
-
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the etiological agent for the global coronavirus disease-19 (COVID-19) pandemic[1], mainly affects the respiratory system in humans but can also cause multi-system damage[2-5]. Neurological symptoms such as loss of taste and smell, headaches, dizziness, cognitive decline, seizures, and impaired consciousness are common in patients with COVID-19[6-8]; however, the underlying mechanisms remain unclear.
Human angiotensin-converting enzyme 2 (hACE2) acts as the major host receptor for SARS-CoV-2 and binds to the viral spike protein[9]. K18-hACE2 transgenic mice expressing hACE2 driven by the K18 promoter were specifically designed to study the pathogenesis of SARS-CoV-2 infection[10]. hACE2 expressed in K18-hACE2 mice is used widely in SARS-CoV-2-related studies, as its binding affinity is higher than that of the receptor in wild-type mice[10]. Studies have reported that infection with SARS-CoV-2 in 6-week-old K18-hACE2 mice results in severe neurological disease, which closely mimics the severe form of human COVID-19[11].
With the COVID-19 pandemic, several variants of concern have emerged[12,13]. The beta variant, also known as B.1.351, was first identified in South Africa in December 2020 and has spread globally since then. The spike (S) protein of this variant contains at least nine amino acid mutations, including N501Y and E484K, in the receptor-binding domain, which enhance infectivity and contribute to immune evasion. Moreover, the affinity between the SARS-CoV-2 S protein of the beta variant and mouse ACE2 was high, enabling infection not only in K18-hACE2 mice but also directly in wild-type C57BL/6 mice, which closely resembled the mild form of human COVID-19[14,15]. Currently, data regarding the post-acute neurological sequelae and mechanisms underlying SARS-CoV-2 beta variant infection are limited.
With the advancement of gene sequencing technology, transcriptomic sequencing has provided a new avenue for studying the molecular pathogenesis of diseases. In this study, we used the SARS-CoV-2 beta variant strain to infect K18-hACE2 and C57BL/6 mice and applied transcriptomic analysis to investigate the main pathways and potential key factors contributing to the pathological damage caused by the beta variant strain in different brain tissues. This study will improve our understanding of the mechanisms underlying the pathogenicity and neurological damage caused by the SARS-CoV-2 beta variant and the therapeutic interventions required to alleviate neurological dysfunction observed in COVID-19.
-
The SARS-CoV-2 beta variant strain, 19nCoV-CDC-Tan-2021GD01, was isolated and preserved at the Emergency Response Center of the National Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention. Vero cells (CCL-81) were cultured in minimum Eagle's medium supplemented with 10% fetal bovine serum at 37 °C under 5% CO2. The titer was determined in Vero cells by 50% tissue culture infectious dose (TCID50) method. All the infection experiments were conducted in the Animal Biosafety Level 3 (ABSL-3) laboratory of the National Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention.
-
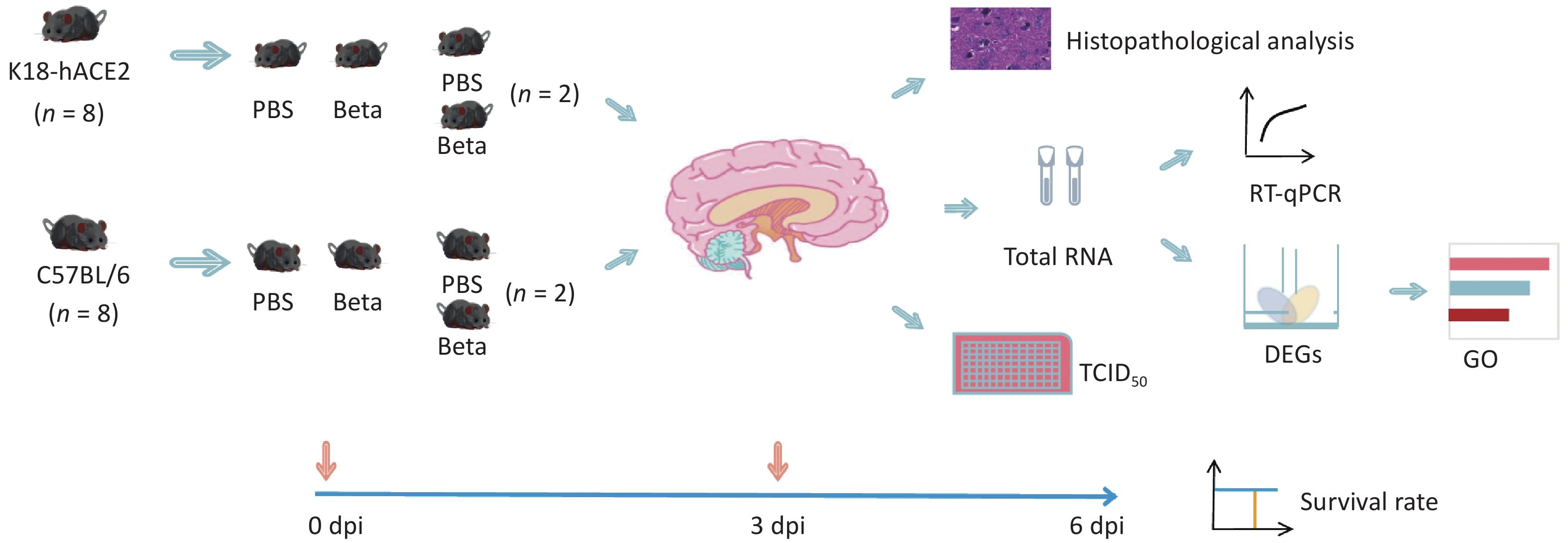
Six-to-eight-week-old female K18-hACE2 and C57BL/6 mice were purchased from Sibfu Biotechnology Co. Ltd. (Beijing, China). All animal experiments conformed to the standards for use and care of laboratory animals. All animal experiments were conducted in the ABSL-3 laboratory of the National Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, and approved by the Animal Ethics and Usage Committee of the National Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention (ethics permit no. 20220106001). To induce anesthesia, sodium pentobarbital was administered at a dose of 60 mg/kg, the mice were infected via the intranasal route with 5 × 104 TCID50 SARS-CoV-2 beta variant in 50 μL per mouse, with eight mice per group and a total of four groups. Changes in the body weight and survival rates of mice were monitored continuously until 6 days post-infection (dpi). Viral replication, pathological changes, and gene expression alterations in the brain tissue of two randomly selected mice of each group were analyzed on the 3 dpi (Figure 1). Total RNA was extracted from the brain tissues for transcriptomic analysis.
Figure 1. Experimental workflow. DEGs, differentially expressed genes; GO, gene ontology; RT-qPCR, reverse transcription-quantitative polymerase chain reaction; PBS, Phosphate buffered saline; TCID50, 50% tissue culture infectious dose.
For SARS-CoV-2 RNA detection in mice tissues using reverse transcription-quantitative polymerase chain reaction (RT-qPCR), the supernatant from centrifugation of K18-hACE2 and C57BL/6 mouse brain tissues at 4000 rpm, 4 °C for 5 min was collected. Nucleic acid was extracted using a Xi'an Tianlong automated nucleic acid extractor, and fluorescence quantitative PCR detection was performed using the AgPath-IDTM one-step RT-PCR premix (Thermo Fisher, China). The primer and probe sequences were as follows: SARS-CoV-2 ORF1ab (forward: 5'-CCCTGTGGGTTTTACACTTAA-3'; reverse: 5'-ACGATTGCATCAGCTGA-3'; Probe: FAM-CCGTCTGCGGTATGTGGAAAGGTTATGG-BHQ1).
For infectious SARS-CoV-2 titration, the brain tissue homogenate was serially diluted 10-fold from 10-2 to 10-5, resulting in four dilution levels. Subsequently, 100 μL of the diluted brain tissue suspension were inoculated into a 96-well plate containing cultured Vero cells. Each dilution was replicated in eight wells, and normal cells were used as controls. Cytopathic effects were observed daily, and the TCID50 was calculated using the Reed–Muench formula on the fourth day after inoculation. Hematoxylin and eosin staining was used for the pathological evaluation of lung and brain tissues.
-
Total RNA was extracted from the brain tissues of K18-hACE2 and C57BL/6 mice using the TRIzol reagent (Thermo Fisher)[16]. The concentration and purity of the RNA were determined using a Nanodrop (Thermo Fisher). RNA integrity was assessed using agarose gel electrophoresis. The RNA integrity number (RIN) was measured using an Agilent 5400 instrument, with a minimum RIN value of eight meeting the library construction criteria. Total RNA was sent to Novogene (Beijing, China), where transcriptome library construction and deep sequencing were performed using the Illumina Nova PE150 platform. The reference genome and annotation files were downloaded from the National Center for Biotechnology Information. The reference genome (mm10) index was constructed using HISAT2 (v2.0.5), and the clean paired-end reads were aligned to the reference genome using HISAT2[17]. Finally, feature counts were used to count each transcript[18]. Differential expression analysis was performed using DESeq2[19], with the criteria of |log2(fold change)| > 1 and false discovery rate < 0.05 to identify differentially expressed genes (DEGs). Subsequently, the DEGs were subjected to GO functional enrichment analysis. The RNA-seq data relevant to this study were deposited at the National Genomics Data Center with the GSA accession number, CRA014776.
-
The HiScript II One Step qRT-PCR SYBR Green kit (Vazyme, Nanjing, China) was used to amplify the RNA samples. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as the internal reference. The primer sequences were provided in the Supplementary Table S1. The 2^-ΔΔCt method (Livak method) was used for calculating gene expression. Each measurement was performed thrice independently.
-
Proteins extracted from mouse brains were separated on Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis and transferred onto poly(vinylidene fluoride) membranes. Subsequently, the membranes were incubated with primary antibodies targeting specific proteins: ZBP1 (1:1,000; 13285-1-AP, Proteintech), pMLKL (1:1,000; D6E3G, Cell Signaling Technology), CASP4/11 (1:1,000; 14340S, Cell Signaling Technology), GSDMD (1:1,000; 39754S, Cell Signaling Technology), GSDMD-NT (1:1,000; 10137S, Cell Signaling Technology), LCN2 (1:1,000; 26991-1-AP, Proteintech), and β-actin (1:1,000; 4967S, Cell Signaling Technology). Following primary antibody incubation, the membranes were treated with a secondary antibody conjugated with goat anti-rabbit IgG-HRP (1:10,000; ZB-2301, ZSGB-BIO). The protein bands were visualized by scanning the membranes using the Amersham™ ImageQuant™ 800.
-
Multiple group comparisons were performed using one-way analysis of variance (ANOVA). Two-group comparisons were performed using student's t-test. Differences were considered statistically significant if P-value < 0.05. Significance levels are: *P < 0.05; **P < 0.01; ***P < 0.001. Graphs were generated using the GraphPad Prism 9 software, and image processing was performed using Adobe Illustrator.
-
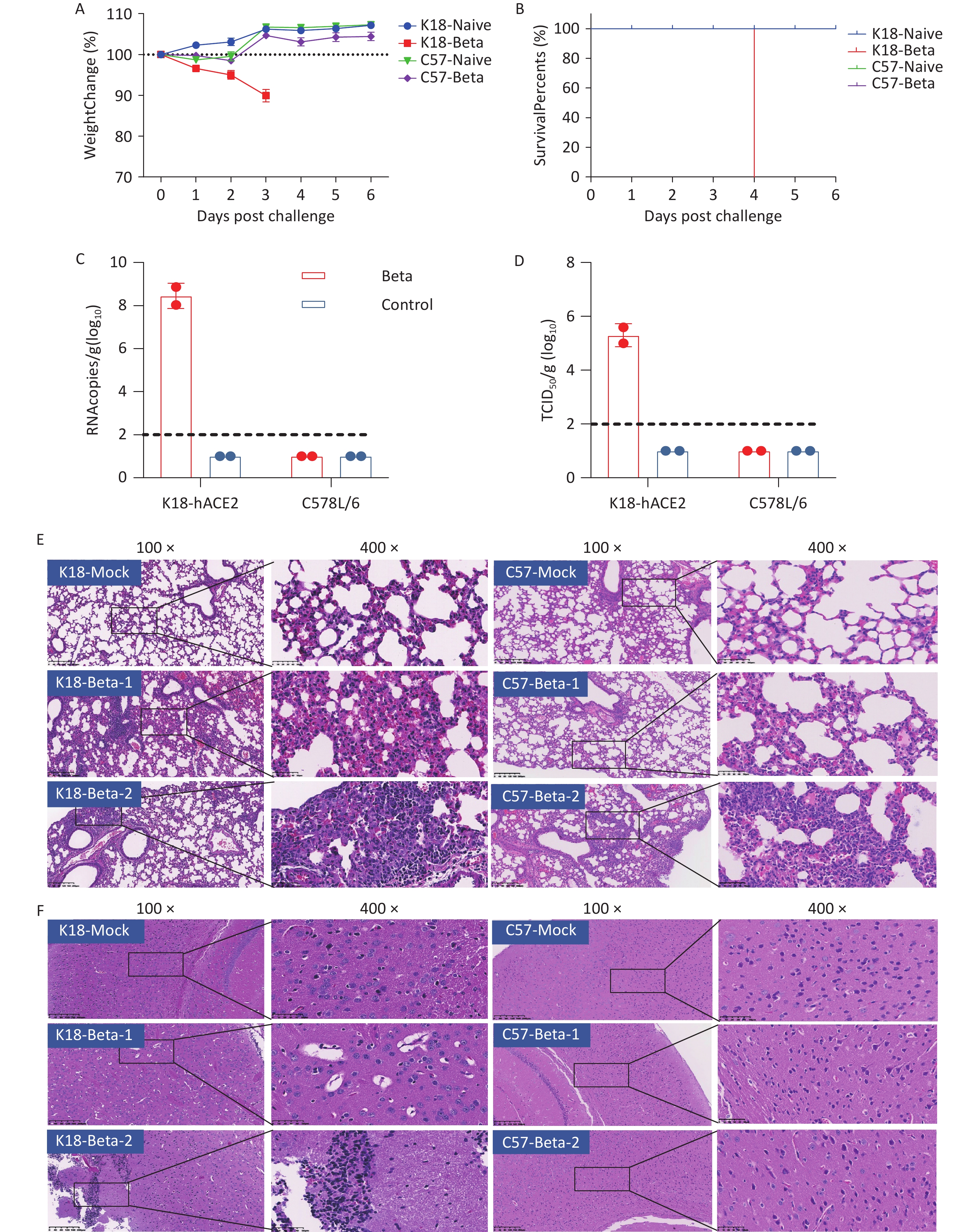
To simulate the clinical symptoms or pathogenic response in patients with COVID-19, either K18-hACE2 or C57BL/6 mice were infected with 5 × 104 TCID50 of the SARS-CoV-2 beta variant via the intranasal route. Changes in the body weight and survival status of the infected and control mice were recorded daily. Only K18-hACE2 mice with SARS-CoV-2 beta infection exhibited body weight loss, with a reduction of approximately 10% on the 3rd dpi (Figure 2A). Survival rate assessments indicated that the infected K18-hACE2 mice died on the 4th dpi owing to the lethal clinical symptoms following the beta variant strain infection (Figure 2B).
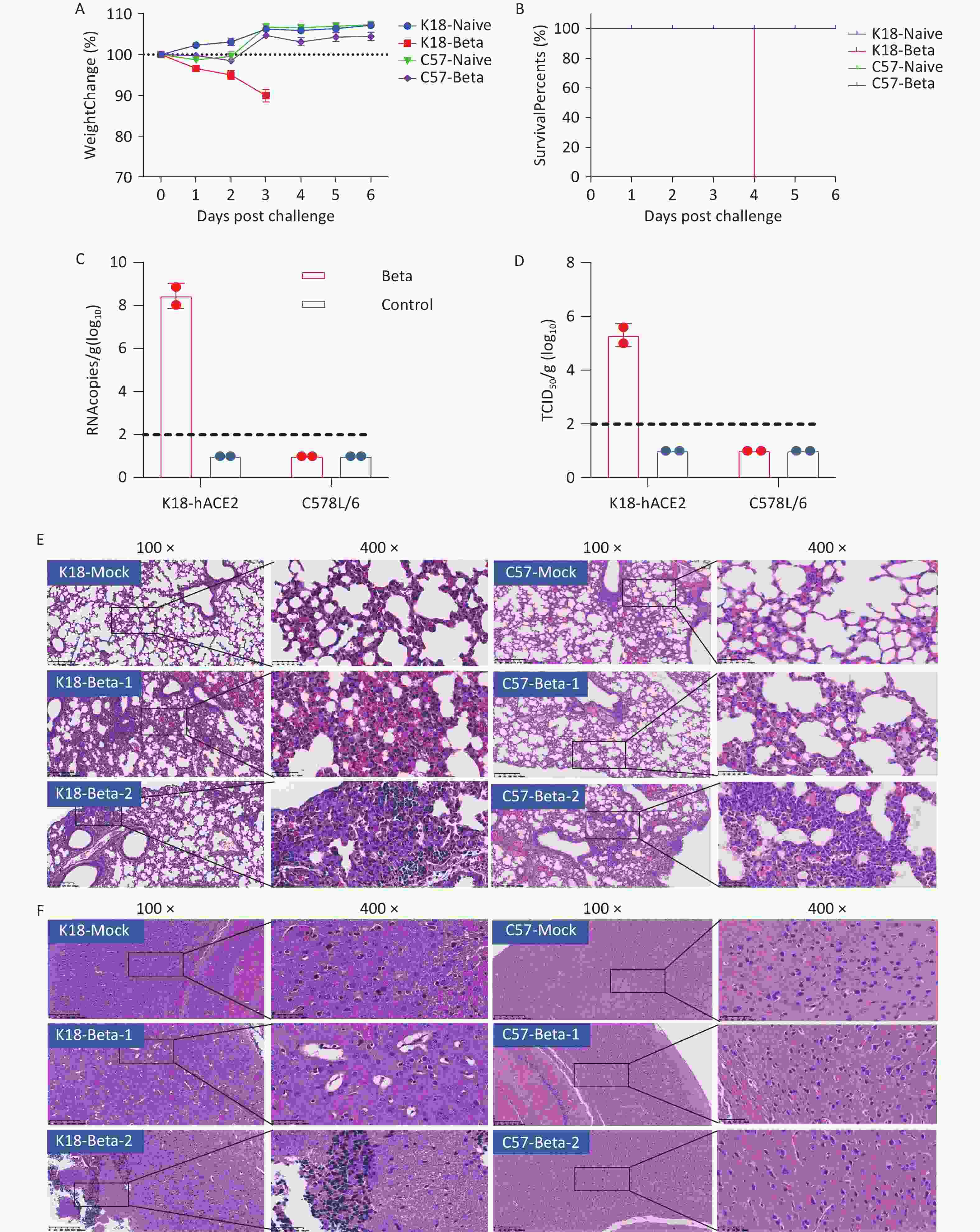
Figure 2. Pathological damage in the lung and brain tissue of mice infected with the SARS-CoV-2 beta variant. (A) Changes in the body weight of K18-hACE2 and C57BL/6 mice after beta variant infection. (B) Survival rate of K18-hACE2 and C57BL/6 mice after beta variant infection. (C) Copy of SARS-CoV-2 RNA in the lungs and brains of mice detected using RT-PCR. (D) Infectious SARS-CoV-2 titration in the lungs and brains of mice. (E) Pathological sections of the lung tissue of mice. (F) Pathological sections of the brain tissue of mice. SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
To evaluate viral replication and pathological damage in K18-hACE2 and C57BL/6 mice following infection with the beta variant, lung and brain tissues were collected from mice 3 days after infection (two mice per group). Viral RNA was detected in the brain tissue with an average copy number of 108.4 copies/g, and the viral titer was 105.3 TCID50/g in the infected K18-hACE2 mice. However, neither viral RNA nor infectious virus were detected in the brain tissue of infected C57BL/6 mice (Figure 2C and 2D). Both strains of mice infected with the beta variant exhibited pathological changes in lung tissues, including the presence of necrotic alveolar epithelial cells, lymphocytes, and macrophages. Degeneration and necrosis were observed in bronchial and bronchiolar epithelial cells. These findings indicated that the beta variant was capable of infecting both K18-hACE2 and C57BL/6 mice, causing typical pathological changes in the lung tissue (Figure 2E). However, only the K18-hACE2 mice exhibited pathological changes in the brain tissue, whereas the C57BL/6 group did not show any signs of pathological damage (Figure 2F). The major pathological changes in the brain tissue of the infected K18-hACE2 mice involved the appearance of focal neuronal degeneration, necrosis, and glial cell proliferation, which led to the formation of glial nodules and hypertrophy of certain astrocytes (Figure 2F). Proliferating activated microglial cells infiltrated the degenerating and necrotic neurons, indicative of typical encephalitic pathology.
In summary, the beta variant of SARS-CoV-2 infected the lung tissues of K18-hACE2 and C57BL/6 mice but caused pathological damage only in the brain tissue of K18-hACE2 mice, ultimately resulting in mortality.
-
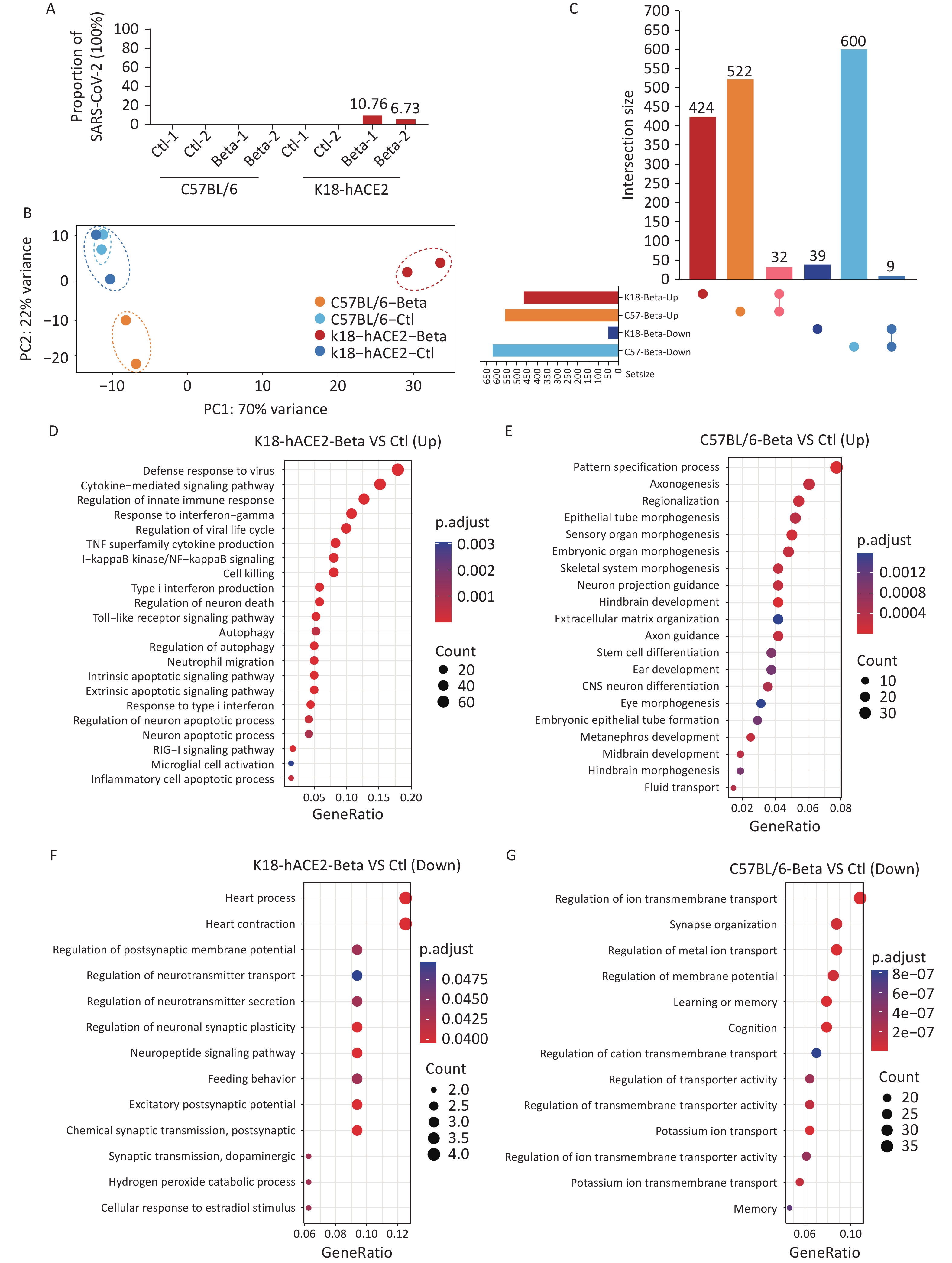
To investigate the potential molecular mechanisms underlying the brain tissue damage caused by the beta variant infection in K18-hACE2 mice, transcriptomic analysis was performed using brain tissue samples. Sequencing reads corresponding to the SARS-CoV-2 genome were not identified in the brain tissues of C57BL/6 mice. However, in the brain samples from two K18-hACE2 mice, the percentages of SARS-CoV-2 sequencing reads out of the total sequencing reads were 10.76% and 6.73%, respectively (Figure 3A). Compared with the control, the infected K18-hACE2 and C57BL/6 mice exhibited distinct transcriptional features (Figure 3B). Although no significant pathological changes were observed in the brain tissue of C57BL/6 mice infected with the beta variant, significant alterations were observed at the transcriptional level. In total, 1,163 DEGs were identified in the infected C57BL/6 mice, with 554 upregulated and 609 downregulated genes. In the infected K18-hACE2 mice, 504 DEGs were screened, including 456 upregulated and 48 downregulated genes. Notably, most of the DEGs responsible for transcriptional changes in K18-hACE2 and C57BL/6 mouse brain tissues were unique to those groups, with an overlap of only 32 upregulated genes and 9 downregulated genes (Figure 3C). These commonly upregulated genes mainly participated in response to interferons, regulation of immunity, and hydrolytic enzyme activity, whereas the commonly downregulated genes were involved in regulation of system activity.
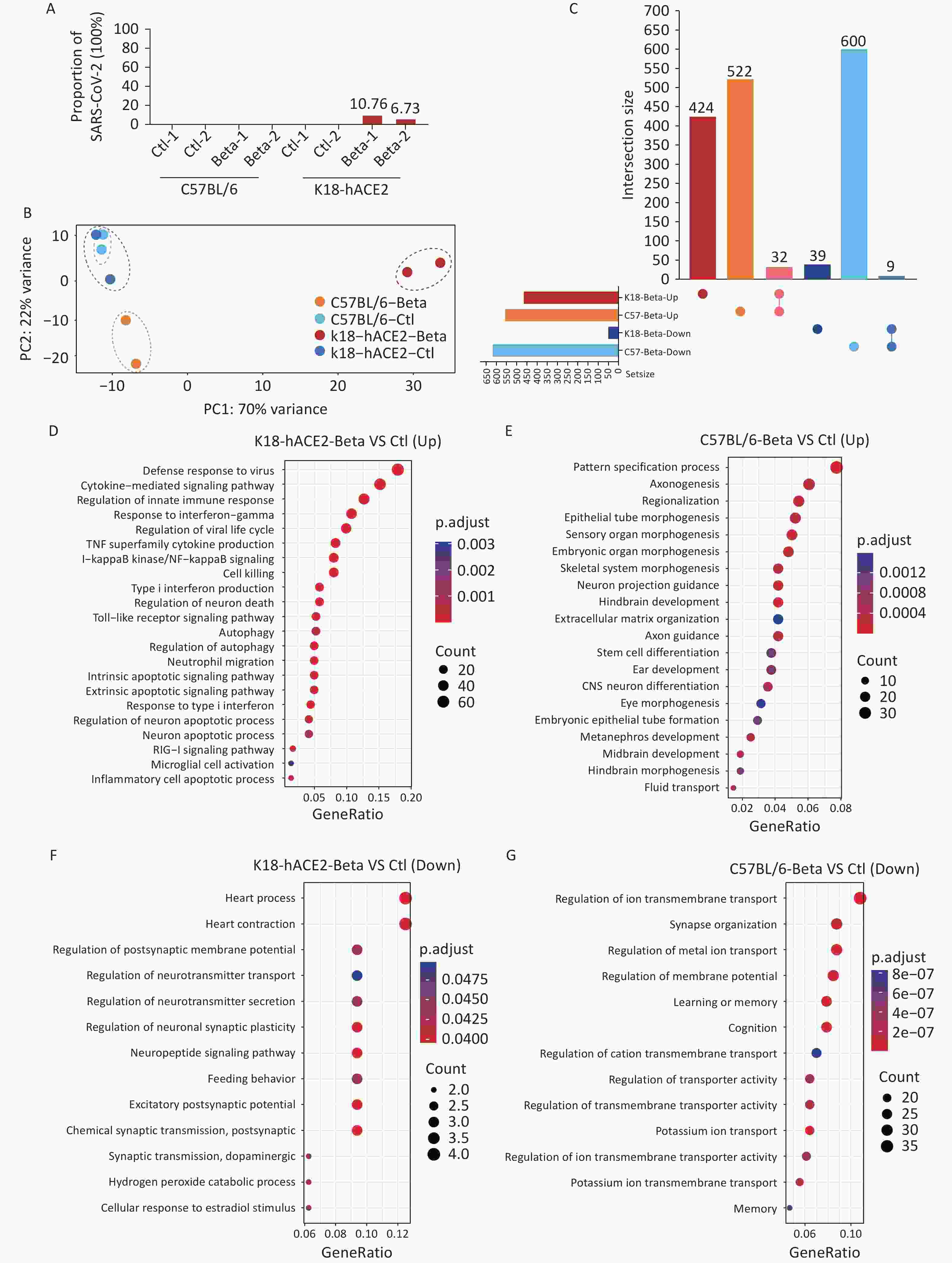
Figure 3. Transcriptomic analysis of the brain tissues of K18-hACE2 and C57BL/6 mice infected with the SARS-CoV-2 beta variant. (A) Proportion of the SARS-CoV-2 genome. (B) Principal component analysis. (C) Upset plot of upregulated and downregulated genes in the brain tissues of K18-hACE2 and C57BL/6 mice after beta variant infection. (D-E) Enriched biological processes of upregulated genes in the brain tissues of K18-hACE2 and C57BL/6 mice after beta variant infection. (F-G) Enriched biological processes of downregulated genes in the brain tissues of K18-hACE2 and C57BL/6 mice after beta variant infection. SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
GO enrichment analysis was performed to investigate the differential gene functions elicited by the beta variant infection in the brain tissues of the infected K18-hACE2 and C57BL/6 mice. Infection with the beta variant activated pathways associated with neural differentiation and neuronal projections in the brain tissue of C57BL/6 mice (Figure 3E). Conversely, it inhibited pathways related to ion transmembrane transport, synaptic organization, and learning and memory regulation (Figure 3G). In K18-hACE2 mice, genes related to pathways linked to the innate immune response, cytokine-mediated signaling, type I interferon signaling, microglial cell activation, neuronal apoptosis, and autophagy were significantly upregulated (Figure 3D). In contrast, the genes downregulated in K18-hACE2 mice were enriched in pathways involved in the regulation of neurotransmitter secretion and neuronal synaptic plasticity (Figure 3F). These results suggested that although the beta variant did not induce pathological changes in the brain tissue of C57BL/6 mice, it induced molecular changes at the transcriptional level in the host. In contrast, invasion of the beta variant into the K18-hACE2 mouse brain tissue possibly activated the innate immune response and inflammatory reactions, leading to pathological damage.
-
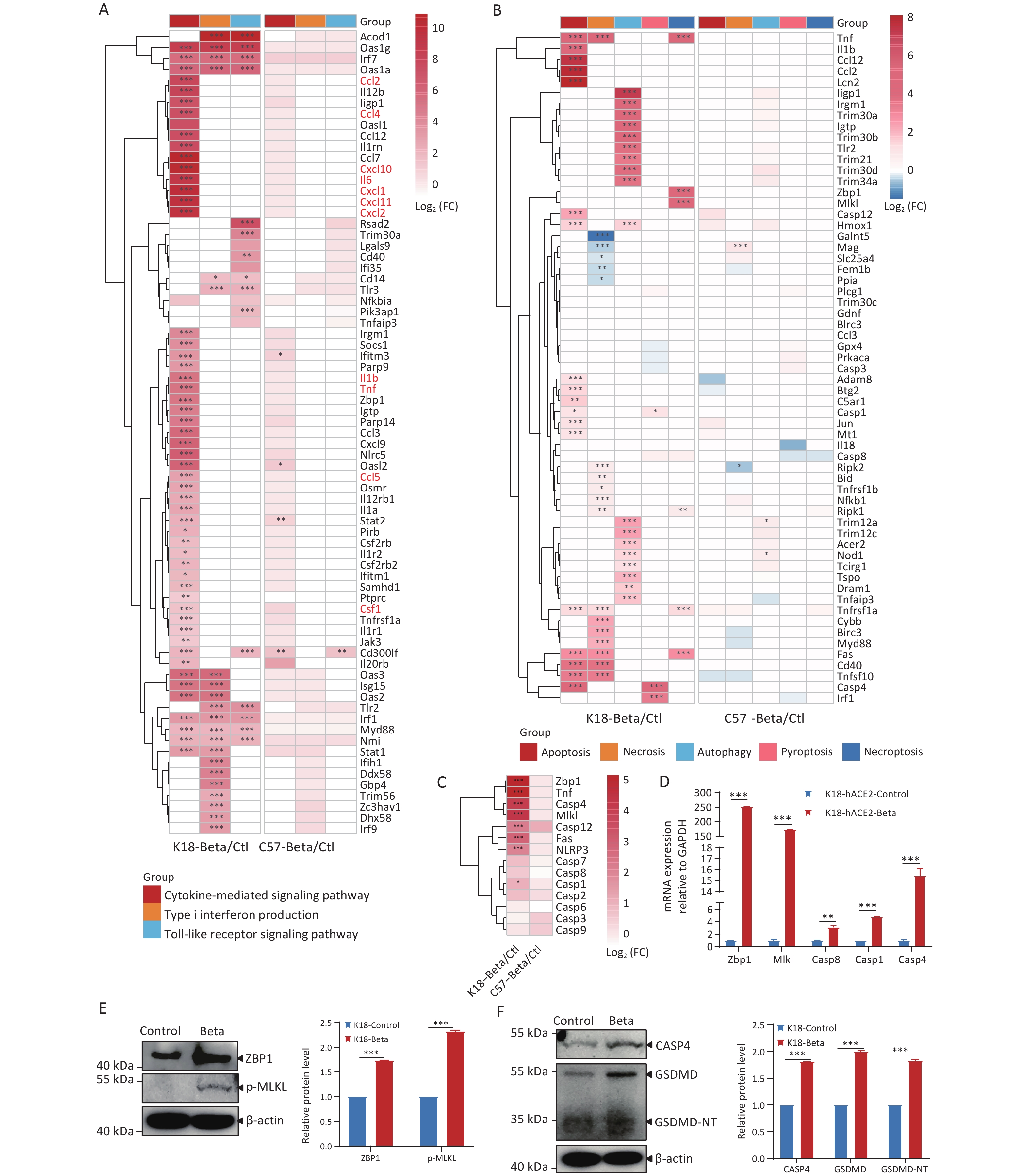
To investigate the key cytokines involved in brain inflammation induced by the infection of K18-hACE2 mice with the beta variant, we performed an in-depth analysis of the DEGs related to inflammatory response pathways triggered after viral infection in the host (Figure 4A). The brain tissue of K18-hACE2 mice exhibited elevated expression of a series of pro-inflammatory cytokines (interleukin [IL]-1β, IL-6, and tumor necrosis factor [TNF]) and chemokines (chemokine ligand [CCL]-2, CCL-4, CXC motif chemokine ligand [CXCL]-1, CXCL-10, CXCL-11, and CXCL-12). Additionally, antiviral genes, including interferon-stimulated gene 15, 2'-5'-oligoadenylate synthetase 2, tripartite motif-containing protein 30a, and zinc finger CCCH-type antiviral protein 1 were upregulated. In contrast, genes related to these pathways were not significantly expressed in the brain tissues of C57BL/6 mice following viral infection. These results suggested that the activation of type I interferon and Toll-like receptor pathways, as well as the upregulation of pro-inflammatory and chemotactic factors in the cytokine-mediated signaling pathway, may be crucial in triggering brain inflammation following infection of K18-hACE2 mice with the beta variant.
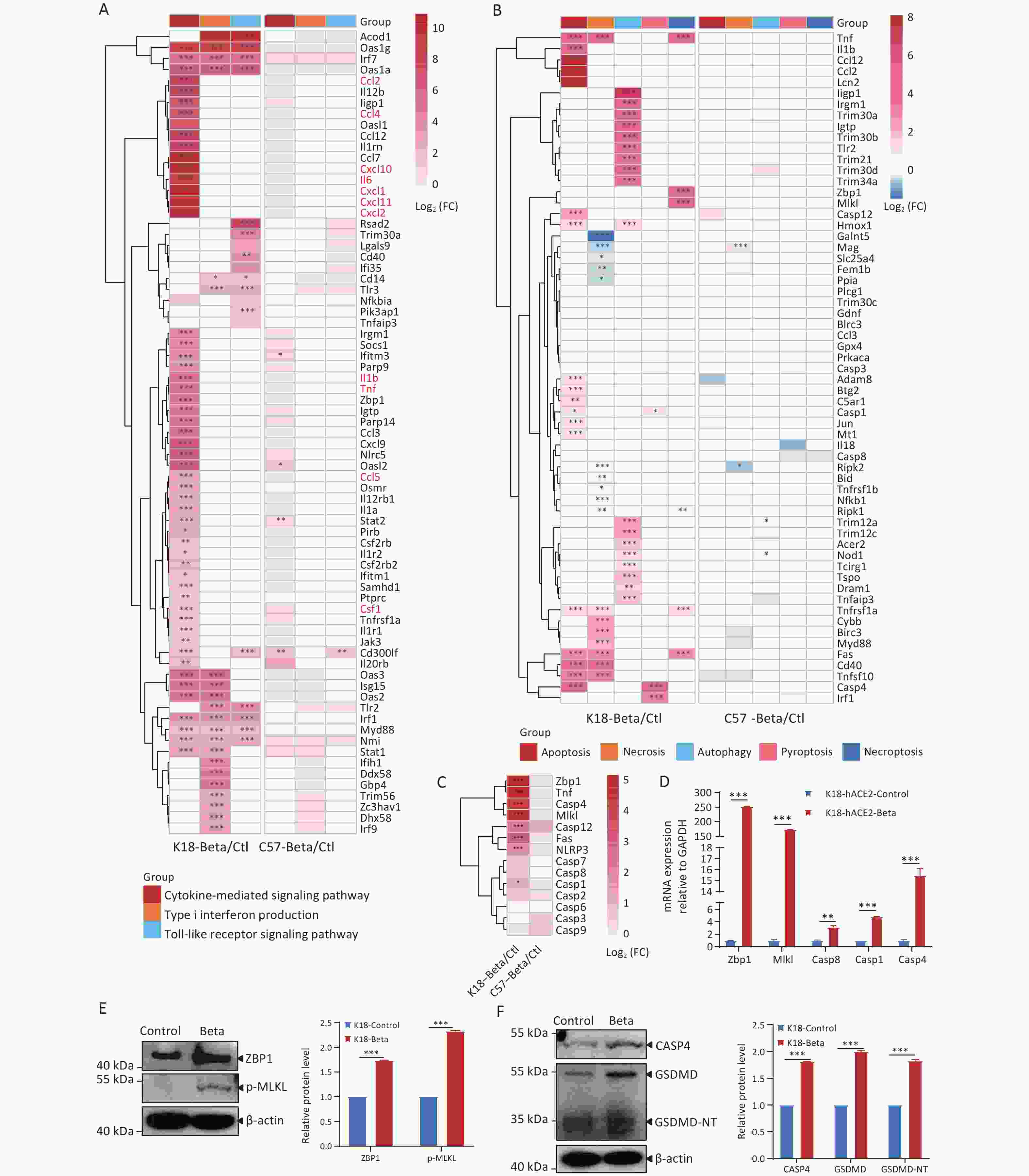
Figure 4. Comparison of differentially expressed genes (DEGs) in the brain tissues of K18-hACE2 and C57BL/6 mice infected with the beta variant of SARS-CoV-2.
In addition, neuropathological analysis of the K18-hACE2 mouse brain tissue revealed that infection with the beta variant led to neuronal death. To investigate the molecular mechanisms underlying neuronal death, we analyzed the DEGs involved in processes such as apoptosis, necrosis, autophagy, pyroptosis, and necroptosis (Figure 4B). Following the infection of K18-hACE2 mice with the beta variant, several key genes were activated, including those involved in apoptosis (Tnf, Fas, Casp1, Casp4, and Casp12) and autophagy (Iigp1, Irgm1, Trim30a, Igtp, Trim30b, and Tlr2). Furthermore, the upregulation of genes involved in the ZBP1/MLKL-regulated necroptosis pathway, such as Zbp1, Mlkl, and Casp8, was also observed (Figure 4B). A member of the TNF superfamily, Tnf, and its receptor, Fas, recruit downstream effector caspases upon receptor activation, leading to apoptosis[20,21]. Analysis of caspase-related genes revealed high expression of Casp4, a key factor in the pyroptosis pathway (Figure 4C), as well as upregulation of the inflammasome-related genes, Casp1 and Il1b (Figure 4A and 4C). The expression trends of Zbp1, Mlkl, Casp1, Casp4, and Casp8 were consistent with the results of RNA-seq using RT-qPCR (Figure 4D). The activation of necroptotic and pyroptotic pathways was further validated through immunoblotting, demonstrating elevated levels of ZBP1 and phosphorylated MLKL proteins (Figure 4E), as well as increased expression levels of CASP4, GSDMD, and GSDMD-NT proteins (Figure 4F). Following infection with the beta variant, multiple cytokines were activated in K18-hACE2 mice, resulting in inflammatory responses. The infection also led to the upregulation of factors including Zbp1, Mlkl, and Casp4, which contributed to cell apoptosis and pyroptosis.
-
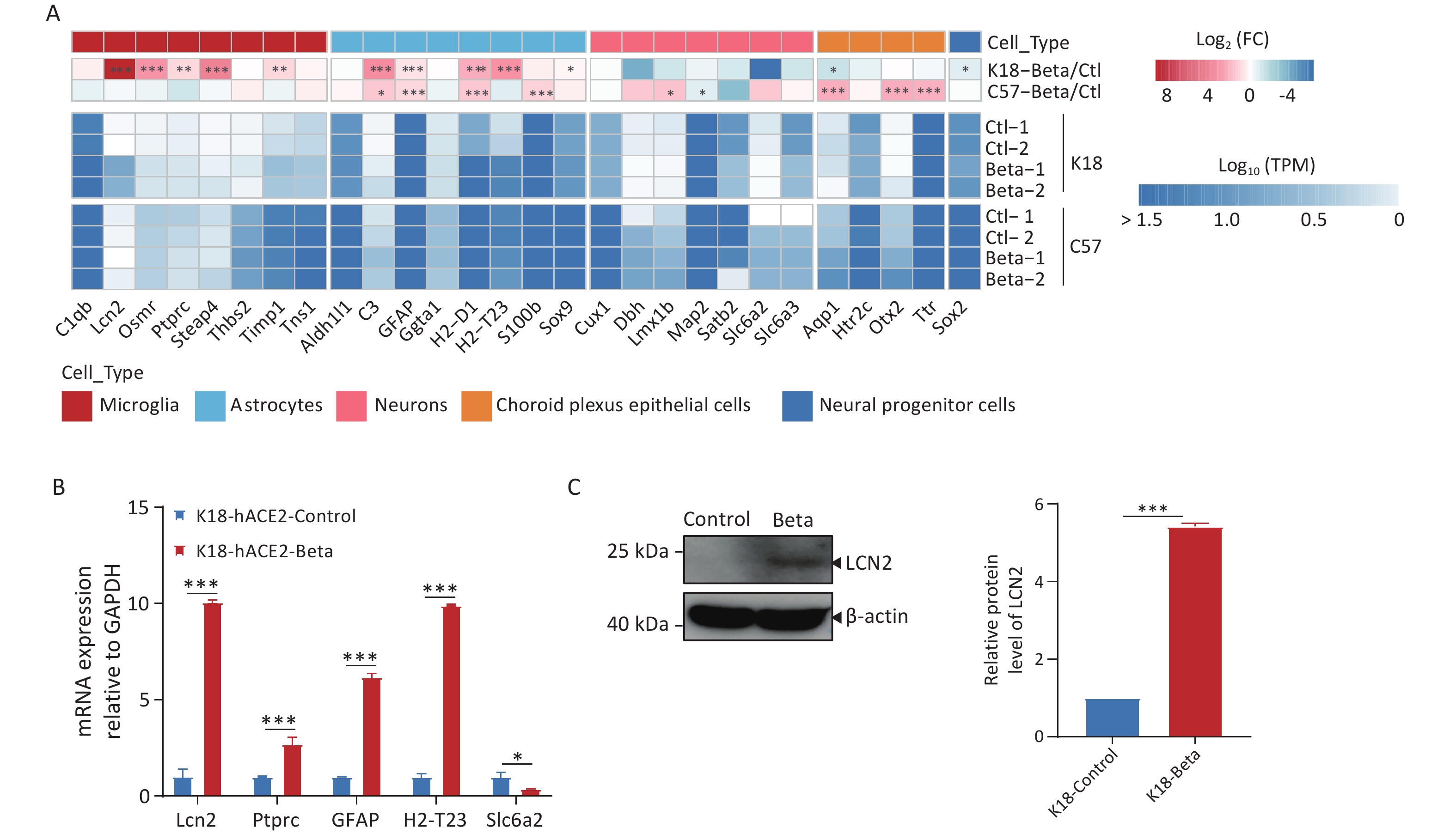
SARS-CoV-2 can infect various types of neuronal cells, including olfactory, sensory, cortical, and dopaminergic cells, as well as epithelial cells of the choroid plexus[22]. Infection of K18-hACE2 mice with the beta variant increased microglial cell proliferation, astrocyte hypertrophy, and neuronal death (Figure 2F). We conducted a molecular-level analysis of neuronal cell markers to identify the mechanisms underlying the pathological changes in the tissue (Figure 5A). Following the infection of C57BL/6 mice with the beta variant, the expression of most marker genes did not change significantly in the brain tissue, indicating the maintenance of normal brain activity. However, in the infected K18-hACE2 mice, markers associated with microglial cells (Lcn2, Ptprc, and Osmr) and astrocytes (C3, H2-t23, H2-d1, and Gfap) were significantly upregulated, suggesting that viral infection may activate microglial and astrocytic cells, which have been reported to be involved in the apoptotic pathway and exert direct neurotoxic effects, or to indirectly induce brain damage by reinforcing neuroinflammation, disrupting the blood-brain barrier (BBB), and enhancing inflammatory infiltration[23]. Downregulation of the neuronal cell markers, Dbh, Slc6a2, and Satb2, was indicative of neuronal damage. We performed RT-qPCR analysis to validate the expression of genes associated with neuronal cell markers (Figure 5B), the results of which agreed with those of RNA-seq. Simultaneously, immunoblotting was employed to validate the upregulation of LCN2 protein expression (Figure 5C).
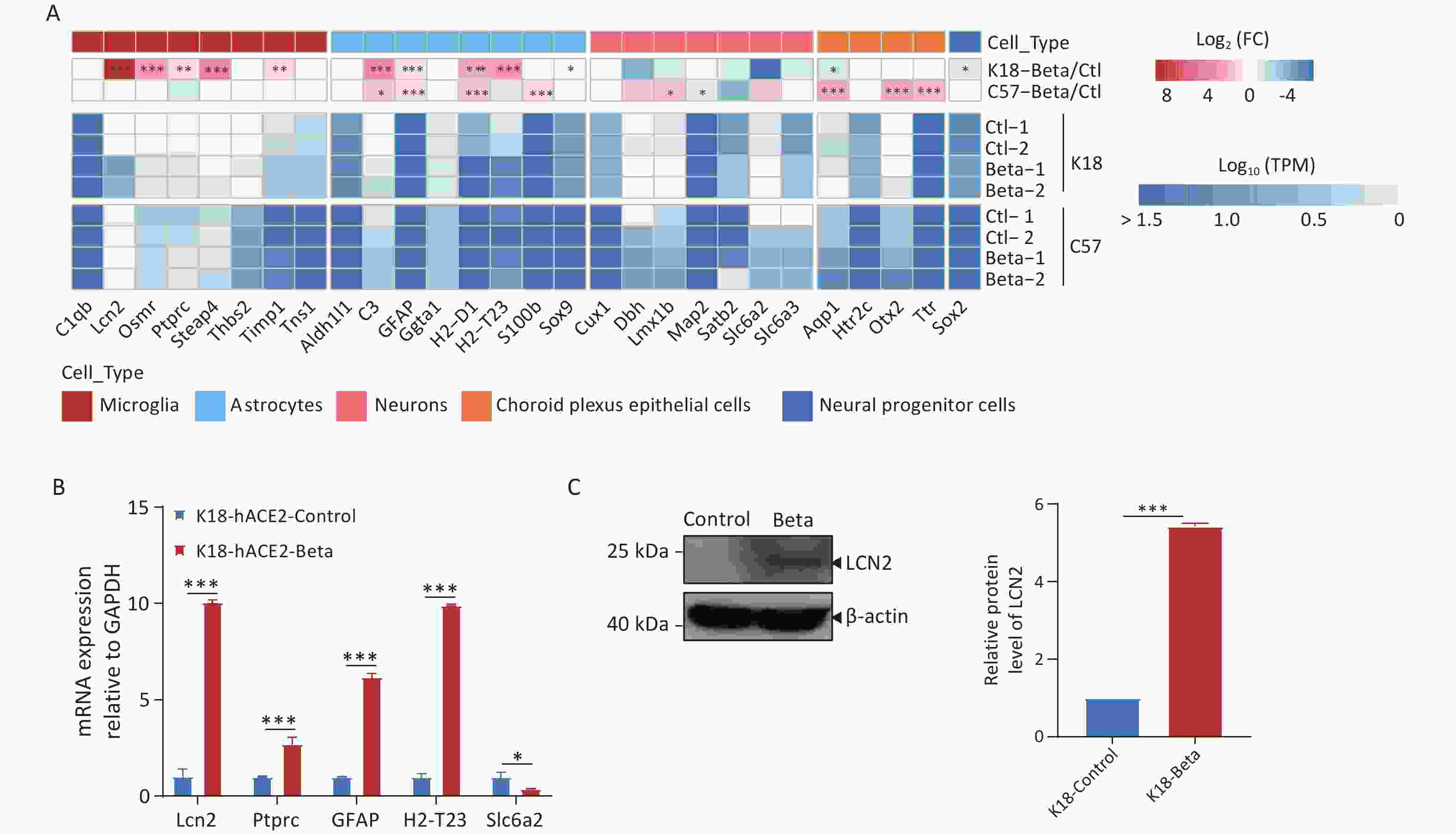
Figure 5. Changes in neuronal cell markers in the brain tissues of K18-hACE2 and C57BL/6 mice infected with the beta variant of SARS-CoV-2. (A) Heatmap showing the fold change and expression levels (TPM) of neuronal cell markers. The asterisks indicate statistically significant dysregulated genes (*FDR < 0.05; **FDR < 0.01; ***FDR < 0.001). (B) RT-qPCR analysis of neuronal cell markers in K18-hACE2 mice infected with the beta variant. *P < 0.05; **P < 0.01; ***P < 0.001. (C) Protein extracts from control and SARS-CoV-2 infected brain tissues were subjected to immunoblotting using LCN2, or β-actin antibodies. FDR, false discovery rate; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2
Based on the pathological findings and the results of transcriptome analysis in this study, we hypothesized that the beta variant primarily infected neuronal cells in mice, simultaneously activating microglial and astrocytic cells to secrete factors such as LCN2, thereby triggering multiple programmed cell death pathways, ultimately leading to neuronal damage and death.
-
COVID-19 primarily affects the respiratory system, which can cause potentially fatal systemic complications, often involving the central nervous system. Whole-body expression of hACE2 in K18-hACE2 mice facilitates the spread of SARS-CoV-2 to multiple tissues, enhancing viral infectivity. To improve our understanding regarding the impact of viral infection on the nervous system, K18-hACE2 and C57BL/6 mice infected with the beta variant strain were used to investigate the role of neuroinflammation in disease progression and its potential molecular basis using histopathological and RNA transcriptomic analyses. To our knowledge, this is the first comparative RNA-seq/transcriptomic analysis of brain tissue from K18-hACE2 and C57BL/6 mice infected with the beta variant of SARS-CoV-2.
Previous studies have reported the detection of SARS-CoV-2 in the brains of patients with COVID-19[23]. Additionally, SARS-CoV-2 replicates and induces cell death in human neural stem cells and brain organoids[24]. Studies have also shown that the beta variant strain can infect the brains of non-human primates and K18-hACE2 mice[11,25]. In this study, we observed that following infection with the beta variant strain, infectious viruses replicated in both the lung and brain tissues of K18-hACE2 mice, whereas viral replication and apparent pathological changes were not observed in the brains of C57BL/6 mice. However, significant transcriptional changes were detected in the brains of both K18-hACE2 and C57BL/6 mice after inoculation with the beta variant strain. GO enrichment analysis of DEGs revealed that pathways involved in neuronal differentiation and neuron projection guidance in the central nervous system were activated in the beta variant strain-infected C57BL/6 mice, whereas pathways related to ion transmembrane transport regulation, synaptic organization, and learning and memory regulation were suppressed. This indicated that although infection of the C57BL/6 mice with the beta variant strain did not elicit a significant inflammatory response in the brain, it indirectly influenced brain function in mice with SARS-CoV-2 infection.
Moreover, enrichment analysis indicated that infection of K18-hACE2 mice with the beta variant strain activated the innate immune response robustly. Excessive activation of the immune system may lead to a cytokine storm. Previous studies have demonstrated high levels of pro-inflammatory cytokines and chemokines, including TNF, IL-1β, IL-6, IL-10, CCL-3, CCL-4, CCL-20, CXCL-2, CXCL-3, CXCL-8, and CXCL-10, in the lungs and bronchoalveolar lavage fluid samples of patients with SARS-CoV-2 infection[8,26]. Similarly, we detected elevated expression of pro-inflammatory factors (IL-1β, IL-6, and TNF) and chemokines (CCL-2, CCL-4, CXCL-1, CXCL-10, CXCL-11, and CXCL-12), which may stimulate neuroglial cells to induce neuroinflammation and activate downstream cell death signaling pathways in neurons, thereby causing neuronal damage. A comparative analysis of cytokine expression profiles was performed between two groups of samples infected with SARS-CoV-2. In K18-hACE2 mice, the top five most significantly upregulated cytokines are Acod1, Ccl7, Cxcl10, Cxcl1, and Cxcl11. In contrast, the top five most upregulated cytokines in C57BL/6 mice are Il20rb, Cd300lf, Oasl2, Irf7, and Ifitm3. Among these, Cxcl10 is known to be critical for recruiting inflammatory cells and has been associated with inducing brain tissue damage in mice (or humans). As a chemokine, Cxcl10 plays a key role in the immune response by directing various immune cells to the site of infection[27]. This finding partially explains why the beta variant of SARS-CoV-2 caused more severe brain tissue damage in K18-hACE2 mice.
Multiple viruses have been reported to activate death receptors via different mechanisms, thereby mediating cell apoptosis and necrosis[28]. Previous studies have reported that SARS-CoV-2 activates the NLRP3 inflammasome in humans, thereby promoting inflammatory responses and worsening disease progression[29]. Previous studies have confirmed that SARS-CoV-2 infects mouse neurons and activates the ZBP1/MLKL-regulated necroptotic pathway[30]. In this study, we observed the upregulation of Zbp1, Mlkl, and Casp8, while the expression of Casp4 and Casp1, genes associated with necroptosis, increased. Previous studies have demonstrated that Casp4 plays a crucial role in initiating inflammatory cell death and is involved in the activation of the NLRP3 inflammasome and CASP1[31]. CASP4, an intracellular receptor of lipopolysaccharide, induces gasdermin D cleavage, leading to pyroptosis and activation of the non-classical NLRP3 inflammasome, which further results in CASP1 activation[32-34]. Pyroptosis, a form of programmed cell death, depends on CASP1 and is accompanied by the release of numerous pro-inflammatory factors. Viral infection-induced type I interferons or cytokines may trigger NF-κB signaling and promote the expression of Casp4/11[35]. Following SARS-CoV-2 infection, the expression levels of human Casp4 and mouse Casp11 are upregulated. Experimental observations in Casp11-deficient mice showed that the absence of Casp11 can prevent severe diseases caused by SARS-CoV-2 infection without affecting viral replication or clearance[36]. These findings suggest that the beta variant may induce pyroptosis in mouse brain tissue to some extent; however, the exact mechanism via which Casp4 causes brain damage requires further investigation. Therefore, in neuroinflammation and SARS-CoV-2-induced brain injury, pharmacological or genetic inhibition of analogous cell death pathways (e.g., ZBP1-MLKL axis for necroptosis, CASP4-GSDMD axis for pyroptosis) may serve as potential therapeutic strategies to mitigate neuronal damage and neuroinflammatory cascades.
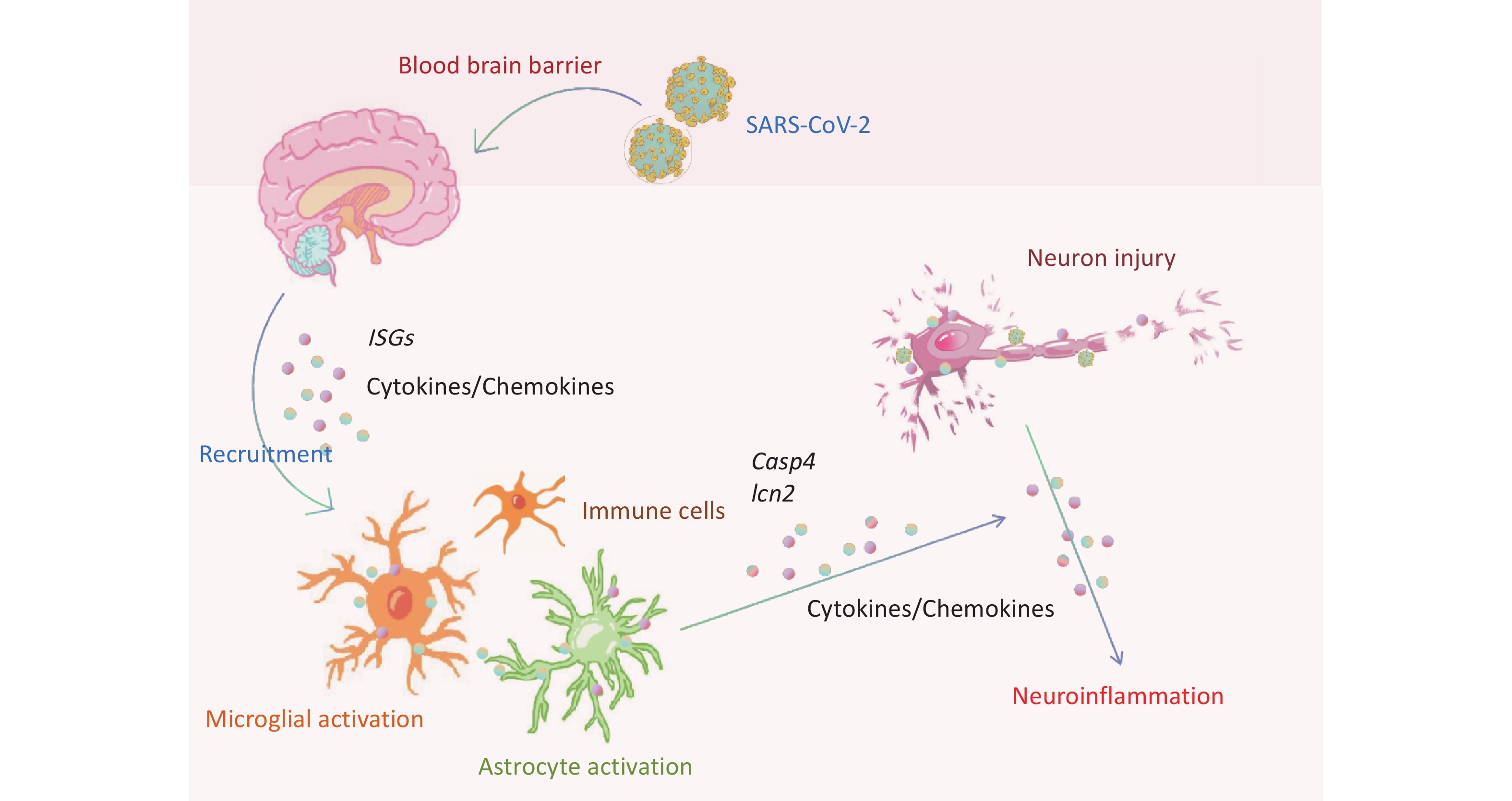
Compared with the brains of C57BL/6 mice infected with the beta variant, those of K18-hACE2 mice exhibited activation of microglial cells and regulation of neuronal death pathways. Villadiego et al. demonstrated the presence of SARS-CoV-2 in neurons after infection of K18-hACE2 mice, whereas the virus was not detected in microglial cells or astrocytes. Additionally, on the 6th dpi, signs of activation were observed in the microglial cells[37]. The release of cytokines and chemokines from activated microglial cells induces astrocyte activation and further pro-inflammatory mediator release, exacerbating the neuroinflammatory response[38]. Astrocytes play various roles in normal physiology and disease states, particularly in maintaining BBB integrity and regulating neuronal function, potentially acting as a target for the virus[37]. Viral infection-induced astrocyte activation may affect metabolic function, thereby indirectly influencing neuronal survival[39]. LCN2 enhances inflammasome activation by activating astrocyte/microglial cells and neutrophil infiltration[40]. Inflammatory stimuli regulate LCN2 expression in microglial cells[41]. LCN2 secretion by reactive astrocytes may accelerate the death of neighboring neurons[42]. We also observed upregulation of Lcn2 during microglial cell activation. These findings suggested that following infection in K18-hACE2 mice, the beta variant may primarily infect mouse neurons and further activate apoptotic and pyroptotic pathways by recruiting neuroglial cells to release factors such as LCN2 and CASP4, exacerbating the neuronal damage (Figure 6).
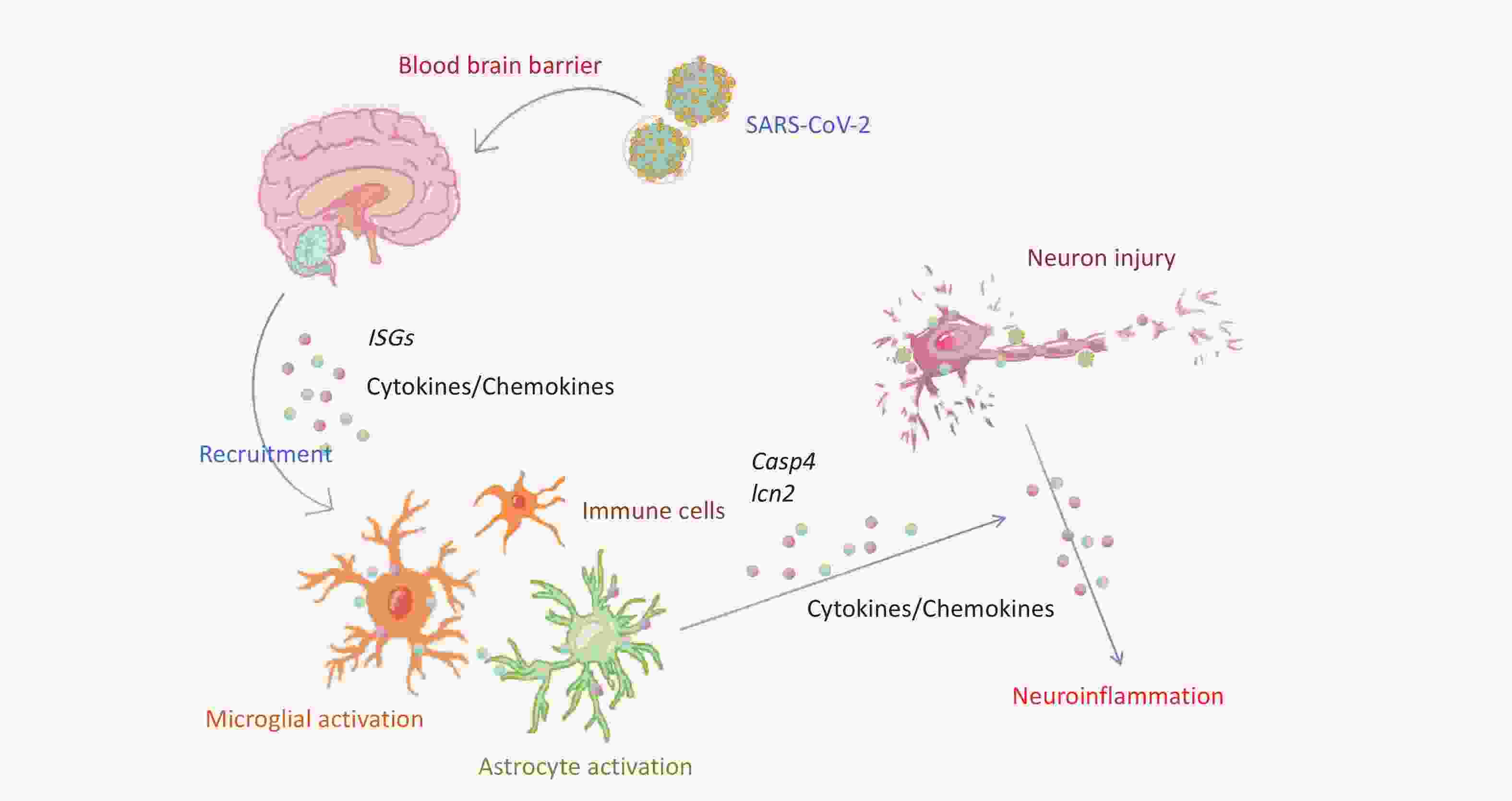
Figure 6. Schematic representation of neuroinflammation and neuronal death in K18-hACE2 mice induced by SARS-CoV-2 beta variant infection. ISGs, interferon-stimulated genes; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
Health status[43] and immune memory post-vaccination[44] play a role in SARS-CoV-2 infection and may potentially contribute to tissue impairment. Therefore, this study has some limitations. First, the small sample size and focus on only one time point was considered. Although the third day post-infection represents an early stage of COVID-19 infection, brain damage is a progressive process. Fully understanding the mechanisms of disease progression and associated genetic changes at different stages post-infection would require a larger sample size. Second, using bulk brain tissue for transcriptomic sequencing limited our ability to screen for gene specificity. Future research should address these issues using single-cell sequencing technology. Clarifying how SARS-CoV-2 penetrates the BBB to infiltrate the central nervous system is crucial for advancing research and developing therapeutic interventions against viral encephalitis. However, our current investigation does not address this specific area of research. Recently, Yoon et al. reported the NLRP3-GSDMD axis activation could break the BBB[45]. However, the influence of SARS-CoV-2 neuro-invasion still requires further studies.
Recent neuropathological analyses by Solomon et al. encompassing nearly 900 COVID-19 autopsy cases, emphasize cerebrovascular pathologies and microglia-predominant inflammation as hallmark features of SARS-CoV-2-associated neurological injury[46]. Nevertheless, akin to the findings from this study, unified mechanisms underlying acute COVID-19 or post-acute neurological sequelae remain elusive. Integrating microstructural and molecular findings from brain tissue into clinical disease frameworks is critical to establishing best practices and prioritizing research directions for COVID-19-related neurological disorders.
-
The beta variant of SARS-CoV-2 infection in C57BL/6 mice indirectly affected the brain functional pathways, while infection in K18-hACE2 mice led to pathological damage in the brain. The beta variant activated genes related to brain tissue inflammation and apoptosis/pyroptosis. Furthermore, we speculated that the beta variant may infect and kill neuronal cells in K18-hACE2 mice, recruiting and activating immune cells such as microglia and astrocytes, upregulating key factors such as Casp4 and Lcn2, triggering multiple programmed cell death pathways, and causing pathological damage to the brain tissue. These findings suggest that the key factors identified in this study may play crucial roles in neuroinflammation, thereby providing new research directions for treating neuroinflammation in patients with SARS-CoV-2 infection.
Pathogenicity and Transcriptomic Profiling Revealed Activation of Apoptosis and Pyroptosis in Brain of Mice Infected with the Beta Variant of SARS-CoV-2
doi: 10.3967/bes2025.106
- Received Date: 2024-11-25
- Accepted Date: 2025-03-11
-
Key words:
- Beta variant of SARS-CoV-2 /
- Encephalitis /
- Neuronal injury /
- Transcriptomics /
- Cell death
Abstract:
The authors declare no conflicts of interest.
All animal experiments were conducted in the ABSL-3 laboratory of the National Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, and were approved by the Animal Ethics and Usage Committee of the National Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention (ethics permit NO.20220106001).
&These authors contributed equally to this work.
| Citation: | Han Li, Baoying Huang, Gaoqian Zhang, Fei Ye, Li Zhao, Weibang Huo, Zhongxian Zhang, Wen Wang, Wenling Wang, Xiaoling Shen, Changcheng Wu, Wenjie Tan. Pathogenicity and Transcriptomic Profiling Revealed Activation of Apoptosis and Pyroptosis in Brain of Mice Infected with the Beta Variant of SARS-CoV-2[J]. Biomedical and Environmental Sciences. doi: 10.3967/bes2025.106

|

 Quick Links
Quick Links
 DownLoad:
DownLoad: