigate other possible mutations involved in macrolide resistance in M. pneumoniae.Methods The whole genomes of 10 clinical isolates of M. pneumoniae with macrolide resistance were sequenced by Illumina HiSeq2000 platform. The role of the macrolide-specific efflux transporter was assessed by efflux-pump inhibition assays with reserpine and carbonyl cyanide m-chlorophenyl-hydrazone (CCCP).Results A total of 56 single nucleotide polymorphisms (SNPs) were identified in 10 clinical isolates in comparison to the reference strains M129 and FH. Strikingly, 4 of 30 SNPs causing non-synonymous mutations were clustered in macrolide-specific efflux system gene macB encoding macrolide-specific efflux pump protein of the ATP-binding cassette transporter family. In assays of the minimal inhibitory concentrations (MIC) of macrolide antibiotics in the presence of the efflux pump inhibitors caused a significant decrease of MICs, even under detectable levels in some strains.Conclusion Our study suggests that macrolide efflux pump may contribute to macrolide resistance in M. pneumoniae in addition to the common point mutations in 23S rRNA gene.
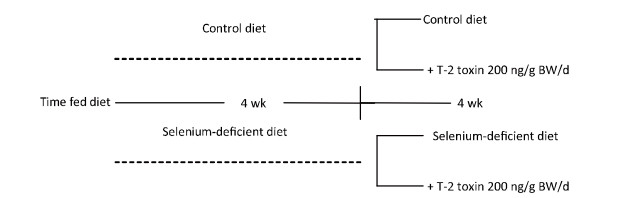
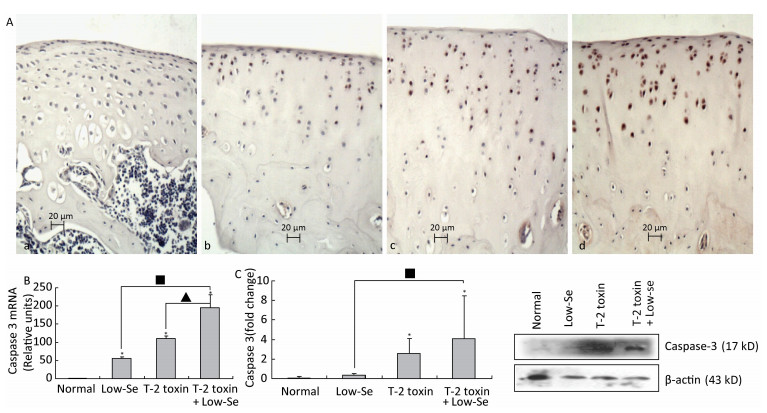
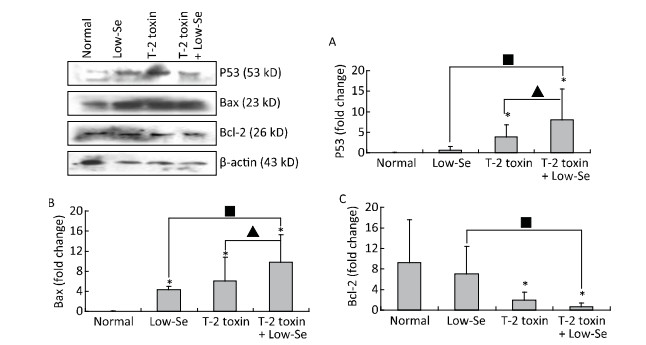
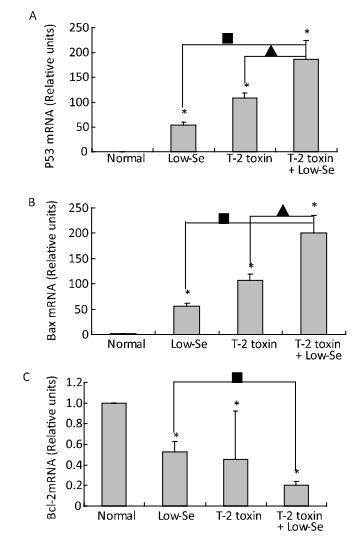
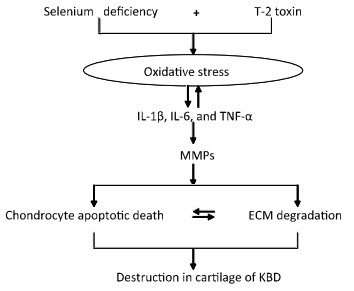
Objective To investigate chondrocyte apoptosis and the expression of biochemical markers associated with apoptosis in Kashin-Beck disease (KBD) and in an established T-2 toxin-and selenium (Se) deficiency-induced rat model.Methods Cartilages were collected from the hand phalanges of five patients with KBD and five healthy children. Sprague-Dawley rats were administered a selenium-deficient diet for 4 weeks prior to T-2 toxin exposure. The apoptotic chondrocytes were observed by terminal deoxynucleotidyl transferase dUTP nick end labeling staining. Caspase-3, p53, Bcl-2, and Bax proteins in the cartilages were visualized by immunohistochemistry, their protein levels were determined by Western blotting, and mRNA levels were determined by real-time reverse transcription polymerase chain reaction.Results Increased chondrocyte apoptosis was observed in the cartilages of children with KBD. Increased apoptotic and caspase-3-stained cells were observed in the cartilages of rats fed with normal and Se-deficient diets plus T-2 toxin exposure compared to those in rats fed with normal and Se-deficient diets. Caspase-3, p53, and Bax proteins and mRNA levels were higher, whereas Bcl-2 levels were lower in rats fed with normal or Se-deficiency diets supplemented with T-2 toxin than the corresponding levels in rats fed with normal diet.Conclusion T-2 toxin under a selenium-deficient nutritional status induces chondrocyte death, which emphasizes the role of chondrocyte apoptosis in cartilage damage and progression of KBD.
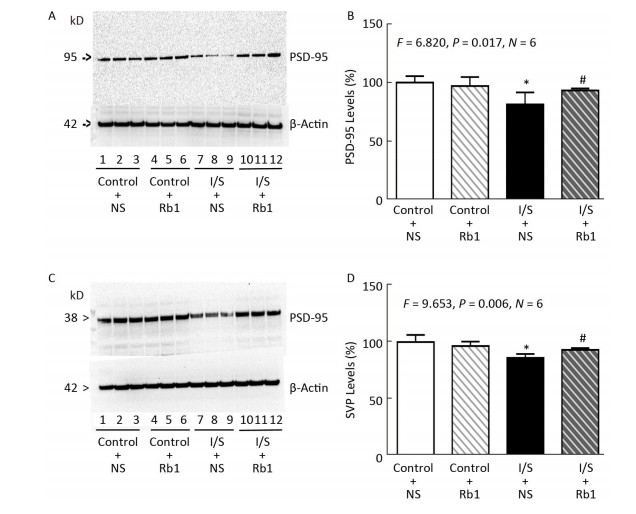
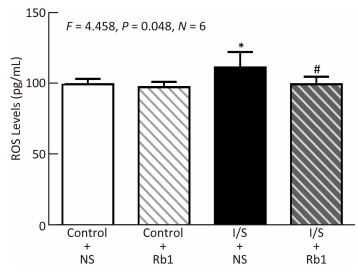
Objective Anesthetic isoflurane plus surgery has been reported to induce cognitive impairment. The underlying mechanism and targeted intervention remain largely to be determined. Ginsenoside Rb1 was reported to be neuroprotective. We therefore set out to determine whether ginsenoside Rb1 can attenuate isoflurane/surgery-induced cognitive dysfunction via inhibiting neuroinflammation and oxidative stress.Methods Five-months-old C57BL/6J female mice were treated with 1.4% isoflurane plus abdominal surgery for two hours. Sixty mg/kg ginsenoside Rb1 were given intraperitoneally from 7 days before surgery. Cognition of the mice were assessed by Barnes Maze. Levels of postsynaptic density-95 and synaptophysin in mice hippocampus were measured by Western blot. Levels of reactive oxygen species, tumor necrosis factor-α and interleukin-6 in mice hippocampus were measured by ELISA.Results Here we show for the first time that the ginsenoside Rb1 treatment attenuated the isoflurane/surgery-induced cognitive impairment. Moreover, ginsenoside Rb1 attenuated the isoflurane/surgery-induced synapse dysfunction. Finally, ginsenoside Rb1 mitigated the isoflurane/surgery-induced elevation levels of reactive oxygen species, tumor necrosis factor-α and interleukin-6 in the mice hippocampus.Conclusion These results suggest that ginsenoside Rb1 may attenuate the isoflurane/surgery-induced cognitive impairment by inhibiting neuroinflammation and oxidative stress pending future studies.
Post-exposure prophylaxis (PEP) has proved to be the most important measure for rabies prevention and control. There is little information regarding adverse reactions to the Essen and 2-1-1 regimens in preschool children (aged 0-6). We reexamined the outcomes of 1, 109 preschool children who were vaccinated using SPEEDA under the Essen regimen between January 2011 and December 2012 and 1, 267 preschool children under the 2-1-1 regimen between January 2013 and December 2014. We find that, in preschool children, the febrile reaction after the first 2-dose injection in the 2-1-1 regimen was significantly higher than that induced by the first 1-dose in the Essen procedure. Thus, we recommend that the Essen regimen should still be used for rabies PEP in preschool children.
We performed molecular identification of clinical isolates of Mycobacterium fortuitum (M. fortuitum) and conducted drug susceptibility testing to analyze the in vitro susceptibility of clinical M. fortuitum isolates and potential molecular mechanism conferring resistance to fluoroquinolone and macrolide drugs. The results showed that moxifloxacin had the highest in vitro activity against M. fortuitum, and most M. fortuitum isolates were resistant to clarithromycin and linezolid in China. The loss of genetic mutation in clarithromycin-and amikacin-resistant isolates indicates that some other intrinsic mechanism conferring clarithromycin and amikacin resistance plays an essential role in M. fortuitum infection.
Kashin-Beck disease (KBD) is an endemic degenerative osteoarthropathy of uncertain etiology. The aim of our study was to identify changes in C-telopeptide of type Ⅱ collagen (CTX-Ⅱ), pyridinoline (PYD), and deoxypyridinoline (DPD) among KBD patients. 54 KBD patients and 78 healthy controls were included this study. Urinary samples were collected and measured by ELISA. The median quantities of PYD, CTX-Ⅱ, and DPD of KBD patients were 1107.73 ng/μmol.cre, 695.11 ng/μmol.cre, and 1342.34 pml/μmol.cre, while the median quantities of healthy controls were 805.59 ng/μmol.cre, 546.47 ng/μmol.cre, and 718.15 pml/μmol.cre, respectively. The differences between KBD patients and healthy controls were statistically significant (Z = 4.405, 3.653, and 3.724; P < 0.001). The higher levels of PYD, CTX-Ⅱ, and DPD detected in KBD patients indicate that they could be used as biomarkers of KBD.
China has a double burden of diabetes mellitus and tuberculosis, and many studies have been carried out on the mutual impact of these two diseases. This paper systematically reviewed studies conducted in China covering the mutual impact of epidemics of diabetes and tuberculosis, the impact of diabetes on multi-drug resistant tuberculosis and on the tuberculosis clinical manifestation and treatment outcome, the yields of bi-directional screening, and economic evaluation for tuberculosis screening among diabetes patients.

























 Quick Links
Quick Links