
-
Tuberculosis (TB), caused by Mycobacterium tuberculosis complex (MTBC) infection, has existed for millennia and remains a major global health problem. Research on the molecular epidemiology of transmission is an effective way to analyze TB transmission[1]. Restriction fragment length polymorphism is a popular molecular technique that has been used to discriminate among clinical MTBC strains in past decades. However, the mycobacterial interspersed repetitive units variable number tandem repeats (MIRU-VNTRs) method, a new polymerase chain reaction-based typing method, determines the size and repeated number of units in each locus by amplifying mycobacterial interspersed repetitive units. Ease of operation, low cost, reproducible results, and high discriminatory power make this method practical for routine use[2]. The performance of MIRU-VNTR genotyping using a panel of 12, 15, or 24 loci has been evaluated, and 15 loci are ideal for molecular epidemiological research[3].
Briefly, sputum culture-positive specimens were collected from June 1, 2018 to August 31, 2019. The specimens were subjected to species identification and phenotypic drug susceptible tests of proportion with Lowenstein-Jensen (LJ) medium according to the operating procedures. The concentrations of isoniazide (INH) and rifampicin (RIF) were 0.2 mg/L and 40 mg/L, respectively. The genomic DNAs of the multi-drug resistant TB (MDR-TB, resistance to both INH and RIF) strains were extracted from fresh sub-cultures. The online MIRU-VNTRplus database (http://www.miru-vntrplus.org/) was used to determine MTBC strain lineage, relatedness, or clustering by matching with another online database[4, 5]. Phylogenetic relationships among strains were investigated by drawing a minimum spanning tree (MST) using web tools available at the MIRU-VNTRplus online database. The MST revealed clonal complexes (CCs) and phylogenetic links between MIRU-VNTR profiles that were different due to genetic variations. To assess genetic relatedness, a dendrogram was produced using the unweighted- pair group method with arithmetic mean algorithm (UPGMA). The Hunter Gaston discriminatory index (HGDI) was calculated to determine the discriminatory power of MIRU-VNTR typing using the formula:
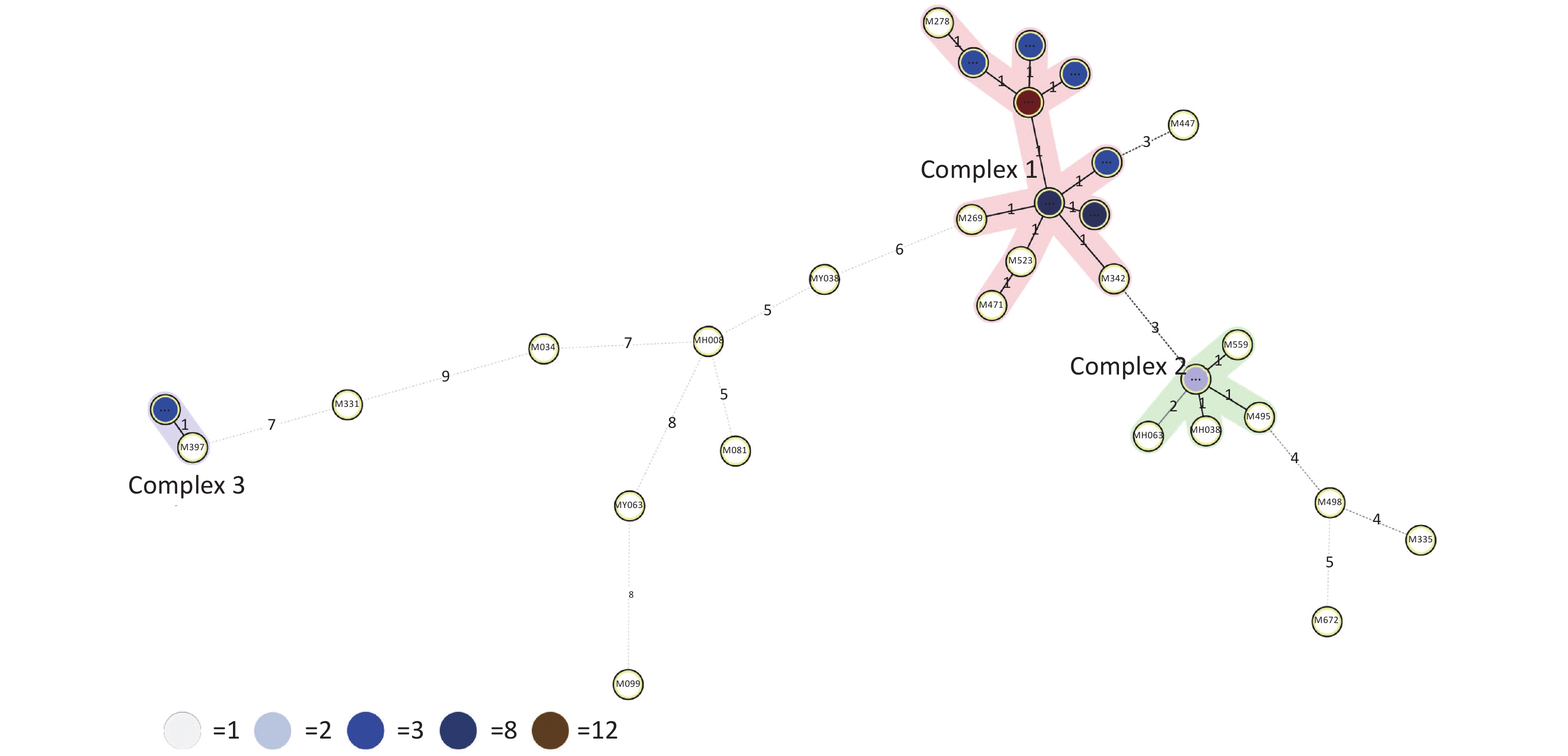
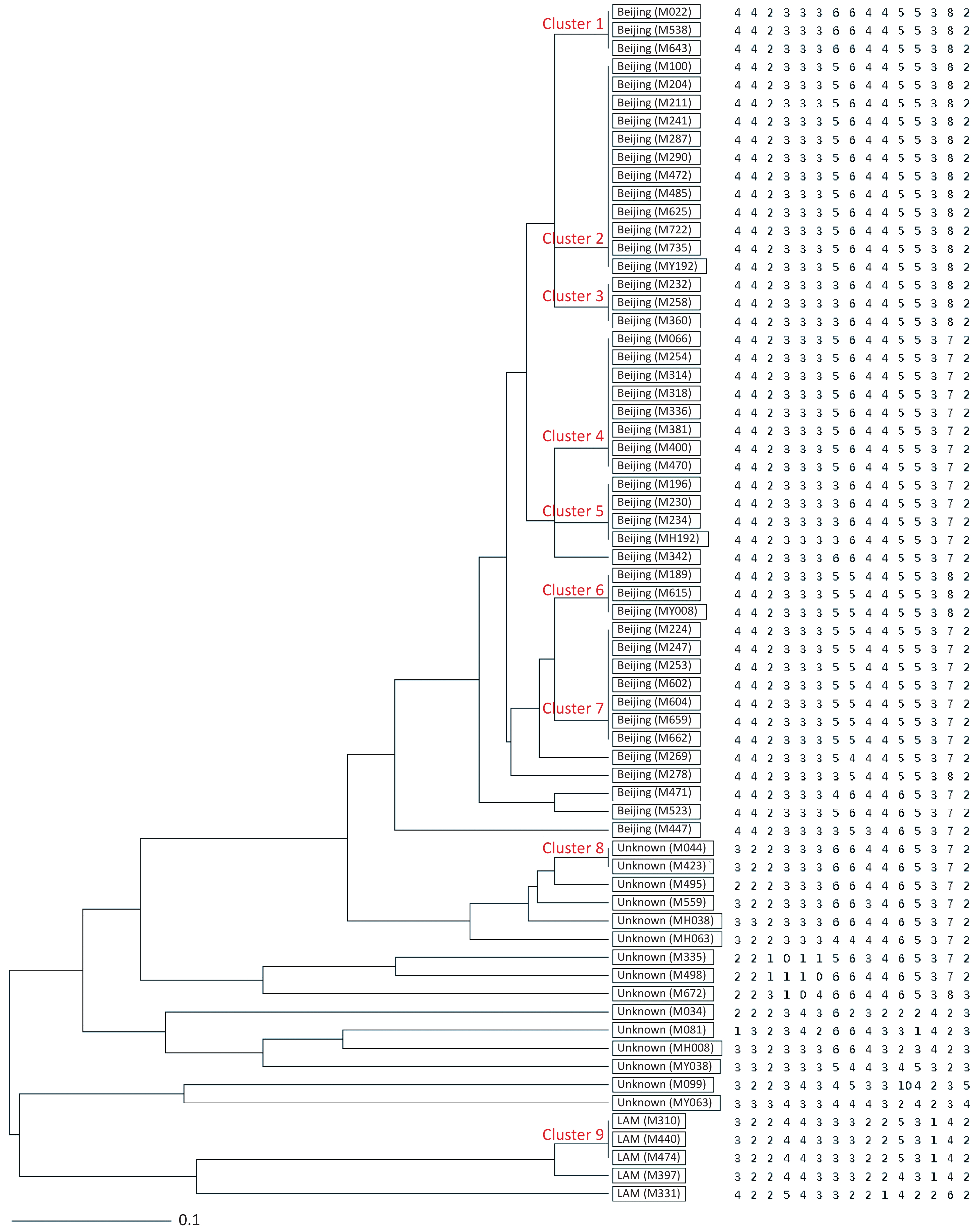
HGDI =
$1 - \dfrac{1}{{N(N - 1)}}\displaystyle\sum\limits_{j = 1}^S {n_j(n_j - 1)}$ (N: total number of strains, S: total number of typings, and nj: number of type j)[6]. A HGDI value of 0.00 indicates that all strains are indistinguishable, whereas a value of 1.00 indicates that all strains are differentiated. Additionally, a value of 0.30–0.60 indicates moderate discriminatory power. A cluster is defined as at least two strains with identical genetic patterns. Clusters are assumed to have arisen from recent transmission, and the clustering rate is used to determine the amount of recent transmission in the population[7]. The patients’ strains with an identical genetic pattern represent an epidemiologically linked cluster, and the minimum estimate of the proportion of TB cases related to recent transmission was calculated as: (nc-c)/N (nc: number of clustered strains, c: number of clusters, and N: total number of strains)[7]. Strains sharing identical MIRU-VNTR patterns were categorized as ‘clusters’, and patterns reported for a single strain are referred to as ‘unique’ using the tools available on the MIRU-VNTRplus database.As a result, 66 MDR-TB strains were identified from 1,873 sputum culture-positive specimens of suspicious TB patients (one specimen per patient). We determined genetic relatedness among the strains based on the UPGMA tree by submitting the strains to the MIRU-VNTRplus database. Among the studied strains, 51 (77.27%, 51/66) were matched with profiles presented in the MIRU-VNTRplus database and were assigned as Beijing (n = 46, 69.70%), followed by Latin-American-Mediterranean (LAM) (n = 5, 7.58%). Fifteen (22.73%, 15/66) strains did not correspond to any profile in the database and were considered unknown patterns, which may have been due to a limitation of the MIRU-VNTRplus website to include them. The unknown patterns might also reveal the circulation of unique and diverse patterns in Hangzhou. Therefore, the problem should be confirmed by spoligotyping or sequencing methods. More than two-thirds of the strains were the Beijing genotype, indicating that it may still be the predominant family in the area. However, previous studies revealed a proportion of 88.9% for the Beijing genotype in Southwest China[8]. Researchers have argued whether 15-locus MIRU-VNTR is optimal for epidemiological analyses in epidemic areas where the Beijing family genotype is predominant[3]. MIRU-VNTR genotyping detected 30 different patterns; 45 strains were distributed in nine clusters, and the remaining strains (n = 21) had unique patterns (Figure 1). The MST was plotted based on the MIRU-VNTR data to describe the genotype classification of the 66 strains (Figure 2). The results revealed that 46 strains belonged to Beijing, which grouped into the largest CC1 with 12 circles marked by bold connecting lines around two central circles and showed the close evolutionary relationship among them. CC2 was closely related to CC1. CC3 was distant from CC1 and CC2 and located in the left hand of the MST. The network of strains in CC1 exhibited compressed patterns in the upper right corner of the MST, and other parts with long branches proposed the existence of relatively ancient transmission from the central node strains in this area. Furthermore, aggregated nodes at CC1 and CC2 with short branches suggested a high rate of active TB transmission in this area.
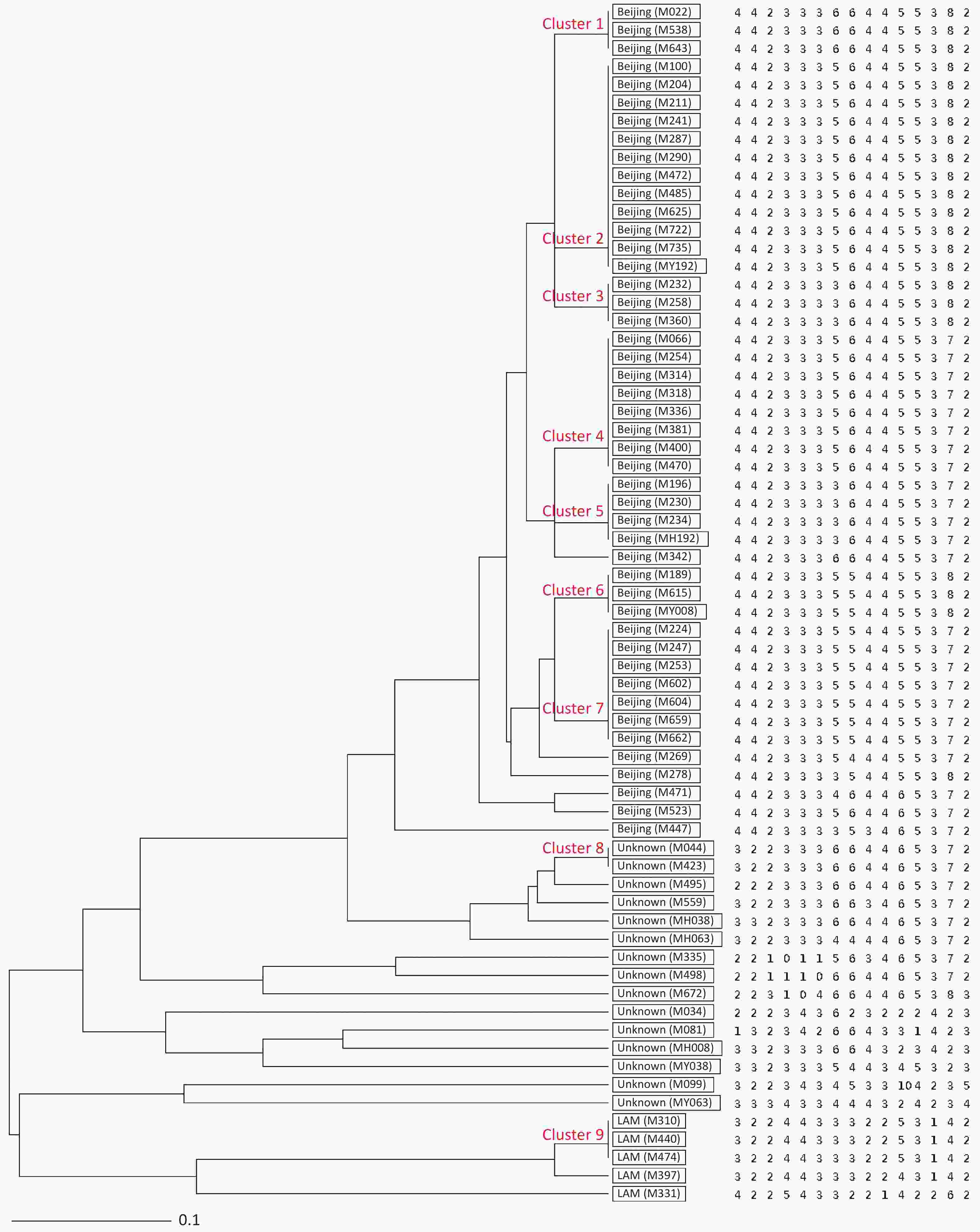
Figure 1. UPGMA tree showing genetic relatedness of 66 MDR-TB isolates isolated from Hangzhou, China based on the 15-locus MIRU-VNTR. Genotype and copy numbers are mentioned for each isolate along with the dendrogram. The arrangement of MIRU-VNTR loci from left to right including: Mtub04, ETR C, MIRU04, MIRU40, MIRU10, MIRU16, Mtub21, QUB11b, ETR A, Mtub30, MIRU26, MIRU31, Mtub39, QUB26 and QUB4156c.
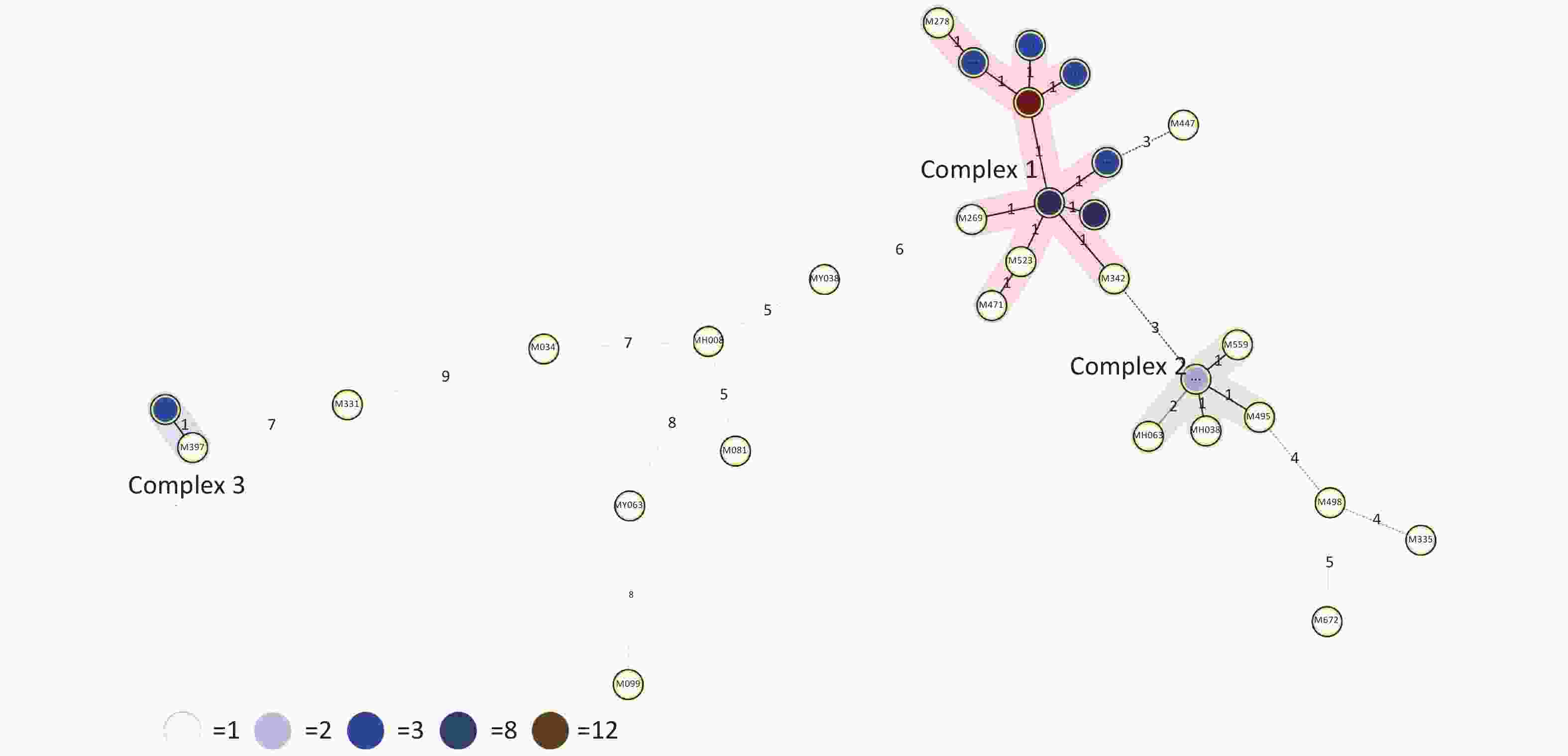
Figure 2. Minimum spanning tree (MST) based on 15-locus MIRU-VNTR demonstrating lineage classification of the 66 MDR-TB. The length of each branch shows the level of variation necessary to go from one allele to another allele. Solid bold and solid-pale lines indicate one and two allele changes, respectively. Dotted lines represent three or more allele changes. The color of circles indicates the number of clinical isolates; white, light blue, blue, dark blue and brown circles indicate 1, 2, 3, 8 and 12 isolates. Circles at the edges of the MST indicate evolutionary distant isolates and genetic diversity among isolates indicated on each branch.
The HGDIs of the 15 loci were 0.117–0.651 after analyzing the allelic polymorphisms (Table 1). The results indicated that Mtub21 and QUB26 had higher discriminatory power (HGDI = 0.651 and 0.645, respectively); Mtub04, ETR C, QUB11b, and MIRU26 had moderate discriminatory power, and the remaining MIRU loci (MIRU 04, 10, 16, 40, 31, ETR A, Mtub 30, 39, and QUB4156c) had low levels of allelic variability (HGDI < 0.30). The proportion of clustered strains in a population generally reflects the level of active transmission[7]. In this study, the minimum estimate of the proportion of TB cases related to recent transmission in Hangzhou was 54.55%, indicating that the total contribution rate to TB transmission efficiency was 54.55%. This value is slightly higher than that Shi et al. reported in Southwest China in 2018[8]. The difference may be related to the different regions involved, the study period, and or the number of MIRU-VNTR loci included. While MIRU-VNTR is still a widely used genotyping tool, whole genome sequencing (WGS) could provide more information for a genotyping analysis of closely related MTBC strains. Nonetheless, WGS remains limited due to the high cost and complex data analysis.
Copy numbers Number of isolates HGDI 0 1 2 3 4 5 6 7 8 9 VNTR sites 15 locus (n = 66) 424 Mtub04 1 5 13 47 0.455 577 ETR C 15 5 46 0.464 580 MIRU04 2 62 2 0.117 802 MIRU40 1 2 57 5 1 0.251 960 MIRU10 1 2 55 8 0.294 1,644 MIRU16 1 1 1 62 1 0.118 1,955 Mtub21 14 4 34 14 0.651 2,163b QUB11b 2 4 4 13 43 0.537 2,165 ETRA 5 5 56 0.273 2,401 Mtub30 1 5 5 55 0.298 2,996 MIRU26 3 1 3 46 12 1 0.484 3,192 MIRU31 1 2 5 2 56 0.276 3,690 Mtub39 4 3 56 3 0.276 4,052 QUB26 4 2 4 1 32 23 0.645 4,156 QUB4156c 59 5 1 1 0.198 Note. HGDI: Hunter Gaston discriminatory index, and HGDI value between 0.30 and 0.60 indicates moderate discriminatory power. Table 1. Allelic diversity of 15-locus MIRU-VNTR
In conclusion, we genotyped 66 MDR-TB strains isolated from a rural area of Hangzhou, China based on 15-locus MIRU-VNTR. The Beijing genotype was the predominant family circulating in this area. A relatively high level of active MDR-TB transmission was detected. These results are evidence that there is a need to modify our strategy and institute better interventions to prevent the transmission of MDR-TB in this area.
Conflicts of Interest No competing interests declared.
Ethical Approval This study was concisely informed, approved, and supported by the patients, and was endorsed by the Hangzhou Center for Disease Control and Prevention. Ethics were respected throughout the entire research period.
Authors’ Contributions JIA Qing Jun contributed to the study design and writing. ZENG Mei Chun provided writing assistance and proofreading. LI Qing Chun collected the data. WU Yi Fei, HUANG Yin Yan, LU Min, and AI Li Yun provided language help. XIE Li contributed to the data analysis and language help.

Funds:
This study was supported by the Independent Declaration Project of Agriculture and Social Development in Hangzhou [Grant number 20191203B143]
Reference 
 Quick Links
Quick Links
 DownLoad:
DownLoad: