
-
Interferon-induced transmembrane protein 3 (IFITM3), first discovered in 1984, is a low molecular weight (15 kD) transmembrane protein belonging to the IFITM family[1]. IFITM3 is known to be involved in various biological processes, such as signal-mediated immune cell regulation, homing, and germ cell maturation[2]. Since it was identified as a host restriction factor against Influenza A virus, West Nile virus, and Dengue virus in 2009, the exact antiviral effects and underlying mechanisms of action of IFITM3 have become a research hotspot[3]. Defective IFITM3 function increases the susceptibility of mice and humans to infection by influenza virus. Ifitm3-/- mice develop serious influenza A virus infections, with symptoms including pathological damage and mortality[4]. In humans, the genetic polymorphism of IFITM3 is closely related to the severity of influenza symptoms. For example, the IFITM3 SNPrs12252-C allele encodes a protein with a 21-amino acid deletion at its N-terminus that lacks the ability to inhibit viral replication; SNPrs34481144-A leads to a mutation in the human IFITM3 promoter that lowers mRNA expression by decreasing IRF3 binding and increasing CTCF binding to the promoter. Both are identified as the risk alleles for severe influenza virus infections[4-6]. In a recent study, SNPs in the promoter region of the IFITM3 gene displayed an association with H1N1 2009 pandemic virus infection[7]. In contrast, several other studies suggest that there is no relationship between influenza severity and SNP rs12252[8,9]. Despite the contradicting reports, IFITM3 is one of the most important known influenza virus restriction factors.
Exact antiviral molecular mechanisms have not been completely elucidated. Recent studies have shown that IFITM3 can block viral entry by modulating several pathways[10]. For example, it restricts the endosomal pathway in the influenza A viral replication cycle after viral uptake, but prior to viral RNA release[11]. In particular, IFITM3 inhibits viral RNA release into the cytoplasm by decreasing the expression of clathrin protein, disrupting intracellular cholesterol homeostasis, and disturbing the acidification function of vacuolar-ATPase[12,13]. A very recent study has shown that IFITM3 directly shuttles the incoming virus particles to lysosomes[14].
The IFITM3 protein expression also affects the body’s adaptive immune response against Influenza A virus by enhancing the survival of lung tissue-resident memory CD8+ T cells during acute influenza infection [15]. Respiratory dendritic cells (DCs) also express IFITM3 to avoid direct viral infection and safeguard virus-specific CD8+ T cell priming[16]. Another recent study reported that human megakaryocytes acquire intrinsic anti-viral immunity through regulated induction of IFITM3[17]. However, as an interferon-induced protein, whether IFITM3 expression is regulated differently in distinct cell types during acute influenza infection remains unclear.
The aim of the present study was to determine the contribution of IFITM3 proteins to immune control of acute influenza infection in vivo. Our results show that IFITM3 alone contributes significantly to the control of influenza in mice and provide further insights into its expression and regulation.
-
Influenza A/PR/8/34 (H1N1) (PR8) was propagated in chicken eggs, and filtered using Stericup 0.22-μm filters (Millipore). Titers were determined by standard plaque assays on Madin-Darby canine kidney cells.
-
Ifitm3-/- mice (background strain C57BL/6) were generated by Beijing Biocytogen Co., Ltd using CRISPR/Cas9 technology. These animals were housed under specific pathogen-free conditions prior to infection. All mice used for experimental research were between 7 and 8 weeks of age. Mice were anesthetized by intraperitoneal injection with 10% sodium Pentobarbital in normal saline (120 μL per 18 g body weight). Anesthetized mice were inoculated intranasally with influenza A virus. All procedures were performed with the approval of the Animal Care and Use Committee of the Chinese center for disease control and prevention.
-
Primary antibodies from commercial sources used in this study are as follows: rabbit anti-IFITM3 (Proteintech); rabbit anti-NF-κB p65, rabbit anti-Phosphor-NF-κB p65, rabbit anti-caspase-3, rabbit anti-cleaved caspase-3, rabbit anti-caspase-7, rabbit anti-beclin-1, and rabbit anti-CCL2 (Cell Signaling Technology); rabbit anti-influenza A virus NP (nucleoprotein) and rabbit anti-Ly6g (GeneTex); mouse anti-SQSTM1/p62 (Abcam); rabbit anti-CD69, and mouse anti-β-actin (Santa Cruz Biotechnology); and rabbit anti-IL-6 (Beyotime Biotechnology); All secondary antibodies were purchased from Santa Cruz Biotechnology.
-
For Western blots, mouse lung tissues were homogenized in RIPA buffer with 1× protease inhibitor (Sigma, P8340). Tissue lysates were diluted in 1x SDS sample buffer and sonicated for 10 s after incubation at 100 °C for 10 min. Total lysate proteins were resolved in 4%–20% Tris-glycine gels (Invitrogen) and blotted to nitrocellulose membranes. Western blots were developed using an ECL Prime kit (GE Health Care). For quantification of Western blot results, each mouse’s lung tissue was analyzed in triplicate. Lung tissues from multiple mice (four to seven mice per group) were analyzed via western blotting. Multiple samples on the same membranes were probed with antibodies to proteins of interest and to β-Actin, which served as a loading control. Signals of immunoreactive bands were quantified with densitometry analysis using the software Image-ProPlus 5.1[18]. Ratios of immunolabeled proteins to β-actin were used to compare relative levels of detected proteins in the same lung tissues from mice on fixed days post-infection.
-
Processed lung lobes were embedded in paraffin and cut into sections of 3–4 μm thickness. One section from each lung was stained with hematoxylin and eosin to assess pathological changes.
-
Pulmonary low-density mononuclear cells from male C57BL/6 mice were separated by lung tissue digestion and density gradient centrifugation. Single-cell suspensions were generated by passing lung cells through 5-mL polystyrene round-bottom tubes with cell-strainer caps. Cells were characterized by flow cytometry as follows: T-lymphocytes—CD4+ or CD8+, activated T-lymphocytes—CD3−CD4+CD69+ or CD3−CD8+CD69+, NKs—NKp46+CD3−CD4−CD8−, activated NKs—NKp46+CD3−CD4−CD8−CD69+, DCs—CD11c+CD11bloLy6gloMHC class II high. We assessed cell viability using cell viability solution (555816, BioLegend). All antibodies were from BD Bioscience, eBioscience, or BioLegend.
For CD107a and IFN-γ detection in NK cells, cells were permeabilized with cytofix/cytoperm buffer (BD Biosciences, San Diego, CA) for 20 min and washed in washing solution. Isotype control Ab was used for each staining combination.
Samples were run on a FACSAria II (BD Bioscience) and visualized using FlowJo V10. Data were analyzed statistically and graphed using Prism 5.0 (GraphPad Software).
-
Pulmonary low-density mononuclear cells from wild type or Ifitm3-/- C57BL/6 mice were obtained on 0, 3, and 5 d after PR8 infection for Protein Digestion and Tandem Mass Tag (TMT) Labeling[19]. Proteins were digested using the filter-aided sample preparation (FASP) method as previously described with slight modifications. Peptides were analyzed using an LTQ Orbitrap Elite mass spectrometer (Thermo Scientific) coupled online to an EasynLC 1000 in data-dependent mode. Peptide and protein identification and quantification were performed with MaxQuant software (version 1.5.3.28)[20]. The UniProt proteome sequences for Mus musculus (including canonical sequences and isoforms) were used for database searching.
-
For KEGG pathway and GO analysis, the Database for Annotation Visualization and Integrated Discovery (DAVID) Functional Annotation tool (http://david.abcc.ncifcrf.gov/home.jsp, DAVID v6.8) was used to identify biological pathways and processes that were significantly enriched in the gene sets. Pathways were identified as significant using default settings[21].
-
GSEA analysis was performed based on the Molecular Signatures Database (MSigDB) v6.2 using detailed steps provided by specifications on the website and/or in the published paper[22].
-
Data were analyzed using SPSS (Version 18.0; SPSS Inc., Chicago, USA). Student’s t-tests were used for comparison of the two groups of mice. *P < 0.05, **P < 0.01, and ***P < 0.001 were considered significant.
-
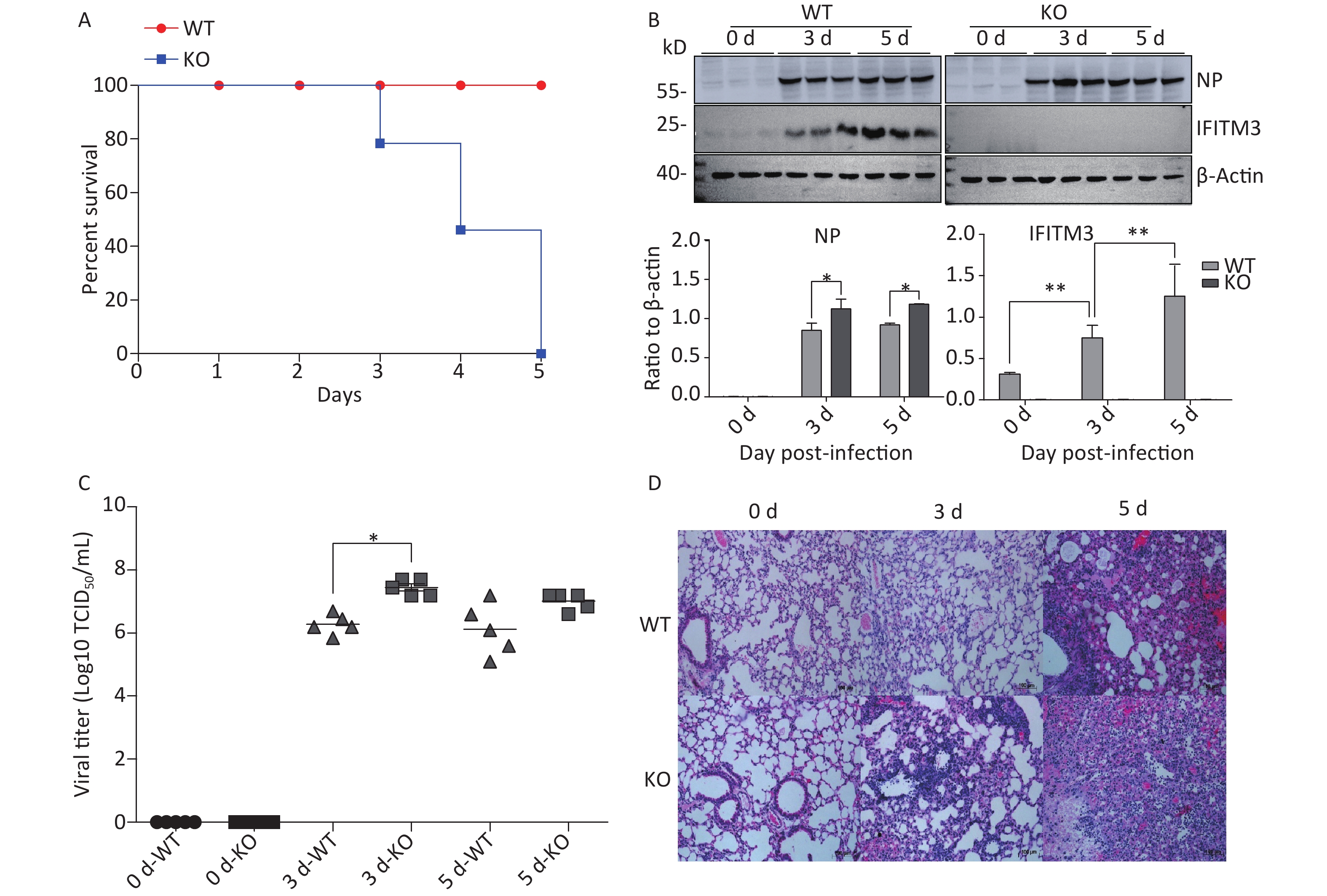
To determine the relevance of IFITM3 in influenza, we challenged Ifitm3-/- and wild type male mice with 50 μL intranasal doses of 104.5TCID50 influenza A/PR/8/34 strain (H1N1) (PR8) (104.5TCID50/mL). The Ifitm3-/- mice exhibited significantly accelerated disease progression and increased mortality compared to wild-type controls (Figure 1A). Western blot analyses of mouse lung tissues were performed at 0, 3, and 5 d post-infection with antibodies against influenza A virus NP (nucleoprotein) and IFITM3. Given that IFITM3 is an interferon-stimulated gene, its protein expression was induced during viral infection in wild-type mice (Figure 1B). Pulmonary viral titers were measured at 0, 3, and 5 d post-infection, and Ifitm3-/- mice challenged with 104.5TCID50 of PR8 had higher virus titers than wt mice (Figure 1C). Pulmonary morphology was assessed by hematoxylin-eosin staining (HE). Our result showed that inflammatory cell infiltration was higher and the prevalence of serious pathological changes was greater in Ifitm3-/- mice (Figure 1D). Together, these results indicated that the IFITM3 limited the severity of acute influenza infection in mice.
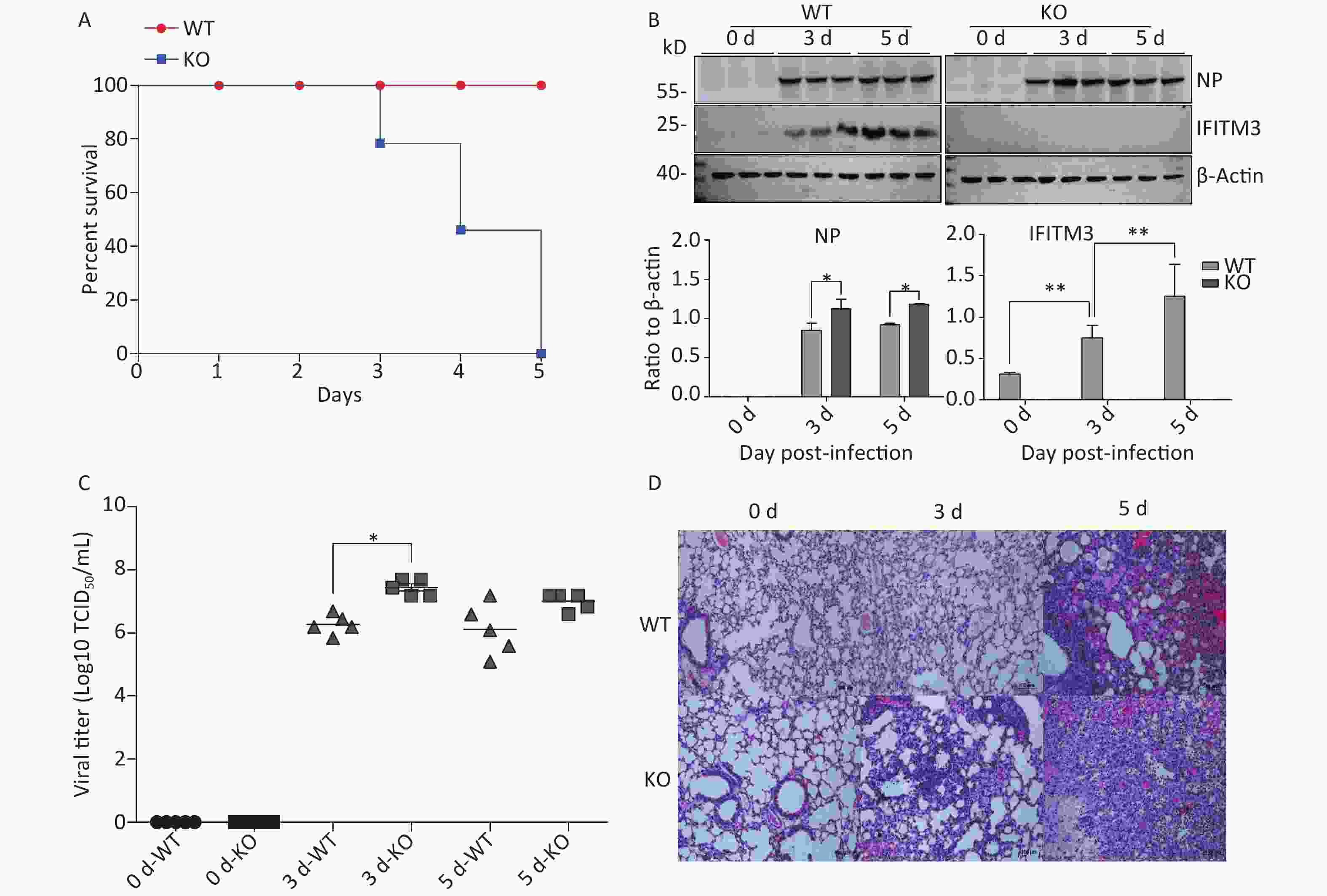
Figure 1. IFITM3 limits the severity of acute influenza in wild type mice. (A) Mortality due to influenza A/PR/8/34 (H1N1) (PR8) infection in mice. Eight-week-old female C57BL/6 mice (n = 20/group) were inoculated intranasally with 104.5TCID50 of PR8. Survival was monitored for 5 d. (B) Western blot analysis of lung tissues from mice at 0, 3, and 5 d after infection with antibodies against Influenza A virus NP (nucleoprotein) and IFITM3. β-Actin was used as the internal control. (C) Pulmonary viral titers were measured at 0, 3, and 5 d post-infection in animals challenged with 104.5TCID50 of PR8. (D) Pulmonary morphology was assessed by hematoxylin-eosin staining. Scale bars: 100 μm. WT: wild-type mice; KO: knock out, Ifitm3-/- mice. *P < 0.05, **P < 0.01.
-
Next, we aimed to explore why Ifitm3-/- mice, but not wild-type mice, died in 3–5 d. Many recent studies have shown that highly pathogenic respiratory viruses cause cytokine storms, leading to immune imbalances and lung damage, resulting in death. Therefore, we examined immunity-related inflammatory, apoptosis-related, and autophagy-related molecules in the lung tissues of both types of mice at different days post-PR8 virus infection. As expected, Ifitm3-/- mice exhibited severe pathological damages in H & E-stained tissues. Similarly, we assessed protein expression levels of total and phosphorylated NF-κB p65, IL-6, and caspase 3, and cleaved caspase 3, p62, and Beclin-1 in the lungs of both types of mice at 0, 3, and 5 days after infection. The expression levels of total NF-κB p65, caspase 3, p62, and Beclin-1 were not different, but phosphorylation of NF-κB p65, IL-6, and the levels of cleaved caspase 3 increased during influenza infection, and higher expression was observed in Ifitm3-/- mice than in wild-type mice at all time points (Figure 2). These results indicated that Ifitm3-/- mice experienced severe inflammatory and apoptotic responses. Upon comparison with many recent studies, we speculate that this could be an important contributor to mortality in infected mice.
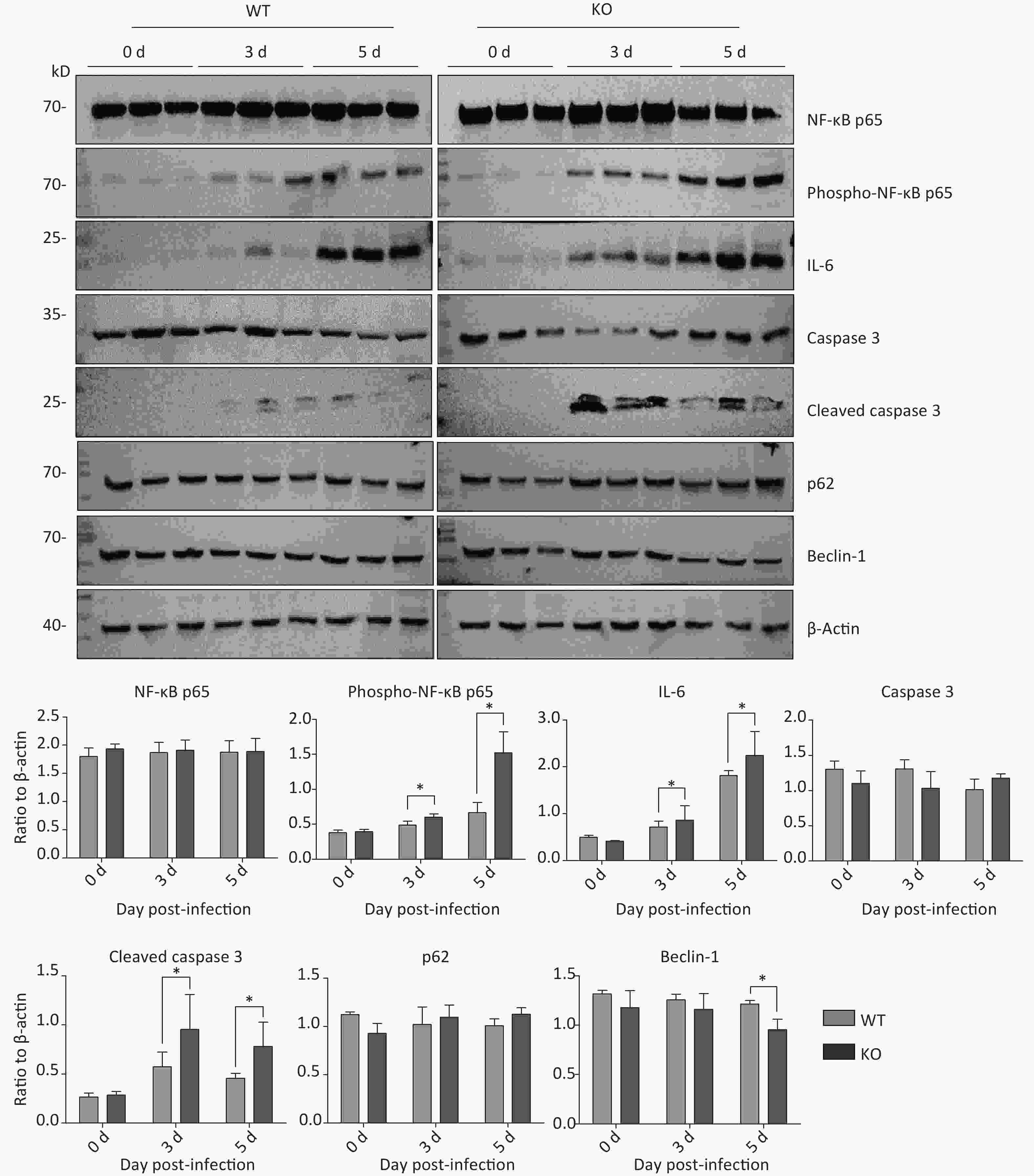
Figure 2. Time-dependent expression of inflammation-, apoptosis-, and autophagy-related proteins in mouse lungs after PR8 infection. Western blot analysis of lung tissues from mice at 0, 3, and 5 d post-infection with antibodies to NF-κB p65, phospho-NF-κB p65, caspase-3, cleaved caspase-3, p62, and beclin-1. β-actin was used as the internal control. Ratios of these six proteins to β-actin were obtained by densitometry and presented as means ± SE. Three or more mouse lungs were examined for each group on each day. WT: wild-type mice; KO: knock out, Ifitm3-/- mice. *P < 0.05.
-
Many studies have revealed that acute influenza infection can lead to immune imbalance in the lungs, causing lung damage. Therefore, we quantitated several activated immune cells, including CD4+ T, CD8+ T, DCs, natural killer (NK) cells, and macrophages in the lungs of both types of mice via flow cytometry at 0, 3, and 5 days post-infection. Levels of most of these activated immune cells did not notably differ between the two mice. However, the proportion of NK cell activation in the lungs of Ifitm3-/- mice was significantly higher than that in wild-type mice (Figure 3).
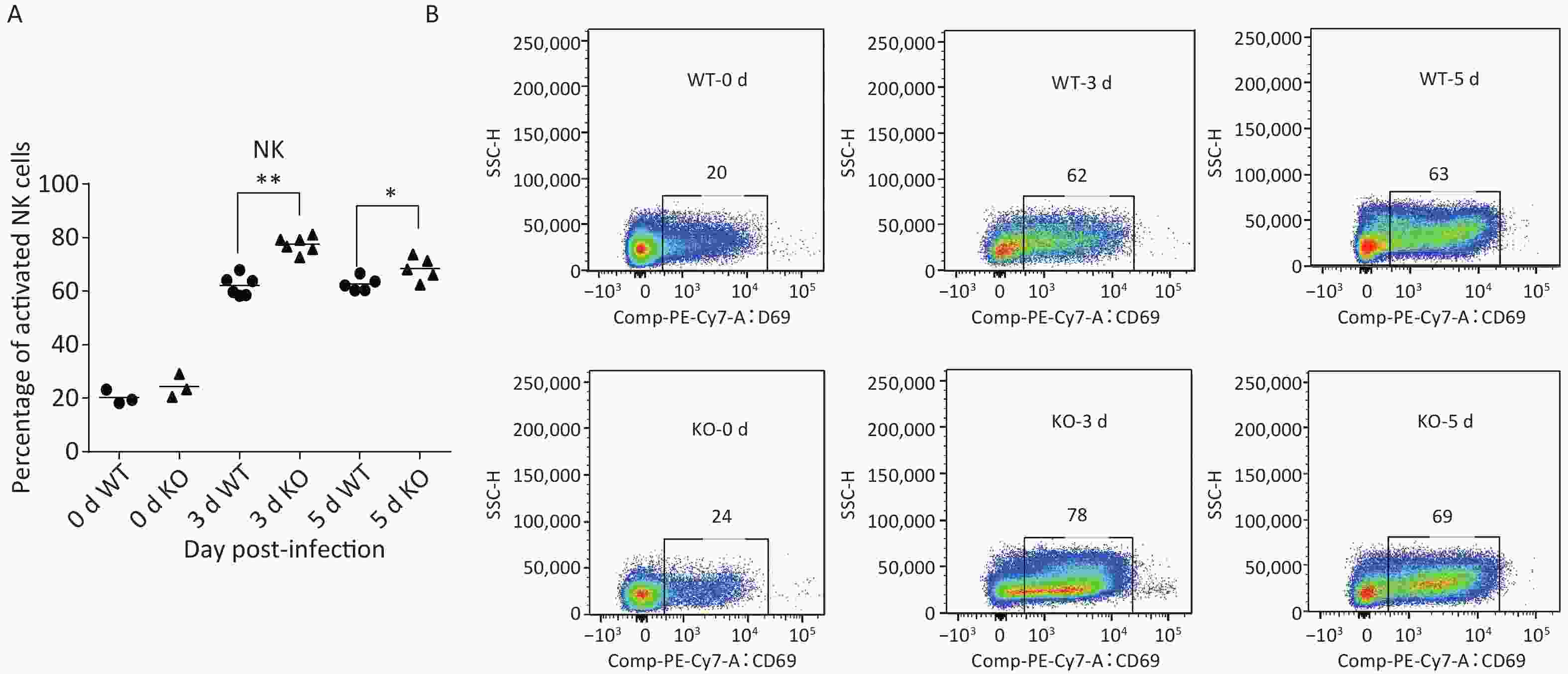
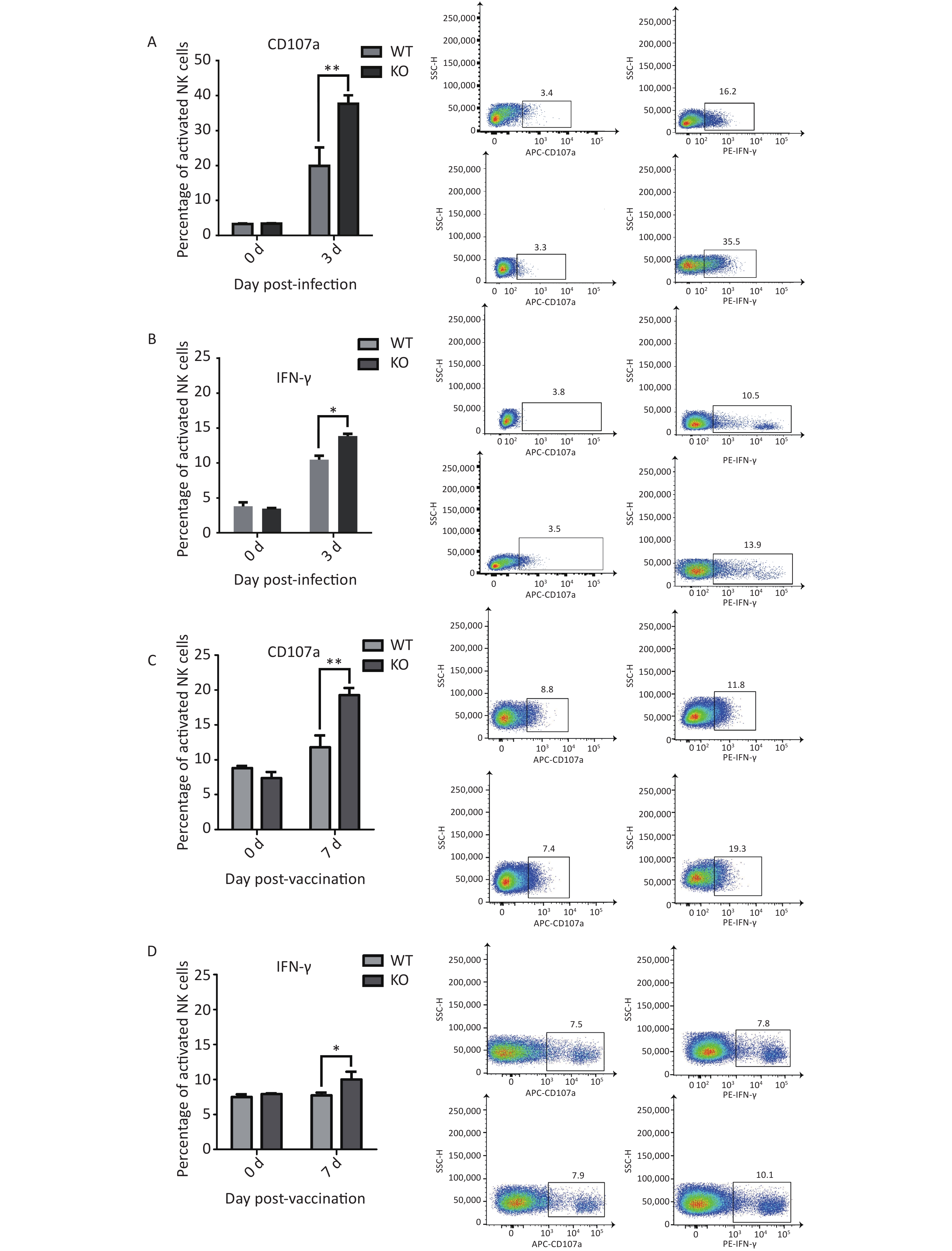
Figure 3. Phenotypes and activation states of natural killer (NK) cells in lungs after PR8 infection. (A) Percentages of activated NK cells at 0, 3, and 5 d after PR8 infection. (B) Flow cytometric analysis of percentages of activated NK cells at 0, 3, and 5 d after PR8 infection, with PE-Cy7-labeled CD69 antibody indicating activated NK cells. Numbers displayed in the figure indicate activated NK cell percentages. SSC-H represents side scatter-height. WT: wild-type mice; KO: knock out, Ifitm3-/- mice. *P < 0.05, **P < 0.01
To evaluate NK cells’ response to influenza infection, we first examined expression of the early activation marker CD69, to characterize the activation ratios of immune cells. We observed an increase in the ratios of active NK cells. Recent studies have shown that migration and activation of NK cells, caused by acute influenza infection, can cause immunopathological damage and even death. Two major antiviral effects of NK cells, lysis of virus-infected cells and IFN-γ production, were confirmed in early stages of acute influenza infection. These two NK cell functions were used to represent functional activation states[23]. We further detected the effector proteins CD107a and IFN-γ from NK cells in the lungs of both types of mice and found significantly elevated activation levels in the Ifitm3-/- mice compared to that in wild-type mice at 3 d post-infection (Figure 4A and B).
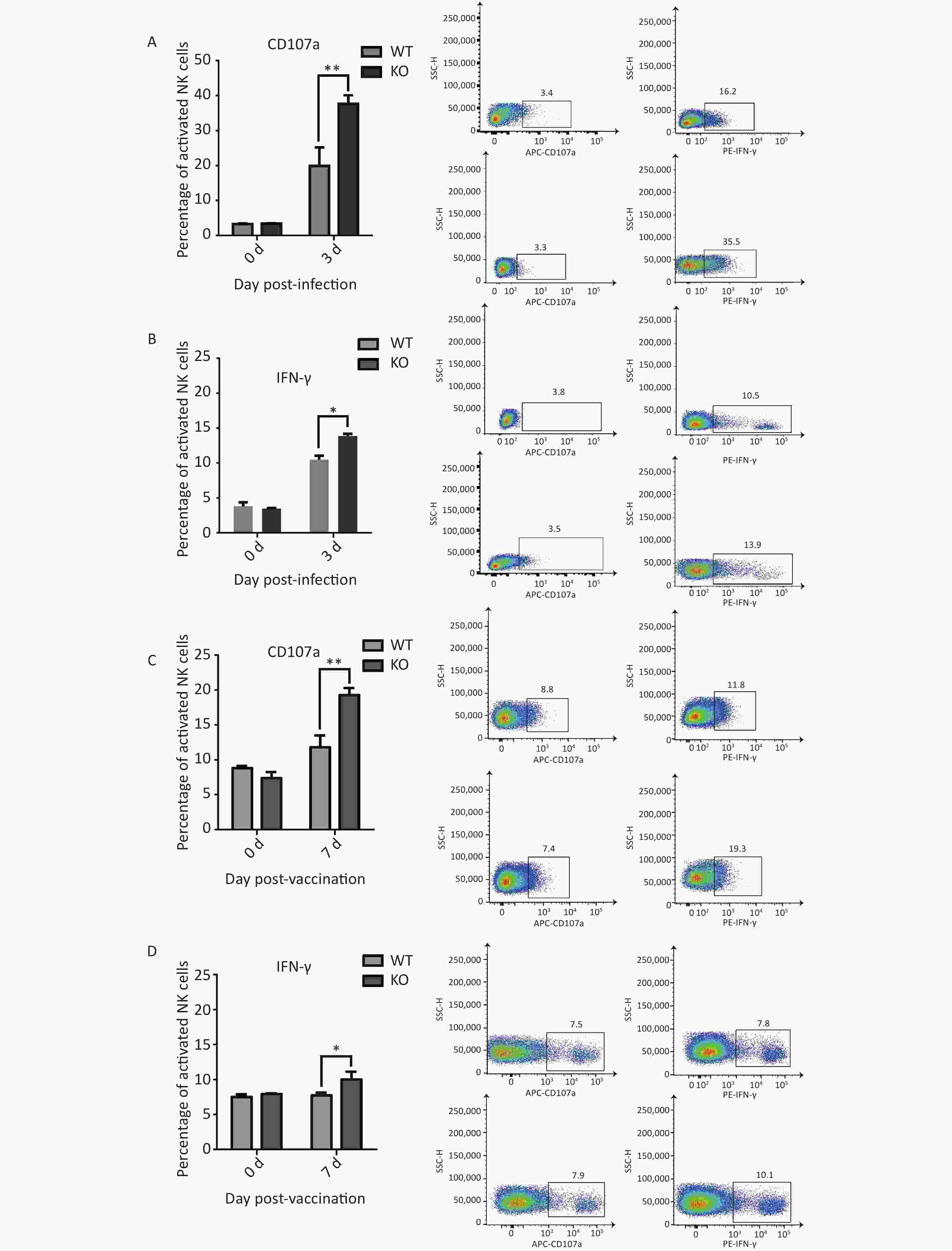
Figure 4. Elevated CD107a and IFN-γ expression in natural killer (NK) cells from Ifitm3-/- mice compared with wild-type mice during PR8 infection or vaccination. (A) Expression of CD107a in NK cells from mouse lungs 3 d post PR8 infection. Percentages of CD107a+ cells are shown. (B) Expression of IFN-γ by mouse lung NK cells 3 d post PR8 infection. Percentages of IFN-γ+ cells are shown. (C) Expression of CD107a in mouse spleen NK cells 7 days post booster immunization. Percentages of CD107a+ cells are shown. (D) Expression of IFN-γ in mouse spleen NK cells 7 d post booster immunization. Percentages of IFN-γ+ cells are shown. SSC-H represents side scatter-height.
We hypothesized that immunopathological damage, caused by an increased post-infection NK cell activation ratio in Ifitm3-/- mouse lung tissue was the major cause of death in mice. However, previous studies have shown that viral loads are higher in infected Ifitm3-/- mice, indicating that influenza virus replicates faster in Ifitm3-/- mice. A higher influenza virus titer will increase NK cell activation. We also found significantly elevated levels of activated effector proteins CD107a and IFN-γ from NK cells in the spleens of Ifitm3-/- mice, compared to wild-type mice 7 d after secondary immunization (Figure 4C and 4D). These results revealed that even in response to the same antigen, ratios of activated CD107a and IFN-γ in NK cells was higher in Ifitm3-/- mice. We speculate that NK cells are more easily activated in an endogenous IFITM3 gene knockout environment. This is could also be an important contributor to mortality in Ifitm3-/- mice.
In this study, lung mononuclear cell proteomics revealed significant differences in influenza-induced enrichment of proteins or molecular pathways, such as inflammation and immune response in Ifitm3-/- mice compared with that in wild-type mice. These results suggest that infected mice that mount a dysregulated antiviral immune response were inadequately protected, and further suggest that atypical host innate immune responses may contribute to lethality. We also explored the significant differences in enriched proteins and biological pathways in lung mononuclear cells in these two types of mice 3 d post-infection via proteomics. Differential expression analysis revealed that 457 proteins were differentially expressed in Ifitm3-/- versus wild-type mice, with a corrected P-value < 0.05. Of the differentially expressed proteins, 303 were up-regulated, while 154 were down-regulated. Heat maps show differential protein expression levels in the two groups (n = 3) (Figure 5A).
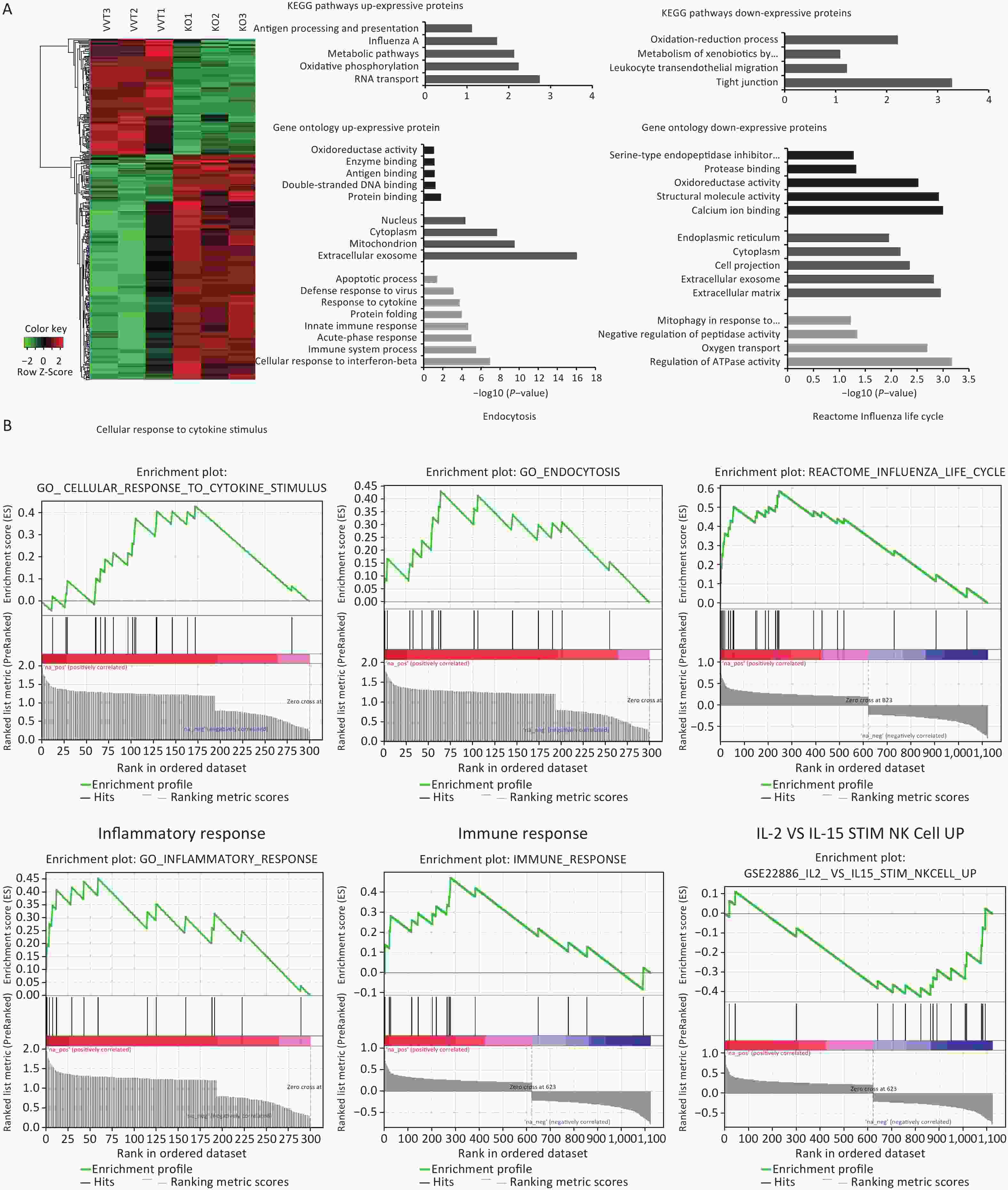
Figure 5. Inflammation and immune response related pathways enriched in lung mononuclear cells assessed by proteomic analyses of Ifitm3-/- and wild-type mice. (A) Hierarchical clustering of proteins differentially expressed in Ifitm3-/- versus wild-type mice. Red represents increased expression, while green represents decreased expression. DAVID bioinformatics tool was used to perform KEGG pathway and GO analysis of differentially expressed proteins. KEGG pathway analysis shows the most significant biological pathways enriched among up- and down-regulated proteins. GO analysis of enriched terms in up- and down-regulated proteins. (B) Gene set enrichment analysis revealed that the corresponding gene sets were differentially expressed in Ifitm3-/- and wild-type mice.
By pathway analysis, we tested whether the differentially expressed proteins were enriched in biological pathways that have potential implications for influenza infected Ifitm3-/- mice. In particular, genes in antigen processing and presentation, and Influenza A virus and leukocyte trans-endothelial migration annotated in the Kyoto Encyclopedia of Genes and Genomes (KEGG) may influence the aberrant innate immune response found in Ifitm3-/- mice (Figure 5A). Gene Ontology (GO) analysis revealed up-regulation of a number of proteins related to innate immune responses, and responses to cytokines (Figure 5A), consistent with a critical role for IFITM3 in intrinsic resistance to influenza viruses. Furthermore, gene set enrichment analysis of significantly differentially expressed proteins revealed several pathways, including cellular response to cytokine stimuli, inflammatory response, and IL-2 and IL-15-stimulated NK cell activation were significantly enriched, indicating that NK cells could induce lung tissue injury (Figure 5B).
We verified the expression of representative proteins CD69, lymphocyte antigen 6 complex locus G6D (Ly6G), caspase 7, and C-C motif chemokine ligand 2 (CCL2) by western blot. The results indicated that the expression of these four proteins increases during influenza virus infections, and expression was higher in Ifitm3-/- mice than in wild-type mice on the same days (Figure 6). These results showed seriously aberrant innate immune responses in lethal infections. Based on these results, we speculate that the death of Ifitm3-/- mice in the acute infection phase could be mainly due to up-regulation of the inflammatory response and immunopathological damage caused by abnormal activation of certain immune cells.
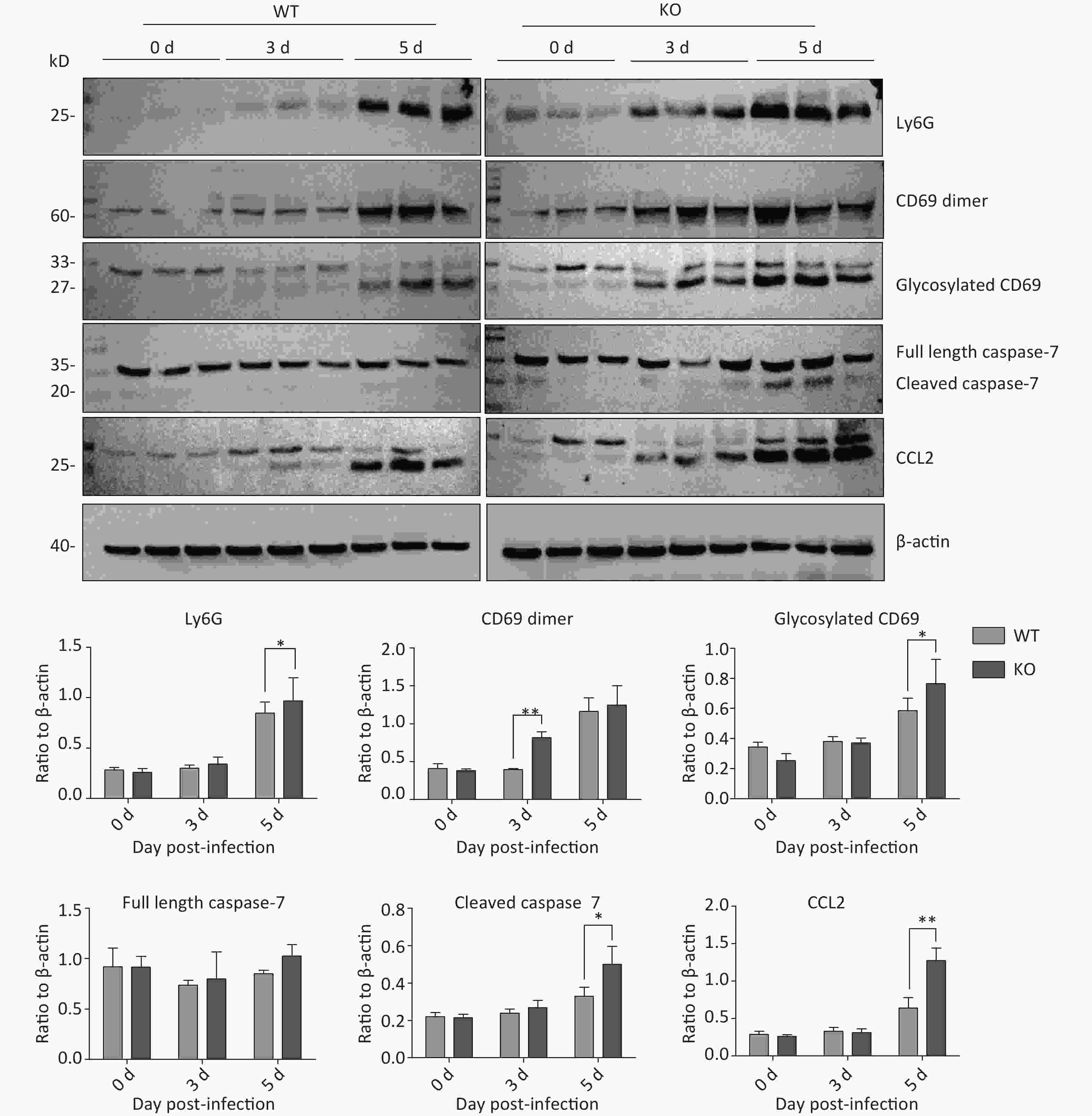
Figure 6. Time-dependent expression of activated natural killer (NK)-cell specific proteins, and inflammation- and apoptosis-related proteins in mouse lungs after PR8 infection. Western blot analyses of mouse lung tissues at 0, 3, and 5 d post-infection, using antibodies to Ly6G, CD69, caspase-7, and CCL2. anti-β-actin was probed as a loading control. Ratios of these six proteins to β-actin were obtained by densitometry and presented as means ± SE. Lungs from 3 or more mice per day per group were examined. WT: wild-type mice; KO: knock out, Ifitm3-/- mice. *P < 0.05, **P < 0.01.
-
IFITM3 belongs to the family of intracellular antiviral proteins discovered recently, and plays a role in inhibiting viral infection, mainly before viral entry into the cytoplasm. Recent studies have shown that IFITM3 not only strongly correlates with severity of influenza virus infection, but also plays an important role in the adaptive immune response to influenza virus infection[24].
Our results showed that Ifitm3-/- mice developed more severe inflammatory and apoptotic responses compared to wild-type mice. We explored the role of IFITM3 in immune cell activation in influenza virus infection. Compared to several other immune cell types, NK cells were activated at significantly higher ratios in the lungs of Ifitm3-/- mice than in wild-type mice. NK cells are large granular lymphocytes, members of the innate immune system, which can secrete cytotoxic granules and cause engagement of death receptors to lyse virus-infected cells, and produce IFN-γ. We used the early activation marker CD69, and the effector proteins CD107a and IFN-γ to characterize the activation and functional states of NK cells[25-27]. NK cells can also produce cytokines and chemokines to attract inflammatory cells to sites of inflammation[28,29]. There is insufficient understanding of the phenotypes and functions of NK cells in the lung during influenza virus infection. Several studies have demonstrated that NK cells might be responsible for enhanced morbidity and mortality during more severe influenza virus infections[30,31].
During acute influenza virus infections, and during immunization, NK cells show higher activation levels in the lungs and spleens of Ifitm3-/- mice, respectively, than in wild type according to expression levels of the early activation marker CD69, and the effector proteins CD107a and IFN-γ. Moreover, IL-2 and IL-15-stimulated NK cell activation was significantly enriched, with cytokine IL-15 acting as a powerful activator of NK cell functions. Based on our results, we speculate that NK cells are more easily activated in an endogenous IFITM3 gene knockout background, causing higher mortality in Ifitm3-/- mice. Our findings are consistent with earlier studies, which have reported that IFITM3 is a critical anti-viral infection protein in host immunity. Further, studies have shown that the IFITM3 plays an important role in maintaining the normal biological functions of several immune cell populations during influenza virus infections, such as protecting respiratory DCs ferrying viral antigen, inducing memory CD8+ T cells, and stimulating the intrinsic anti-viral immunity of human megakaryocytes through regulated induction of IFITM3[15-17].
Our study outcomes suggest that the IFITM3 effector protein is associated with NK cell activation during acute influenza virus infection. We also report the differences in pathways and single gene expression in NK cells in the lung tissues of wild type and Ifitm3-/- mice after PR8 virus infection, and the effects of endogenous IFITM3 deletion on NK cells. However, we could not confirm that the abnormal NK cell activation was the cause of death in Ifitm3-/- mice; this limitation of the current study should be addressed in future.
-
The authors would like to thank the staff of the National Institute for Viral Disease Control and Prevention, the Collaboration Innovation Center for Diagnosis and Treatment of Infectious Diseases, the Chinese Center for Disease Control and Prevention, the Key Laboratory for Medical Virology, and the National Health and Family Planning Commission, Beijing, China.
-
Conceived and designed the experiments: SUN Qiang, LEI Na, and SHU Yue Long. Performed the experiments: SUN Qiang, LEI Na, LU Jian, GAO Rong Bao, LI Zi, LIU Li Qi, SUN Ying, and GUO Jun Feng. Contributed reagents/materials/analysis tools: SUN Qiang and SHU Yue Long. Drafted the manuscript: SUN Qiang and SHU Yue Long. All authors reviewed the manuscript.
Interferon-induced Transmembrane Protein 3 Prevents Acute Influenza Pathogenesis in Mice
doi: 10.3967/bes2020.041
Funds:
This study was supported by the National Science Fund for Distinguished Young Scholars of China [Grant No. 81525017] and National Natural Youth Science Foundation [Grant No. 31900140]
- Received Date: 2019-11-11
- Accepted Date: 2020-02-10
-
Key words:
- IFITM3 /
- Influenza /
- Immune response /
- NK cells
Abstract:
| Citation: | SUN Qiang, LEI Na, LU Jian, GAO Rong Bao, LI Zi, LIU Li Qi, SUN Ying, GUO Jun Feng, WANG Da Yan, SHU Yue Long. Interferon-induced Transmembrane Protein 3 Prevents Acute Influenza Pathogenesis in Mice[J]. Biomedical and Environmental Sciences, 2020, 33(5): 295-305. doi: 10.3967/bes2020.041

|

 Quick Links
Quick Links
 DownLoad:
DownLoad: