
-
Acute lung injury (ALI)/acute respiratory distress syndrome (ARDS) is a severe clinical disorder characterized by widespread inflammation, diffuse alveolar damage, and pulmonary edema, often leading to respiratory failure and death. Despite significant advances in clinical care, ALI/ARDS remains the leading cause of death among intensive care unit patients. Sepsis is the primary risk factor for the development of ALI/ARDS, as excessive inflammatory responses contribute to organ injury and high mortality in critically ill patients. Mitochondrial dysfunction is closely associated with the severity of lung injury and mortality in sepsis-related ARDS, and mitochondrial functional enhancement alleviates sepsis severity in endotoxin-induced ALI, suggesting that its regulation is a potential therapeutic strategy for ALI/ARDS.
Sirtuin 3 (Sirt3), an NAD-dependent deacetylase involved in diverse physiological and pathological processes, facilitates mitochondrial metabolic adaptation to stress by activating cytoplasmic signaling pathways and nuclear gene expression, thereby modulating proliferation, differentiation, and cell death. Sirt3 has been shown to mitigate ALI by reducing oxidative stress and improving mitochondrial metabolism[1]. Ferroptosis, a recently identified form of regulated cell death in ALI, is characterized by iron-dependent lipid peroxidation, reactive oxygen species (ROS) overproduction, and disrupted cellular redox homeostasis. Mitochondria play a central role in ferroptosis, harboring both pro-ferroptotic and anti-ferroptotic machinery. While Sirt3 can suppress ferroptosis by maintaining mitochondrial redox homeostasis[2], the underlying mechanisms remain unclear. Given the close relationship between Sirt3 and mitochondrial metabolism, we hypothesize that Sirt3 activation regulates ferroptosis in alveolar epithelial cells by suppressing their pro-inflammatory phenotype and reprogramming energy metabolism, thereby reducing lipopolysaccharide (LPS)-induced ALI.
To investigate the role of Sirt3 in ALI and its regulatory pathways, an LPS-induced ALI model was established in Sirt3−/− mice and their littermates (Sirt3+/− mice), followed by RNA-seq analysis. A total of 1,926 differentially expressed genes (DEGs) were identified (Supplementary Figure 1A). Sequencing data revealed that the upregulated genes were significantly enriched in inflammatory pathways (Supplementary Figure 1B), whereas the downregulated genes were prominently enriched in glutathione metabolism based on KEGG analysis (Supplementary Figure 1C). Reactome enrichment analysis further demonstrated that the upregulated genes were associated with NF-κB pathway activation and glycolysis regulation via fructose 2,6-bisphosphate metabolism (Supplementary Figure 1D), whereas the downregulated genes were associated with glutathione synthesis/recycling and glycogen synthesis (Supplementary Figure 1E). Protein-protein interaction (PPI) network analysis of ferroptosis-related DEGs in ALI highlighted the involvement of Sirt3, GPX4, and RELA in ferroptosis regulation (Supplementary Figure 2), suggesting a potential connection between Sirt3, the NF-κB pathway, and ferroptosis. Collectively, these findings indicated that Sirt3 may modulate ALI progression by regulating ferroptosis-associated glutathione metabolism and inflammatory responses.
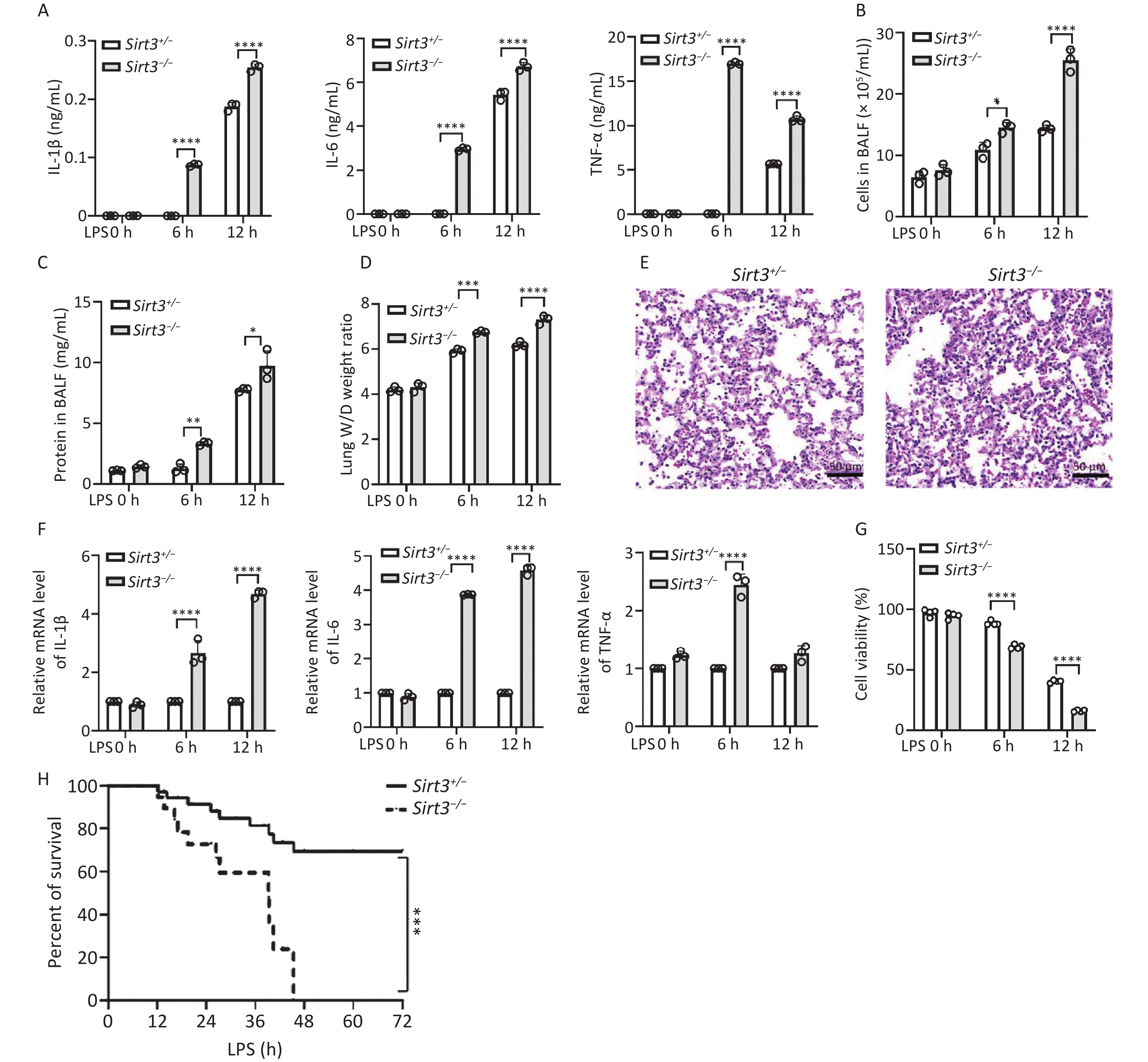
To investigate the function of Sirt3 in inflammatory responses during LPS-induced ALI, we established an LPS-induced ALI mouse model. The production of pro-inflammatory cytokines (IL-6, IL-1β, and TNF-α) in the bronchoalveolar lavage fluid (BALF) of Sirt3−/− mice was notably higher compared with their littermates (Sirt3+/−) in vivo (Figure 1A). Total cell counts in the BALF of Sirt3−/− mice were dramatically increased compared with the control group (Figure 1B). The lung wet/dry (W/D) ratio and BALF protein concentration—two key indicators of pulmonary vascular permeability in ALI/ARDS—were significantly elevated in Sirt3−/− mice (Figures 1C-D). Histological analysis revealed more severe inflammatory infiltration in the lungs of Sirt3−/− mice compared with littermates (Sirt3+/−) in the ALI model (Figure 1E). At the cellular level, mRNA expression of pro-inflammatory cytokines (IL-6, IL-1β, and TNF-α) in Sirt3−/− alveolar epithelial type II cells (AEC II) was significantly higher than in Sirt3+/− AEC II after LPS stimulation (Figure 1F). Furthermore, cell viability of Sirt3−/− alveolar epithelial cells was markedly reduced compared with Sirt3+/− AEC II under LPS treatment (Figure 1G). Consistently, survival rates of Sirt3−/− mice were significantly lower than those of Sirt3+/− mice in LPS-induced ALI (Figure 1H). These results indicated that Sirt3 deficiency exacerbates LPS-induced ALI by amplifying inflammatory responses and promoting alveolar epithelial cell death.
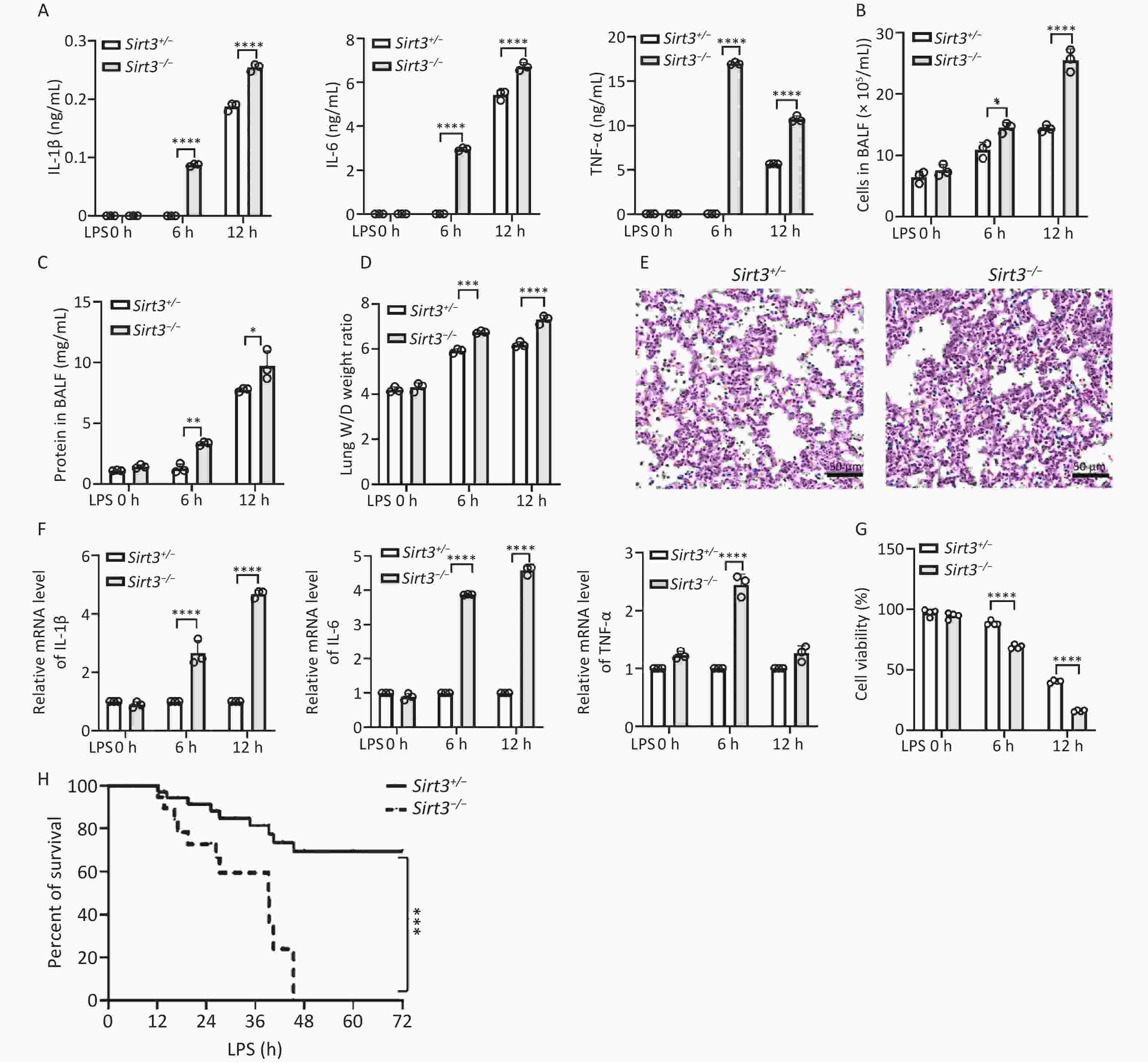
Figure 1. Sirt3 deficiency aggravates LPS- induced lung injury. (A) Pro-inflammatory cytokines (IL-6, IL-1β and TNF-ɑ) in BALF were detected by ELISA in ALI mice (12 h for intratracheal infusion of LPS). (B) Total cells in BALF were counted by an automated cell counter in ALI mice (12 h for intratracheal infusion of LPS). (C) Protein concentrations in BALF were tested using BCA protein assay kit in ALI mice (12 h for intratracheal infusion of LPS). (D) Lung W/D ratio. (A–D, n = 3 mice per group) (E) Lung tissues in ALI mice were stained using HE staining. (F) IL-6, IL-1β and TNF-ɑ in vitro were detected by qRT-PCR (6 h after LPS stimulating). (G) Viability of Sirt3+/−, Sirt3−/− AECⅡ with LPS was detected by CCK8 Assay (6 h after LPS stimulating). Data were presented as mean ± SD of three independent experiments. (H) Survival rate in ALI mice was analyzed by Kaplan Meier on GraphPad Prism 10 (n = 6 mice per group). (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
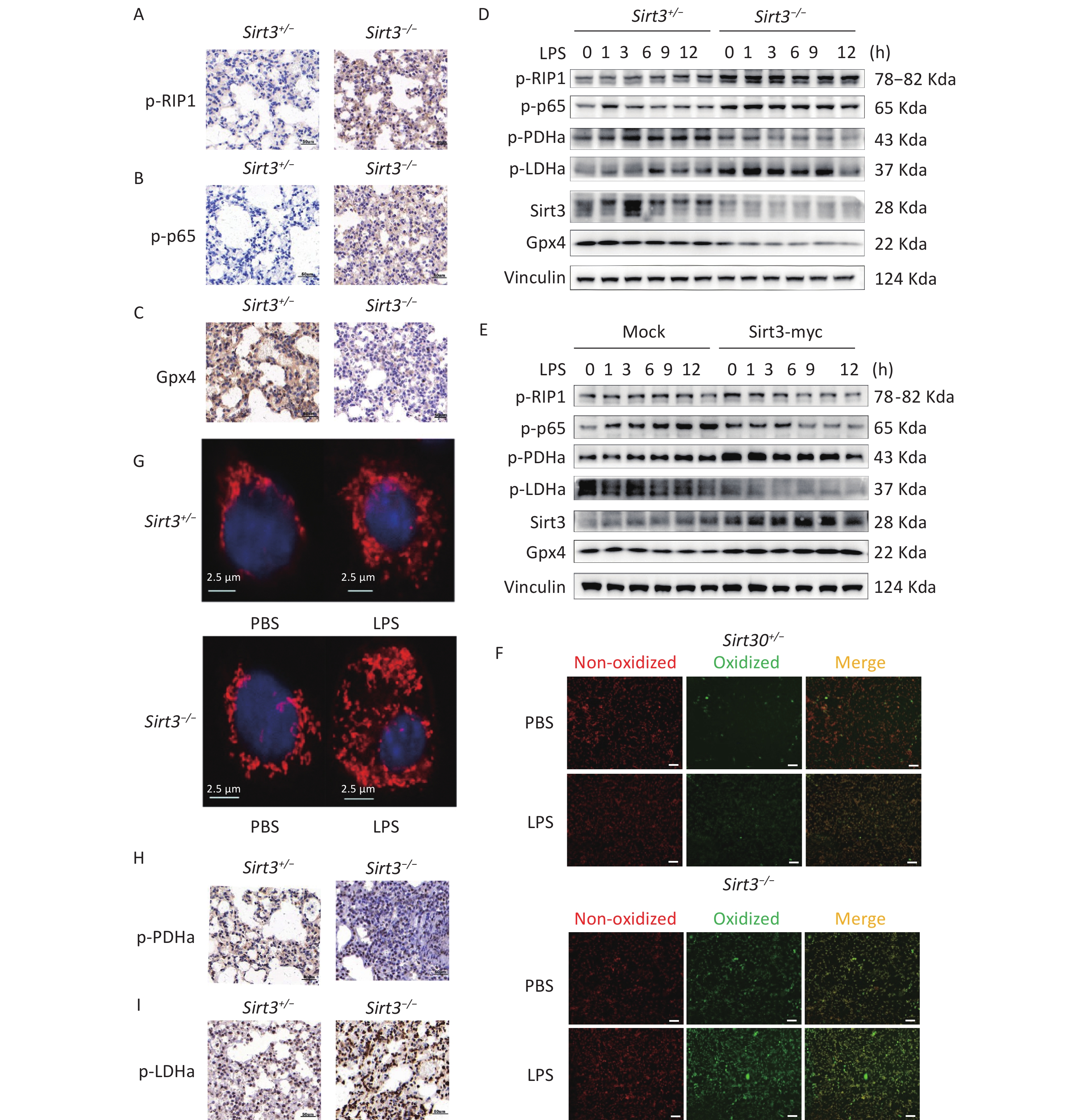
Receptor-interacting protein kinase 1 (RIP1) is a critical effector of inflammatory responses and cell death pathways. It regulates various forms of cell death, including apoptosis and necroptosis. NF-κB pathway activation induces the transcription of inflammation-related factors such as IL-1β, IL-6, and TNF-α. Moreover, numerous studies have demonstrated that NF-κB may activate antiapoptotic pathways while paradoxically participating in cell death regulation. Therefore, we investigated the activation of RIP1, NF-κB, and the expression of ferroptosis marker Gpx4. Compared with control (Sirt3+/−) mice, Sirt3−/− mice exhibited increased phosphorylation of RIP1 (p-RIP1) and p65 (p-p65) along with reduced Gpx4 expression in lung tissue (Figure 2A–C). This pattern was recapitulated in alveolar epithelial cells: Sirt3−/− cells showed enhanced p-RIP1 and p-p65 activation but decreased Gpx4 expression compared to Sirt3+/− AEC II (Figure 2D). Importantly, Sirt3 overexpression in the alveolar epithelial cell line MLE12 significantly attenuated p-RIP1 and p-p65 activation, while restoring Gpx4 expression (Figure 2E). Given that Gpx4 inactivation induced by glutathione depletion drives ferroptosis and previous studies having established the association between NF-κB activation and Gpx4 suppression[3], our findings demonstrated that Sirt3 deficiency activates NF-κB signaling to downregulate Gpx4 expression, thereby promoting ferroptosis in alveolar epithelial cells.
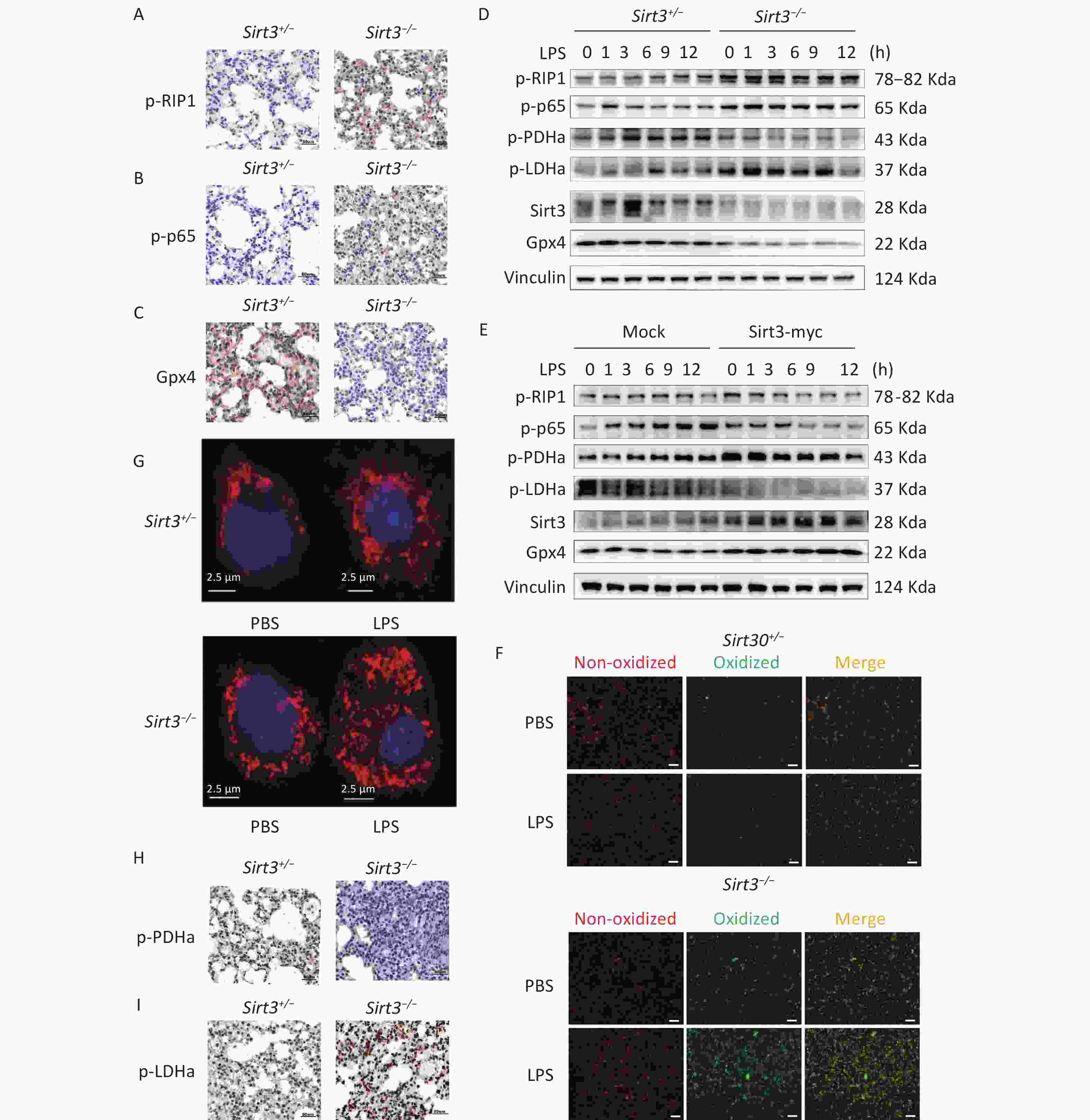
Figure 2. Sirt3 suppresses ferroptosis and inflammation via inhibition of aerobic glycolysis. (A–C) Immunohistochemical staining of p-RIP1, p-p65, Gpx4 in lung tissues of Sirt3+/− and Sirt3−/− littermates in ALI model. (D) Western blot for p-RIP1, p-p65, p-LDHa, p-PDHa, Gpx4 and Sirt3 in Sirt3+/− and Sirt3−/− AEC Ⅱ. (E) Western blot for p-RIP1, p-p65, p-LDHa, p-PDHa, Gpx4 and Sirt3 expression in Sirt3 overexpressed MLE-12 cells. Data presented three independent experiments. (F) Lipid peroxidation levels detected by Lipid Peroxidation Kit in Sirt3+/− and Sirt3−/− AEC Ⅱ (6 h after PBS or 1 µg/ml LPS stimulation). Images were photographed using a fluorescence microscope, Scale bar = 100 µm. (G) Mito Tracker Red CMXRos for Sirt3+/− and Sirt3−/− AEC Ⅱ (6 h after PBS or 1 µg/ml LPS stimulation). Images were photographed using a laser confocal microscope. (H-I) Immunohistochemical staining of p-PDHa, p-LDHa in lung tissues of Sirt3+/− and Sirt3−/− littermates in ALI model.
LPS stimulation induces ROS production in mitochondria. Although ROS play a major role in inflammatory processes, ferroptosis is primarily initiated through ROS-driven lipid peroxidation and iron overload. Notably, ROS interact with NF-κB signaling pathways through multiple mechanisms. The glutathione antioxidant system, which comprises glutathione (GSH), glutathione peroxidase (GPX), and related enzymes, serves as a key regulator of lipid peroxidation by preventing excessive ROS generation. To elucidate how Sirt3 deficiency exacerbates ferroptosis in LPS-induced ALI, we analyzed cell membrane lipid peroxidation and ROS production. Notably, Sirt3−/− alveolar epithelial cells exhibited significantly enhanced lipid peroxidation compared to Sirt3+/− controls during LPS-induced ALI (Figure 2F). Intriguingly, LPS stimulation increased ROS production in both groups, with markedly higher potential observed in Sirt3−/− cells than in Sirt3+/− cells (Figure 2G). Collectively, these findings suggested that Sirt3 suppresses ferroptosis via ROS modulation. These observations are further supported by evidence that mitochondrial ROS overproduction disrupts endothelial barrier function and directly triggers ferroptosis[4]. Sirt3 is known to enhance ROS detoxification via SOD2 deacetylation[5], and its deficiency likely exacerbates LPS-induced ROS accumulation, leading to redox imbalance and ferroptosis[6].
Mitochondria play a pivotal role in regulating cellular metabolism, signal transduction, and cell death pathways. Dysfunctional mitochondria drive the shift from mitochondrial respiration to glycolysis in cancer cells. However, the relationship between mitochondrial dysfunction and glycolysis in ferroptosis remains unclear. To determine how Sirt3 modulates glycolytic metabolism and ROS dynamics during LPS stimulation, we assessed the activity of two key metabolic enzymes: Tricarboxylic acid cycle regulator pyruvate dehydrogenase A (PDHa) and glycolytic enzyme Lactate dehydrogenase A (LDHa). In vivo analyses revealed that Sirt3−/− mice exhibited reduced p-PDHa activation but significantly elevated p-LDHa activation compared to Sirt3+/− controls, indicating enhanced aerobic glycolysis in Sirt3−/− lungs (Figure 2H-I). Consistently, Sirt3−/− alveolar epithelial cells recapitulated this metabolic pattern (Figure 2D). Conversely, Sirt3 overexpression in vitro increased p-PDHa expression, while suppressing p-LDHa expression (Figure 2E). These findings demonstrated that Sirt3 attenuates aerobic glycolysis and ferroptosis. The metabolic reprogramming toward glycolysis under Sirt3 deficiency aligns with studies showing NF-κB activation drives LDHA-dependent glycolysis[7]. Such shifts deplete antioxidants (e.g., GSH) and amplify ROS production, to create a permissive environment for ferroptosis[8,9].
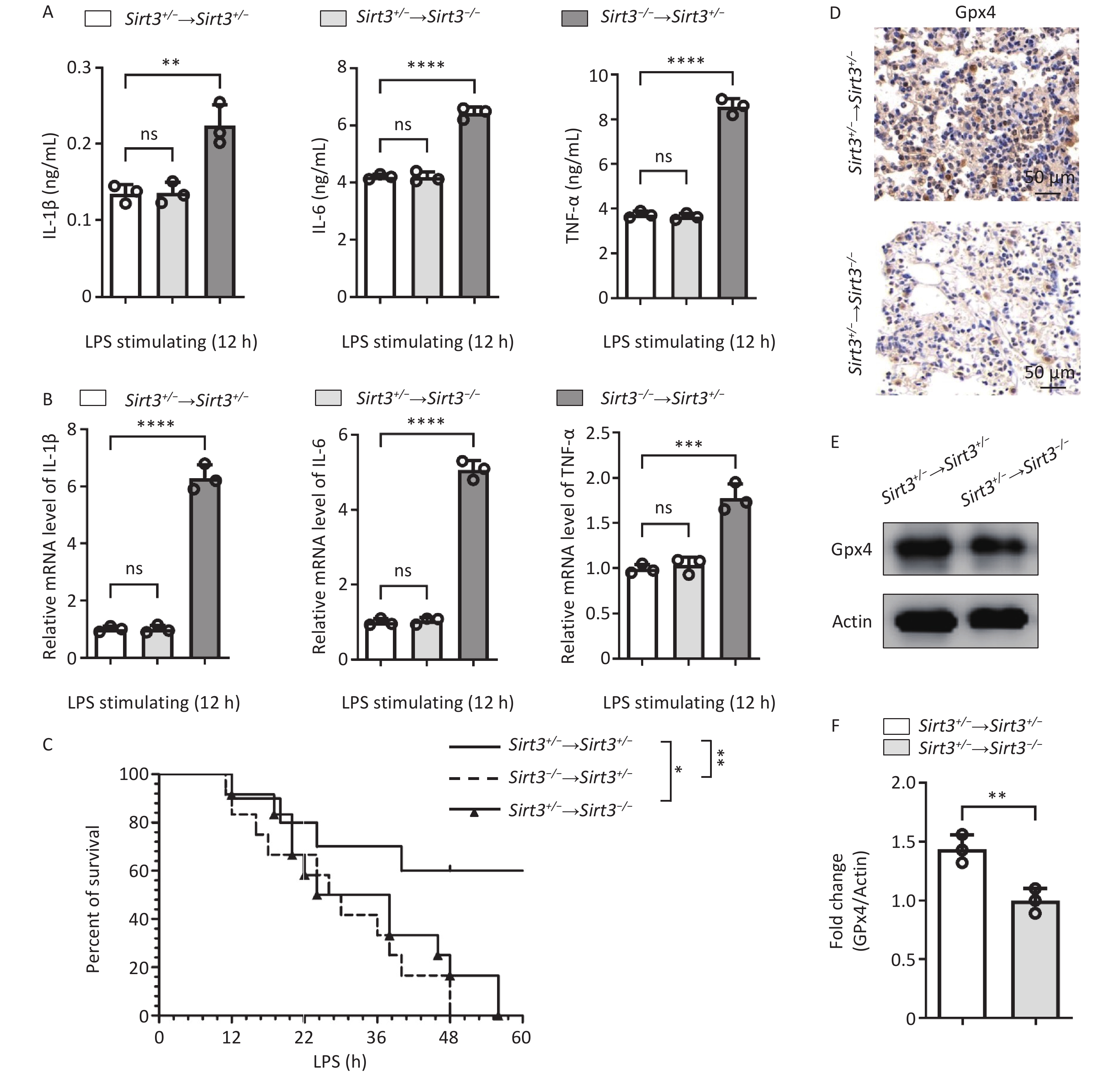
Our study identified Sirt3 as an inhibitor of ROS-induced ferroptosis through the regulation of inflammatory responses. Sirt3 is widely expressed in both blood-derived cells, such as macrophages and neutrophils, and organ-derived cells, such as lung epithelial cells. To explore the role of Sirt3 in different cell types, we reconstructed the bone marrow cells transplantation experiments. Therefore, we used an allogeneic bone marrow transplantation (BMT) mouse model to observe the changes in the protective effect of Sirt3. The Sirt3−/− and Sirt3+/− bone marrow cells were transplanted to Sirt3+/− mice irradiated with 8.5 Grey. LPS-induced pro-inflammatory cytokines (IL-6, IL-1β and TNF-ɑ) were significantly higher in Sirt3−/− → Sirt3+/− group, compared with Sirt3+/− → Sirt3+/− group (Figure 3A). Quantitative PCR analysis of lung tissue showed similar results (Figure 3B). The survival rate in vivo of the Sirt3−/− → Sirt3+/− mice was lower than that of the Sirt3+/− → Sirt3+/− mice in LPS-induced ALI (Figure 3C). These results showed that Sirt3 inhibits inflammation in blood-derived cells. Then we transplanted bone marrow of Sirt3+/− to Sirt3−/− or Sirt3+/− mice to further determine the role of Sirt3 on ferroptosis in lung epithelial cells. The Sirt3+/− → Sirt3−/− group showed less Gpx4 expression than the Sirt3+/− → Sirt3+/− group, indicating a higher ferroptosis level (Figure 3D–F). The production of inflammatory cytokines (IL-6, IL- 1β and TNF-ɑ) in the Sirt3+/− → Sirt3−/− group was not significantly higher than that in the Sirt3+/− → Sirt3+/− group (Figure 3A-B), which indicated that Sirt3 majorly regulates ferroptosis in lung epithelial cells. The Sirt3+/− → Sirt3−/− mice also showed lower survival rates than the Sirt3+/− → Sirt3+/− group and a later onset of death compared to the Sirt3−/− → Sirt3+/− group in LPS-induced ALI (Figure 3C). These results suggested that Sirt3 inhibits ferroptosis in lung epithelial cells and decreases inflammation in blood-derived cells.
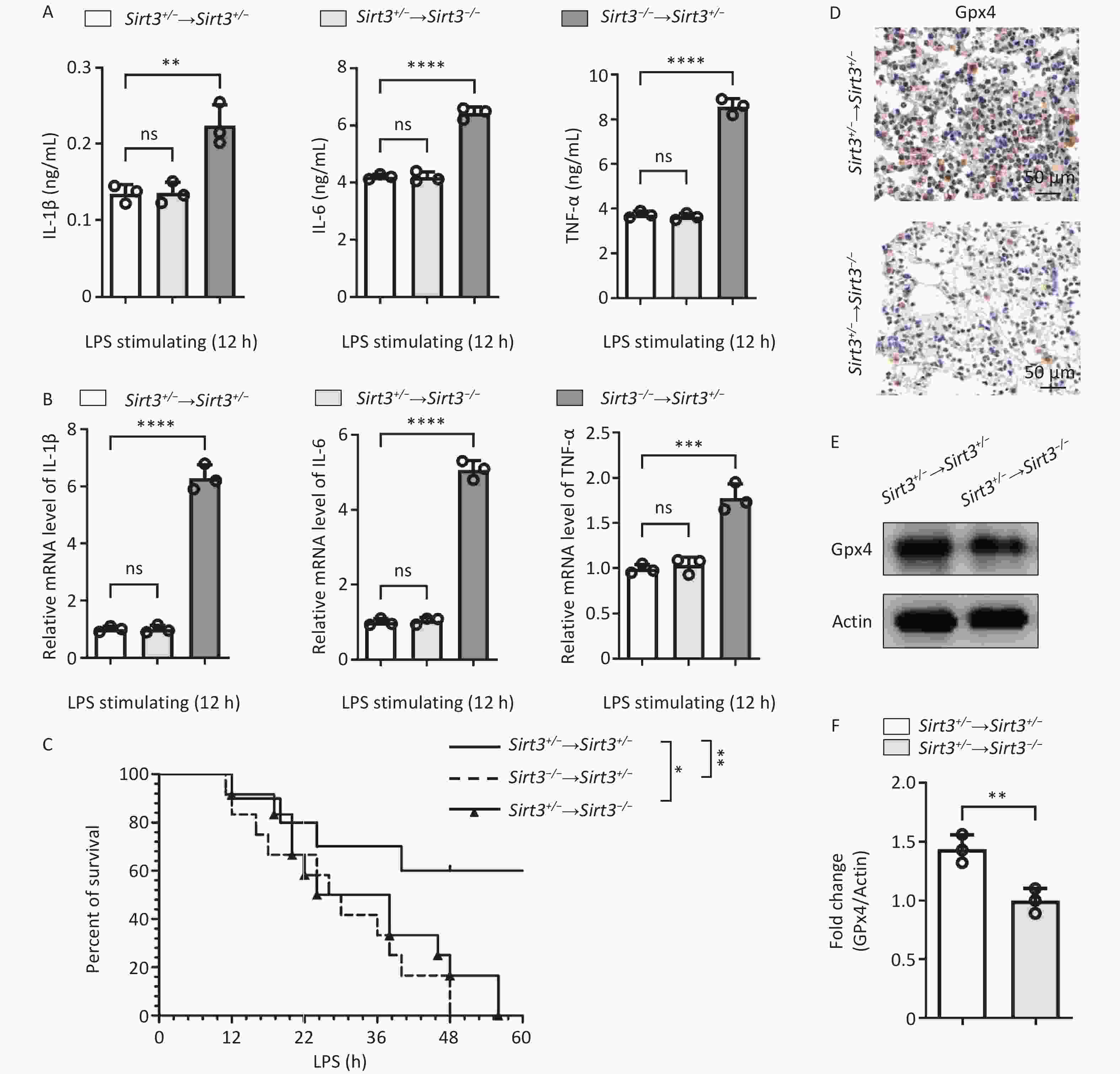
Figure 3. Sirt3 decreases inflammation in blood-derived cells and inhibits ferroptosis in lung epithelia cells. (A) Pro-inflammatory cytokines (IL-6, IL-1β and TNF-ɑ) in BALF were detected by ELISA in ALI mice (12 h for intratracheal infusion of LPS) after BMT. (B) IL-6, IL-1β, and TNF-ɑ in lung tissue from ALI mice after BMT were detected by qRT-PCR. (A–B, n = 3 mice per group) Data were presented as mean ± SD of three independent experiments. (C) Survival rate in ALI mice was analyzed by Kaplan Meier on GraphPad Prism 10 (n = 10 mice per group). (D) Immunohistochemical staining of Gpx4 in lung tissues of ALI mice (12 h for intratracheal infusion of LPS) after BMT. (E) Western blot for Gpx4 in lung tissues of ALI mice (12 h for intratracheal infusion of LPS) after BMT. Data presented three independent experiments. (F) Subsequent semi-quantitative analysis of Gpx4 expression in lung tissues of ALI mice after BMT. Results are expressed as protein/Actin ratio. (*P < 0.05,**P < 0.01, ***P < 0.001, ****P < 0.0001).
This study revealed a novel mechanism by which Sirt3 alleviates LPS-induced ALI through the regulation of ferroptosis in alveolar epithelial cells. The main findings of this study were as follows: (1) Sirt3 deficiency promotes inflammation in LPS-induced alveolar epithelial cells and lung parenchymal injury; (2) Sirt3 deficiency leads to LPS-induced ferroptosis in alveolar epithelial cells through activation of the NF-κB signaling pathway; (3) Sirt3 deficiency promotes LPS-induced ROS production, leading to ferroptosis in alveolar epithelial cells, accompanied by a reversal of cellular energy metabolism toward glycolysis; (4) Sirt3 functions in both blood- and organ-derived cells. In summary, Sirt3 regulates ferroptosis and inflammation in alveolar epithelial cells to reduce LPS-induced ALI via NF-κB signaling pathways and glycolytic metabolism. Our results provided new insights into the mechanism by which Sirt3 links glycolytic reprogramming, ferroptosis, and inflammation in LPS-induced ALI, outlining novel mechanisms and drug targets.
Sirtuin 3 Attenuates Acute Lung Injury by Decreasing Ferroptosis and Inflammation through Inhibiting Aerobic Glycolysis
doi: 10.3967/bes2025.110
- Received Date: 2025-02-16
- Accepted Date: 2025-07-28
The authors declare that they have no competing interests.
All animal experiments were performed in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals and approved by the Scientific Investigation Board of the Naval Medical University. This study did not involve human participants.
&These authors contributed equally to this work.
| Citation: | Kewei Qin, Qingqing Ji, Weijun Luo, Wenqian Li, Bingbing Hao, Haiyan Zheng, Chaofeng Han, Jian Lou, Liming Zhao, Xingying He. Sirtuin 3 Attenuates Acute Lung Injury by Decreasing Ferroptosis and Inflammation through Inhibiting Aerobic Glycolysis[J]. Biomedical and Environmental Sciences. doi: 10.3967/bes2025.110

|

 Quick Links
Quick Links
 DownLoad:
DownLoad: