
-
Human adenovirus (HAdV) is a non-enveloped virus with a linear double-stranded DNA genome of approximately 36,000 base pairs (bp) that encodes 40 proteins. Its three major capsid proteins are hexon, penton base, and fiber[1]. HAdVs are classified into 51 serotypes based on serum neutralization (SN) and hemagglutination inhibition (HI) tests, with type-specific sera targeting hexon and fiber proteins, respectively[2]. Polymerase chain reaction (PCR)-based molecular genotyping of the three major capsid protein genes or whole-genome sequencing has identified 52–116 genotypes. According to the Human Adenovirus Working Group's March 2024 update (http://hadvwg.gmu.edu/), all types are classified into seven species (A–G). HAdV genotypes 53–116 have been confirmed as recombinant HAdVs that cause inconsistent serological results in the SN and HI tests when detecting antigenic determinants of hexon and fiber proteins, thereby leading to inaccurate typing results. A prominent example is HAdV-B55, which was previously identified as HAdV-B11 for decades. Bioinformatic analysis of its complete genome sequence revealed that the genomic backbone originated from HAdV-B14, whereas the partial hexon gene was derived from HAdV-B11[3]. Consequently, the Human Adenovirus Working Group recommends that accurate HAdV typing relies on the gene sequences of the three capsid proteins and, when possible, whole-genome sequencing[4].
Different HAdV types exhibit strong tropism for different tissues[5]. Species B, C, and E are primarily associated with acute respiratory infections (ARIs), species A and F are mainly related to gastrointestinal manifestations, and species D is the most common pathogen of adenoviral keratoconjunctivitis, a major cause of ocular morbidity[6-8]. Epidemiological studies have indicated that HAdV-B3 and HAdV-B7 are the predominant types responsible for acute lower respiratory infections (LRIs) in children under 5 years of age worldwide. In China, an epidemiological investigation of HAdV infections in hospitalized children with LRIs in Beijing from 2015 to 2019 revealed that HAdV-B3 and HAdV-B7 were the primary types[9]. Clinical evidence has shown that HAdV-B7 infections cause more severe pneumonia than HAdV-B3 infections, resulting in longer hospital stays and a higher risk of respiratory failure[10,11].
Intra- or inter-species recombination is the principal force driving HAdV genetic evolution[12], potentially leading to altered virulence, enhanced transmissibility, modified tissue tropism, and novel disease spectra. Recombination events between HAdV-B11 and HAdV-B14 led to the formation of the highly pathogenic and infectious genotype HAdV-B55, which caused multiple severe ARIs outbreaks in China and was responsible for severe pneumonia in the South Korean military[13,14]. Compared to the HAdV-B7 prototype strain (GenBank No. AY594255), the HAdV-B7d strain contains the L1 52/55 kDa DNA packaging protein from HAdV-B16, which is associated with more severe illnesses and higher fatality rates in pediatric inpatients than HAdV-B3 and HAdV-C strains[15,16]. An outbreak of ARIs among infants in Lisbon, Portugal, was reportedly caused by a recombinant strain containing the hexon gene of HAdV-B7 and the fiber gene of HAdV-B3[17]. The Human Adenovirus Working Group (http://hadvwg.gmu.edu/) reported a genome sequence deposited in GenBank (No. OR853835) and was designated as HAdV-B114, which contains a backbone of HAdV-B3 and a penton base gene from HAdV-B7. However, the effects of these recombination events are unclear.
Clinical specimens were retrospectively collected from children with ARIs in Beijing between 2022 and 2023 to monitor the molecular epidemiology of HAdVs. These speciments were screened for HAdV using a capillary electrophoresis-based multiplex PCR (CEMP) assay, followed by HAdV typing based on penton base, hexon, and fiber gene sequences. For specimens exhibiting inconsistent typing results, cloned penton base, hexon, and fiber gene sequences in the pGEM-T Easy recombinant vector were analyzed to differentiate recombinant HAdV infections from coinfections involving different HAdV types. Metagenomic next-generation sequencing (mNGS) was performed to characterize the genomes of recombinant HAdVs in children with ARIs in Beijing.
-
Clinical respiratory specimens, including throat swabs, nasopharyngeal swabs, nasopharyngeal aspirates, and bronchoalveolar lavage fluids from pediatric patients, were included in the study according to the following eligibility criteria: (1) diagnosis of ARIs presenting with signs and symptoms of fever, cough, chills, expectoration, nasal congestion, sore throat, chest pain, tachypnea, or abnormal pulmonary breath sounds[18], (2) hospitalization at the Children’s Hospital Affiliated with the Capital Institute of Pediatrics between January 2022 and December 2023, and (3) age between 1 month and 16 years. The exclusion criteria were as follows: (1) patients with more than one sample collected during a single hospitalization, where only the first sample was included, and (2) readmission within 7 days of discharge. Upon arrival at the laboratory, each clinical specimen was handled in a Class II biosafety cabinet. The specimens were immediately processed using 2.5 mL of viral transport medium (Yocon Biotechnology Co., Ltd., Beijing, China) and centrifuged at 500 × g for 10 min. A portion of the supernatant was used for viral nucleic acid extraction, and the remainder was stored at –80°C for future use.
The minimum sample size required to accurately represent the HAdV infection status among children in Beijing was estimated to be 1,825. This calculation was based on a 5% positivity rate (4.34%–5.64%) for HAdV in children with ARIs in Beijing and an allowable error of 1%[19,20].
$$ n = \frac{u_{1-\alpha/2}^2 \pi (1-\pi)}{\delta^2} = \frac{1.96^2 \times 0.05 \times (1-0.05)}{0.01^2} \approx 1,825 $$ (1) -
Respiratory specimens were screened using a CEMP assay (Ningbo HEALTH Gene Technologies Ltd., Ningbo, China)[21]. Total nucleic acid (DNA and RNA) was extracted from 140 µL of supernatant from each specimen using the QIAamp MinElute Virus Spin Kit (Qiagen GmbH, Germany), following the manufacturer’s instructions. Subsequently, according to the CEMP assay protocol, nucleic acids were amplified in a reaction mixture containing a PCR enzyme, 0.25 µmol/L of each of the 15 primer pairs targeting specific pathogens, dNTPs, MgCl2, and buffer. Amplified products were analyzed by capillary electrophoresis using the GeXP capillary electrophoresis system (Sciex, Concord, ON, Canada). A total of 15 primer pairs were designed to detect 13 respiratory pathogens in human DNA and RNA samples. Fluorescence signals from 15 labeled PCR products were measured to identify pathogen-specific amplicons. Based on the kit instructions, the amplicon positions for the detected pathogens were as follows: influenza virus (Flu) A, 105 nucleotides (nt) (2009 H1N1, 163.3 nt; H3N2, 244.9 nt), Flu B, 212.7 nt, HAdV, 110.2/113.9 nt (indicating different subtypes), human bocavirus (HBoV), 121.6 nt, human rhinovirus (HRV), 129.6 nt, human parainfluenza virus (PIV), 181.6 nt, chlamydia (Ch), 190.5 nt, human metapneumovirus (HMPV), 202.8 nt, Mycoplasma pneumoniae (Mp), 217 nt, human coronavirus (HCoV), 265.1 nt, and respiratory syncytial virus (RSV), 280.3 nt[21]. The remaining nucleic acids were stored at –20 °C for future use.
-
For specimens confirmed as HAdV-positive by the CEMP assay, the penton base, hexon, and fiber genes were amplified by PCR using the following primer pairs: Penton-F (5’-CTATCAGAACGACCACAGCAACTT-3’) and Penton-R (5’-TCCCGTGATCTGTGAGAGCRG-3’) to amplify a 1,153 bp fragment of the penton base gene, HVR-F (5’-CAGGATGCTTCGGAGTACCTGAG-3’) and HVR-R (5’-TTTCTGAAGTTCCACTCGTAGGTGTA-3’) to amplify a 1,658 bp fragment of the hexon gene, Fiber-F (5’-CCCTCTTCCCAACTCTGGTA-3’) and Fiber-R/CR (5’-GGGGAGGCAAAATAACTACTCG-3’/5’-GAGGTGGCA GGTTGAATACTAG-3’) to amplify predicted amplicons of 1,153 bp and 2,027 bp of the fiber gene in species B and C, respectively, and Fiber-D1 (5’-GATGTCAAATTCCTGGTCCAC-3’) and Fiber-D2 (5’-TACCC GTGCTGGTGTAAAAATC-3’) to amplify a predicted amplicon of 1,205 bp of the fiber gene in species D[22]. PCR conditions were as follows: 94 °C for 5 min, 45 cycles of 94 °C for 30 s, 52 °C for 30 s, 72 °C for 1.5 min, and a final extension at 72 °C for 7 min. PCR products were purified and sequenced using Sanger sequencing (Sino Geno Max Co., Ltd., Beijing, China). All sequences were designated CHN-BJ-Number-Year.
-
Nucleotide sequences of the penton base, hexon, and fiber genes generated by Sino Geno Max were assembled using Sequencher 5.4.5 software and analyzed using BLAST (https://blast.ncbi.nlm.nih.gov) to determine the most closely related HAdV types. Phylogenetic analysis was performed to type the HAdV sequences from this study, using the format [Pn-Hn-Fn], and compared with the penton base, hexon, and fiber genes of reference sequences retrieved from GenBank. The maximum likelihood method with 1,000 bootstrap pseudo-replicates was implemented in MEGA7 software with pairwise deletion for gaps and missing data. Phylogenetic trees of species C were constructed by comparing newly identified sequences with species C reference sequences labeled according to the updated nomenclature (Px1–Px6) to designate primary penton base genetic groups[23].
-
For specimens with inconsistent typing results, the amplified penton base, hexon, and fiber genes were purified and ligated into the pGEM-T Easy vector (TransGen Biotech, Beijing, China) at 4°C overnight for TA cloning. The recombinant DNA was subsequently transformed into competent Escherichia coli (E. coli) DH5α cells. For each gene, at least five recombinant clones were selected and sequenced using the Sanger method. Coinfections involving different HAdV types were identified when the sequences corresponded to more than one type. In the absence of coinfection, recombination events were inferred.
-
For specimens with suspected recombination events, nucleic acids were extracted using the QIAamp MinElute Virus Spin Kit (Qiagen GmbH, Düsseldorf, Germany), following the manufacturer’s instructions. Subsequently, mNGS was performed on a NovaSeq 6000 sequencing platform (Illumina, San Diego, CA, USA) using a 2 × 150 cycle paired-end sequencing protocol with a data yield of 10 GB per library. Sequence data were trimmed using FASTP[24]. The clean reads were mapped to the human reference genome (hg19) using the Bowtie2 software; reads matching the human genome were discarded[25], and the residual reads were assembled using Trinity (version 2.8.4). Metagenomic taxonomic classification was performed using Kraken2 (version 2.0.8-beta)[26,27]. Finally, the genomes were polished using the BLASTn package, and the genome termini were manually inspected and corrected by mapping the reads against HAdV reference sequences in GenBank. The mapped results were manually verified and compared with those of the assembled scaffolds.
-
Potential recombination events among the genome sequences were assessed using the Recombination Detection Program (RDP) version 4[28]. Seven algorithms—RDP, GENECONV, Chimaera, MaxChi, Bootscan, SiScan, and 3Seq—were applied, and recombination was considered valid only when detected by at least three methods with a threshold P-value of 0.05, thereby ensuring the robustness of the results. For high-quality mNGS sequences, SimPlot (version 3.5.1) was used to perform recombination analysis with bootscanning under the following parameters: window size of 1,000 bp, step size of 200 bp, gap stripping, and Kimura two-parameter distance correction. Nucleotide sequences were compared using zPicture, employing a moving overlapping window to calculate percentage identity scores[29].
-
Measurement data with a normal distribution are presented as mean ± standard deviation, whereas skewed data are presented as median and interquartile range. The rates of the two categorical datasets were compared using the chi-square (χ2) test. All statistical analyses were performed using SPSS Statistics software (version 27.0; IBM, Armonk, NY, USA).
-
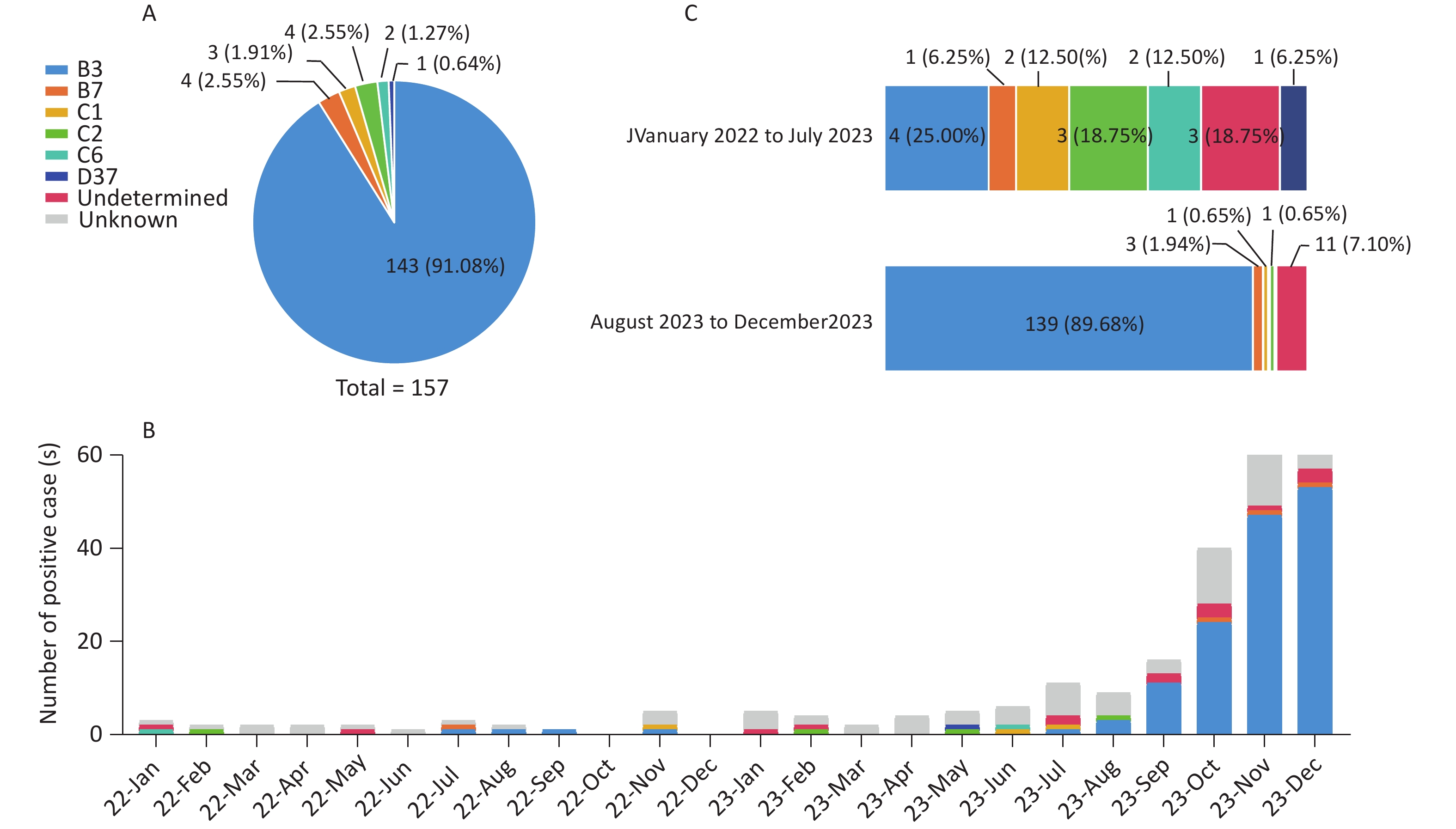
From January 2022 to December 2023, a total of 6,771 pediatric patients with ARIs were enrolled. Using CEMP assays, 277 patients (4.09%; 277/6,771) tested positive for HAdV, including 164 boys and 113 girls, with a median age of 4.9 years (interquartile range, 2.2–7.6 years). The positivity rate of HAdV infection increased significantly from 1.04% (23/2,209) in 2022 to 5.46% (254/4,652) in 2023 (P < 0.001) and peaked in November 2023, with a positivity rate of 13.69% (76/555) (Figure 1). Of the 277 HAdV-positive patients, 121 (43.68%) were infected solely with HAdV, whereas 156 (56.32%) were coinfected with other pathogens, such as Mp (50/156, 32.05%) and HRV (24/156, 15.38%).
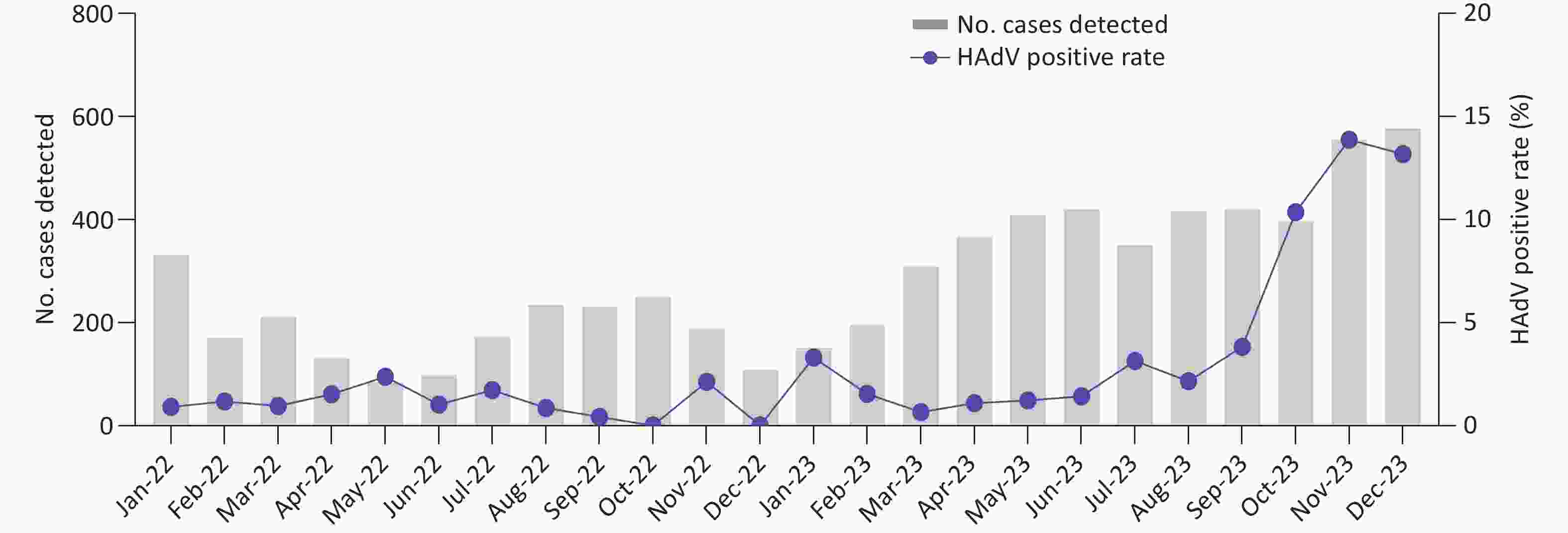
Figure 1. Monthly distribution of acute respiratory tract infection (ARIs) cases collected for HAdV screening and the corresponding positivity rates from January 2022 to December 2023.
-
Of the 277 HAdV-positive patients, 49 (17.69%, 49/277) lacked sequencing results due to low PCR product yields, and 57 (20.57%, 57/277) had only one or two available gene sequences. A total of 106 cases remained untyped. For patients with sequences of all three genes (penton base, hexon, and fiber), phylogenetic analysis identified 157 cases (56.68%, 157/277) belonging to six HAdV types. The most prevalent type was HAdV-B3 [P3H3F3] (143/157, 91.08%), followed by HAdV-B7 [P7H7F7] (4/157, 2.55%), HAdV-C2 [Px1ps2H2F2] (4/157, 2.55%), HAdV-C1 [Px1ps1H1F1] (3/157, 1.91%), HAdV-C6 [Px1ps6H6F6] (2/157, 1.27%), and HAdV-D37 [P37H37F37] (1/157, 0.64%) (Figure 2A). The monthly distribution of HAdV-positive cases showed that HAdV-C was the dominant species from January 2022 to July 2023, during which HAdV positivity rates were low. Beginning in August 2023, HAdV-B3 cases rose sharply, driving a marked increase in overall HAdV positivity rates and rapidly establishing HAdV-B3 as the dominant type (Figure 2B). Its proportion among HAdV types increased significantly from 25.00% (4/16) before August 2023 to 89.68% (139/155) thereafter (P = 0.02) (Figure 2C).
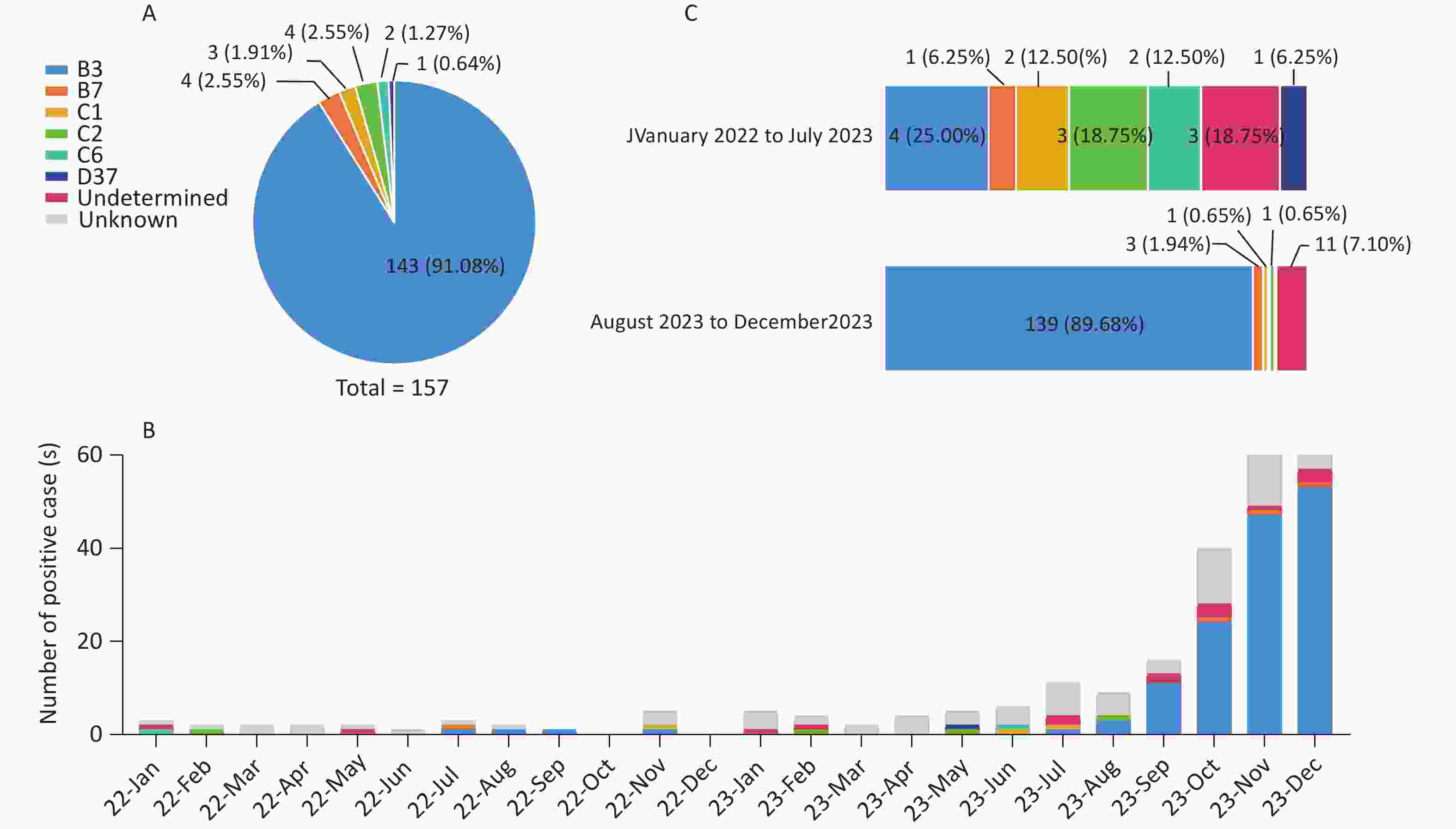
Figure 2. HAdV typing results and distribution of different HAdV types. (A) Successfully typed HAdV cases in a pie chart. (B) Number of positive cases of different HAdV types (2022–2023). (C) Composition of HAdV types in HAdV-positive cases with sequences of penton base, hexon, and fiber genes before and after August 2023.
Additionally, 14 cases (5.05%, 14/277) exhibited inconsistent typing results across the penton base, hexon, and fiber genes, and were classified as undetermined.
-
Among the 14 cases with inconsistent penton base, hexon, and fiber typing results, sequencing of pGEM-T recombinant clones detected no coinfections involving different HAdV types. Recombinant events were suspected in eight cases clustered with HAdV-C and six cases clustered with HAdV-B (Table 1).
Genetic recombinant pattern No. of cases Type of penton base gene Type of hexon gene Type of fiber gene Collection year Supposed type P7H3F3 6 P7 H3 F3 2023 Maybe B114 Px1ps1H2F2 5 Px1ps1 H2 F2 2022, 2023 Maybe C108 Px1ps3H5F5 1 Px1ps3 H5 F5 2023 NA Px2H5F5 1 Px2 H5 F5 2023 NA Px3H1F1 1 Px3 H1 F1 2023 NA Table 1. Genetic recombinant patterns of the 14 undetermined specimens based on penton base, hexon, and fiber gene sequences from children with ARIs in Beijing
Phylogenetic maximum-likelihood trees were constructed for the eight HAdV-C strains based on penton base, hexon, and fiber gene sequences (Figure 3). Fiber gene sequences formed three clades (F1, F2, and F5), hexon gene sequences clustered into three clades (H1, H2, and H5), and penton base gene sequences showed high variability across the four major clades (Px1ps1, Px1ps3, Px2, and Px3).
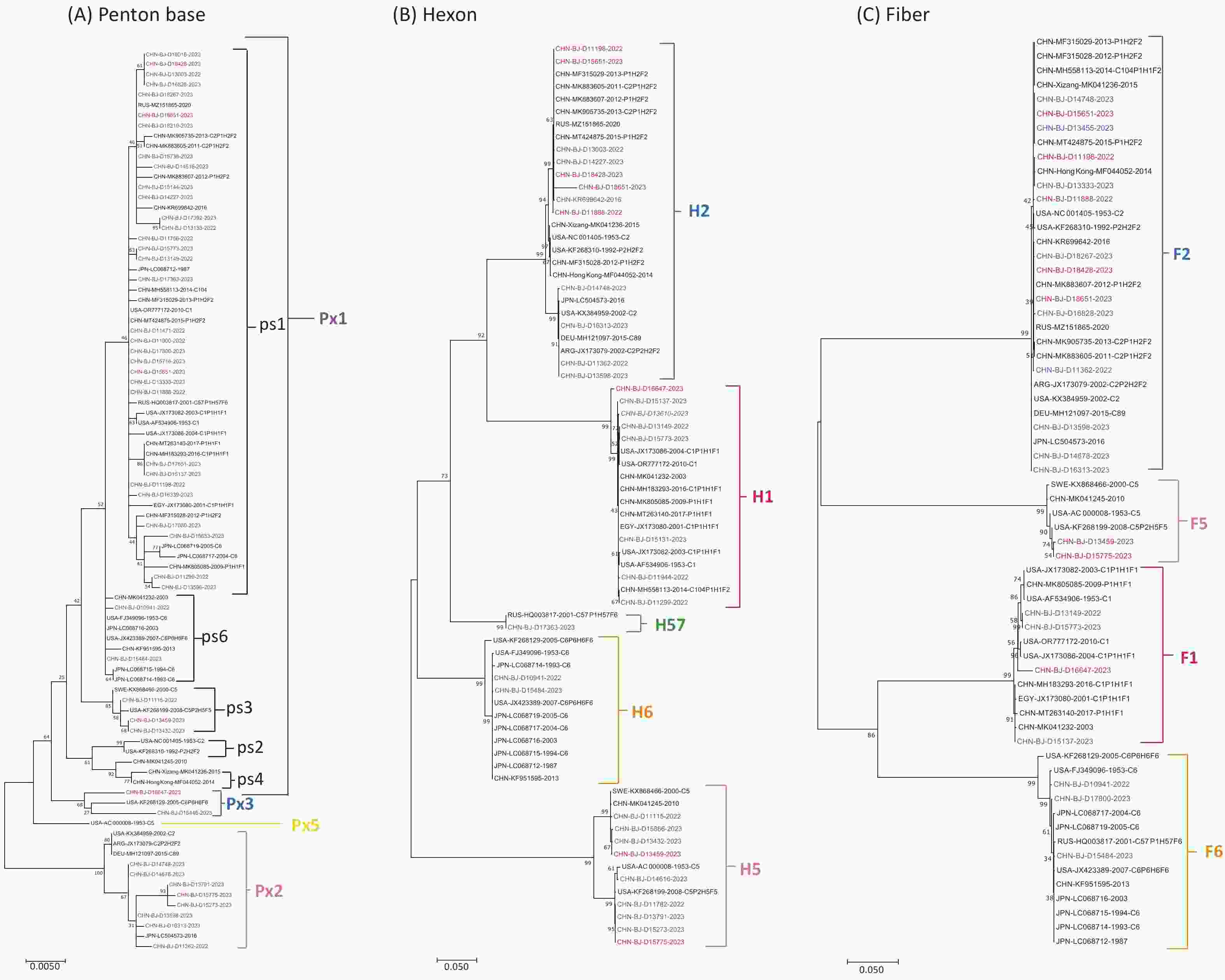
Figure 3. Phylogenetic trees of HAdV-C penton base (A), hexon (B), and fiber genes (C) constructed using the maximum likelihood method in MEGA7 software with the best-fit DNA model and 1,000 replicates. The eight sequences with inconsistent typing results from patients with ARIs in this study are shown in red, other sequences are shown in blue, and reference sequences from GenBank are shown in black. (A) The phylogenetic tree of the penton base gene sequences was divided into the major genetic groups Px1, Px2, Px3, and Px5, and subgroups ps1–ps6 of Px1. (B) The phylogenetic tree of the hexon gene sequences was divided into H1, H2, H5, H6, and H57. (C) The phylogenetic tree of the fiber gene sequences was divided into F1, F2, F5, and F6.
For the six HAdV-B cases, the hexon and fiber gene sequences were typed as HAdV-B3, whereas the penton base gene sequences were typed as HAdV-B7. Subsequently, mNGS was performed to further characterize the genomes.
-
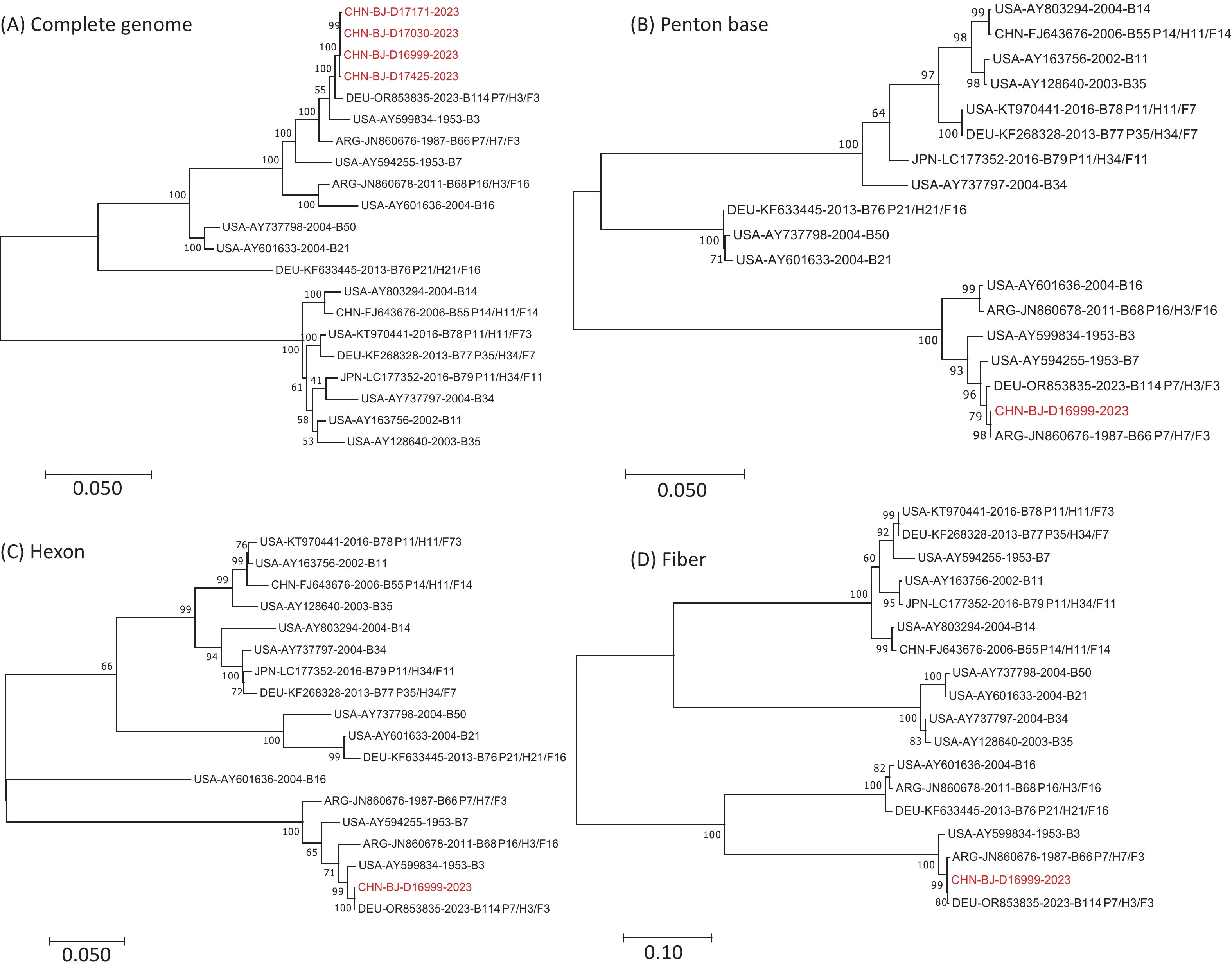
High-quality mNGS sequences were obtained from four of the six patients with suspected HAdV-B recombination events. These sequences were compared phylogenetically with the genomic sequences of 16 prototype HAdV-B strains, including HAdV-B3 (GenBank No. AY599834), HAdV-B7 (GenBank No. AY594255), HAdV-B11 (GenBank No. AY163756), HAdV-B14 (GenBank No. AY803294), HAdV-B16 (GenBank No. AY601636), HAdV-B21 (GenBank No. AY601633), HAdV-B34 (GenBank No. AY737797), HAdV-B50 (GenBank No. AY737798), HAdV-B55 (GenBank No. FJ643676), HAdV-B66 (GenBank No. JN860676), HAdV-B68 (GenBank No. JN860678), HAdV-B76 (GenBank No. KF633445), HAdV-B77 (GenBank No. KF268328), HAdV-B78 (GenBank No. KT970441), HAdV-B79 (GenBank No. LC177352), and HAdV-B114 (GenBank No. OR853835) (Figure 4). The four whole-genome sequences obtained in this study exhibited high sequence similarity and formed a single cluster. Therefore, CHN-BJ-D16999-2023 (D16999) were selected for detailed recombination analysis.
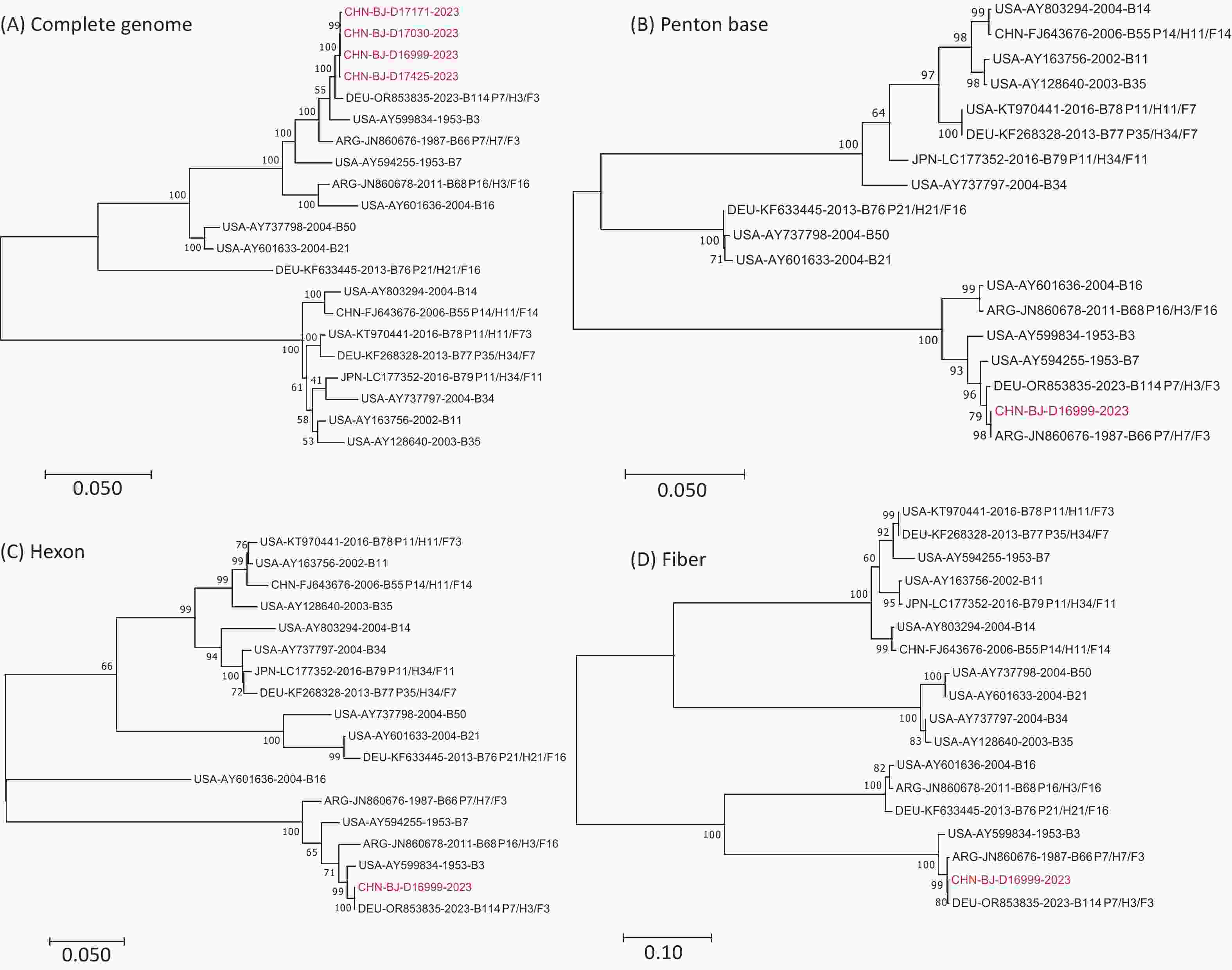
Figure 4. Phylogenetic analysis of complete genomic sequences of four cases (A), penton base (B), hexon (C), and fiber (D) gene sequences of D16999 compared with those of HAdV-B3, -B7, and -B114.
The complete genome sequence of strain D16999, derived from a respiratory specimen of a 3-year-old girl, was 35,253 bp in length, with a GC content of 50.92%, and shared 99.25%, 98.33%, and 97.27% similarity with HAdV-B114 (GenBank No. OR853835), HAdV-B3 (GenBank No. AY599834), and HAdV-B7 (GenBank No. AY594255), respectively. High sequence similarities were observed within the hexon (98.90%) and fiber (98.43%) genes between D16999 and HAdV-B3, and within the penton base gene (99.14%) between D16999 and HAdV-B7 (Table 2). The novel HAdV-B genome of CHN-BJ-D16999-2023 has been submitted to GenBank under accession number PP272026 and will be released after the publication of the manuscript.
Nucleic acid identities (%) compared with that of D16999 Prototype strains whole-genome penton base gene hexon gene fiber gene HAdV-B3 98.33 98.71 98.9 98.43 HAdV-B7 97.27 99.14 95.87 74.69 HAdV-B114 99.25 99.51 100.00 99.90 Table 2. The nucleic acid identities between D16999 and representative prototype HAdV-B types
Phylogenetic analysis of the complete genomic sequences and the penton base, hexon, and fiber gene sequences demonstrated that D16999 was most closely related to HAdV-B3 based on the complete genomic sequence (Figure 4A). The penton base gene sequence shared the highest identity with HAdV-B7, whereas the hexon and fiber genes were most similar to those of HAdV-B3 (Figure 4B–D). This recombination pattern is similar to that of HAdV-B114, which has a HAdV-B3 genomic backbone and a penton base gene derived from HAdV-B7.
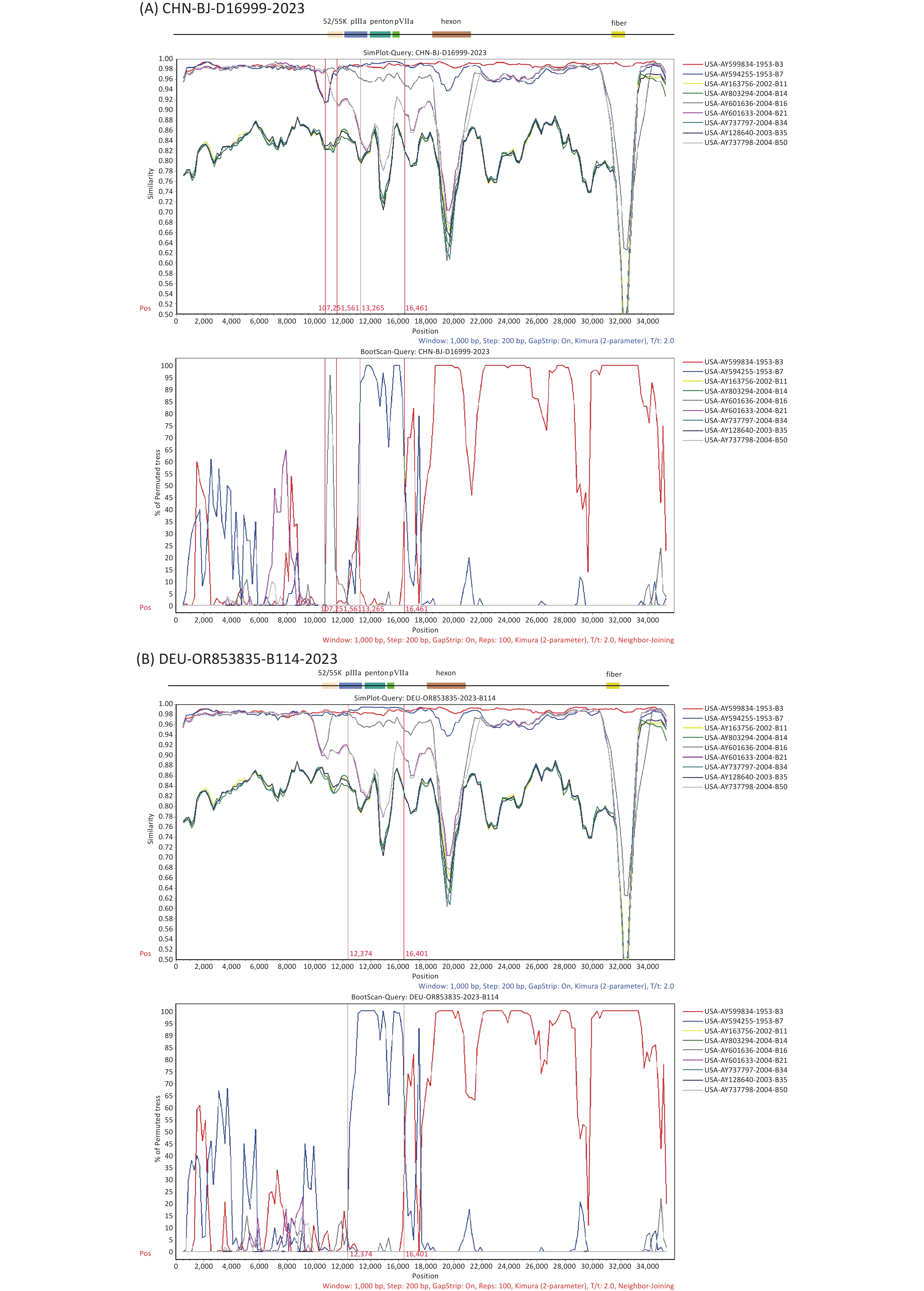
To further explore the breakpoints in the complete genome sequence of D16999 (P7H3F3), recombination analysis was performed using SimPlot software with similarity plotting and bootscanning. The results indicated that gene fragments encoding the partial protein IIIa precursor, penton base, and protein VII precursor were derived from HAdV-B7, using HAdV-B3 as the backbone, with a recombination mechanism similar to that of HAdV-B114. Notably, an additional recombination breakpoint was detected involving gene fragments from HAdV-B16 encoding the L1 52/55 kDa protein, differentiating D16999 from HAdV-B114 (Figure 5A–B).
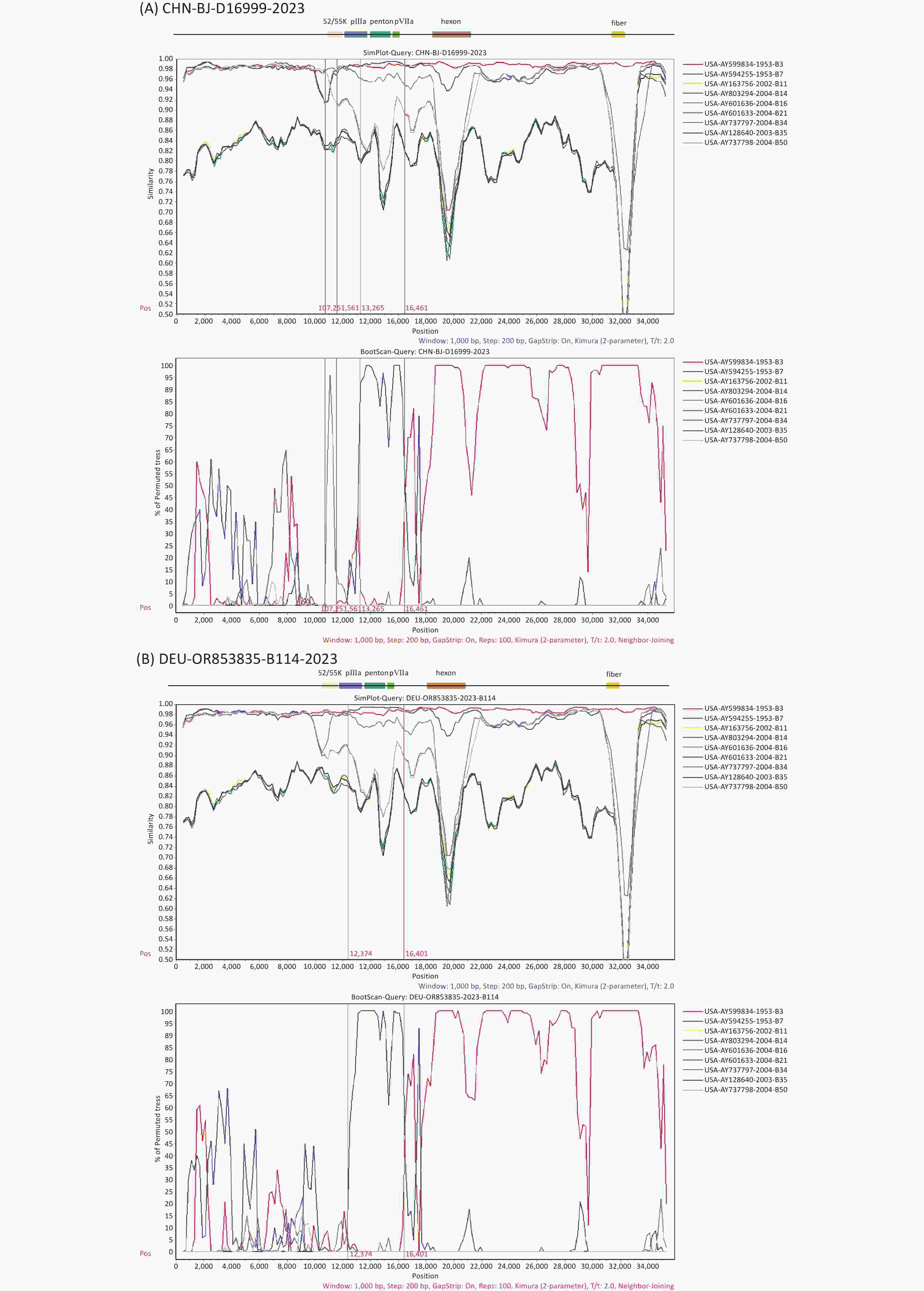
Figure 5. Recombination analysis of the genomic sequences of CHN-BJ-D16999-2023 and HAdV-B114 (GenBank No. OR853835) compared with that of other HAdV-B strains, performed using SimPlot software through similarity plotting and bootscanning with the following parameters: window size of 1,000 nt, step size of 200 nt, the Kimura distance model, and the neighbor-joining tree model. (A) Recombination analysis of the genomic sequences of CHN-BJ-D16999-2023. (B) Recombination analysis of the genomic sequences of HAdV-B114. Gene fragments of protein L1 52/55 kDa (on 10,890–12,026 nt), protein IIIa precursor (on 12,051–13,817 nt), penton base (on 13,905–15,539 nt), and protein VII precursor (on 15,553–16,131 nt) of the prototype strain GB.
-
The coronavirus disease 2019 (COVID-19) pandemic, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in December 2019, has resulted in over 13 million deaths worldwide. To effectively prevent its spread, the strictest nonpharmaceutical interventions (NPIs) were implemented globally, which also influenced the epidemic patterns of other respiratory viruses, including HAdV. In the United States, HAdV prevalence remained low throughout 2020 until early May 2021[30]. In South Korea, the positivity rate of HAdV decreased to 6.1% during the COVID-19 pandemic from 2020 to 2022, then increased to 42.2% in week 34 of 2023, making it the predominant pathogen in the summer of 2023[31]. In Beijing, HAdV-B3 and HAdV-B7 were the predominant HAdV types before NPIs were implemented on January 24, 2020. Following NPIs implementation, the positive rates of HAdV-B3 declined sharply, and the positive rates of HAdV-B7 dropped to zero by 2021. This shift was accompanied by a rise in HAdV-C1, which became the dominant type, marking a transition in the HAdV endemic pattern from species B to species C[32]. In this study, the positivity rate of HAdV infections in pediatric patients with ARIs remained low throughout 2022, under the influence of NPIs. However, after the one-by-one waves of COVID-19, Flu, and RSV epidemics in Beijing in 2023[33,34], the positivity rates of HAdV infections increased from August 2023, peaking in November 2023, with HAdV-B3 as the predominant type. These findings are similar to those reported in several provinces in China, where the prevalence of HAdV species B replaced the circulating species C associated with elevated HAdV activity from October 2023 to August 2024[35]. Similarly, a cohort study by the Johns Hopkins Hospital System reported an increase in HAdV circulation from January to June 2023, with HAdV-B3 emerging as the predominant type[36].
Recombination events are common during HAdV evolution and are often accompanied by base substitutions and insertions/deletions. Homologous recombination involving the penton base, hexon, and fiber genes is considered a major mechanism of HAdV evolution. HAdV recombination events lead to changes in the structure and surface properties of the capsid, ultimately enabling the virus to alter its tissue tropism and evade host immune responses. For example, HAdV-D56—a recombinant virus derived from types causing epidemic keratoconjunctivitis (EKC)—was isolated in France and implicated in the death of a newborn with severe respiratory infections[37]. Recombination is also a critical consideration in adenovirus-based vaccine development, as the potential for homologous recombination between vaccine strains and wild-type adenoviruses has been identified[38]. The detection of HAdV recombination requires a public health monitoring system with high sensitivity and accuracy. Traditional surveillance methods may struggle to promptly identify emerging recombinant HAdVs, highlighting the need for bioinformatics tools to verify recombination events. In this study, four genetic recombination patterns of HAdV-C were identified. Three patterns—Px1ps1H2F2, Px1ps3H5F5, and Px2H5F5— were previously reported in Beijing[39], while a fourth pattern, Px3H1F1, was reported between 2010 and 2014 in Shanxi, China[23]. Although HAdV-C infections usually cause mild disease, more evidence is required to assess the clinical effects of these recombination events.
This study identified a novel recombination pattern in HAdV-B, designated P7H3F3. The genomic sequence of D16999 contained the backbone sequence of HAdV-B3, with gene fragments encoding a partial protein IIIa precursor, penton base, and protein VII precursor derived from HAdV-B7, and a gene fragment encoding the L1 52/55 kD protein derived from HAdV-B16. The recombinant breakpoints within the partial protein IIIa precursor, penton base, and protein VII precursor were identical to those of the prototype strain B114 (GenBank No. OR853835) reported in 2018 in Beijing, China[40]. However, the recombination event in the gene fragment encoding the L1 52/55 kD protein was unique and has not been previously reported for HAdV-B114. The classification of HAdV genotypes by the Human Adenovirus Working Group (http://hadvwg.gmu.edu/) relies on the penton base, hexon, and fiber genes. Furthermore, recombination events that occurred in other gene fragments were considered. For example, HAdV-7d was named after a recombination event in the gene fragment encoding the L1 52/55 kD protein originating from HAdV-B16, which is different from the prototype strain HAdV-B7, also known as HAdV-B7p[15]. Therefore, we propose that the prototype strain HAdV-B114 (GenBank No. OR853835) be designated HAdV-B114p and that the P7H3F3 strain identified in this study be designated HAdV-B114a.
This study had several limitations. First, it was a single-center study, broader nationwide data collection or continuous single-center monitoring is required to accurately determine the prevalence of HAdV infection in children. Second, recombination events in HAdVs are common and can alter their tropism and immunogenicity. In this study, only six patients tested positive for P7H3F3. Additional data are required to elucidate the clinical characteristics of P7H3F3 infection in children.
In conclusion, the epidemic patterns of HAdV in pediatric patients with ARIs in Beijing, China, were influenced not only by NPIs but also by SARS-CoV-2 outbreaks and concurrent epidemics of other respiratory viruses, including Flu and RSV. Notably, a new recombinant genotype, HAdV-B114a, was identified, with sequences similar to those of HAdV-B114, derived from HAdV-B3, -B7, and -B16.
Molecular Characterization of New Recombinant Human Adenoviruses Detected in Children with Acute Respiratory Tract Infections in Beijing, China, 2022–2023
doi: 10.3967/bes2025.105
- Received Date: 2025-01-15
- Accepted Date: 2025-06-30
-
Key words:
- Children /
- Human adenovirus /
- Acute respiratory tract infections /
- Recombination.
Abstract:
No financial or non-financial benefits were received or will be received from any party directly or indirectly related to this study.
This study was approved by the Ethics Committee of the Capital Institute of Pediatrics (approval number: SHERLL2022015).
&These authors contributed equally to this work.
| Citation: | Yinan Guo, Ri De, Fangming Wang, Zhenzhi Han, Liying Liu, Yu Sun, Yao Yao, Xiaolin Ma, Shuang Liu, Chunmei Zhu, Dong Qu, Linqing Zhao. Molecular Characterization of New Recombinant Human Adenoviruses Detected in Children with Acute Respiratory Tract Infections in Beijing, China, 2022–2023[J]. Biomedical and Environmental Sciences. doi: 10.3967/bes2025.105

|

 Quick Links
Quick Links
 DownLoad:
DownLoad: