
HTML
-
Human mesenchymal stem cells are of great significance in tissue repairing due to their advantageous characteristics, such as unlimited in supply, clear in the biological background, and convenient for quality control, genetic modification and scale‐up production[1]. Human hair follicle mesenchymal stem cells (hHF-MSCs) are receiving more and more attention because of their easy availability, low immunogenicity, less ethical dispute in the regeneration of hair follicles and skin repairing[2] as well as their capacity of self-renewal and multipotency[3-5].
Environmental factors, such as exposure to ultraviolet radiation and hair dying agents, are able to affect the microenvironment of hair follicles[6] and result in the damage of hHF-MSCs. One of the mechanisms underlying the damage of hHF-MSCs caused by exposure to UV or hair dying agents is oxidative stress, which will lead to the aging of hair follicles, hair loss or hair whitening[7]. Therefore, it is pivotal to identify the mechanism that will help hHF-MSCs to resist oxidative stress.
NANOG is a transcription factor which is able to help maintaining the capacity of self-renewal and multipotency in stem cells[8, 9]. The results from Andreadis et al.' research demonstrated that NANOG would promote proliferation, delay senescence and increase myogenic differentiation capacity[9-11]. Tanno et al. identified NANOG as the key factor in keeping the self-renewal capacity of cells underwent irradiation damage[12]. Thus, it is hypothesized that NANOG would play a part in protecting hHF-MSCs against oxidative stress.
The signal pathway of PI3K/AKT is implicated in the regulation of cell proliferation, metabolism, aging and oxidative stress which the activation of this pathway will protect cells from the damage of oxidative stress and enhance the cell survival[13-15]. The present study will investigate whether or not NANOG would protect hair follicle mesenchymal stem cells from oxidative stress elicited by hydrogen peroxide (H2O2) through the PI3K/AKT pathway.
-
The study was approved by the Institutional Review Board of School of Public Health at Jilin University.
-
The isolation and culture of hHF-MSCs were performed according to previous studies[4, 16, 17]. The hair follicles were washed with Phosphate-Buffered Saline (PBS) for three times. Then they were transferred into 24-well culture plate which contained one or two follicles per well. The hHF-MSCs were cultured in Dulbecco's Modified Eagle Medium supplemented with F-12 (DMEM/F-12, Gibco by Life technologies, USA, 10% fetal bovine serum (FBS, Gibco, USA) and 2 ng/mL bFGF (Sino Biological Inc, China) at 37 ℃, 5% CO2. The medium was refreshed every 3 days. The cells were digested with 0.25% trypsin (Biosharp, China) when the cell confluence was approximately 80%. Flow cytometry were used to detect the expression of cell surface markers, including CD44 (eBioscience, USA), CD73 (eBioscience, USA), CD90 (eBioscience, USA), CD105 (eBioscience, USA) and CD31 (eBioscience, USA). The assays were performed as described previously[4].
-
For adipogenic differentiation assay, hHF-MSCs were cultured in adipogenic differentiation medium which contained High Glucose Dulbecco's Modified Eagle Medium (DMEM; Gibco, USA) supplemented with 10% FBS (Gibco, USA), 1 μmol/L dexamethasone (Sigma-Aldrich, USA), 0.5 mmol/L isobutyl-methylxanthine (Sigma-Aldrich, USA), 10 μmol/L insulin (Sigma-Aldrich, USA), and 200 μmol/L indomethacin (Sigma-Aldrich, USA). The formation of acellular lipid droplets in hHF-MSCs after two-week culture stained with Oil red O (Sigma-Aldrich, USA) were observed by inversion fluorescence microscope (Leica, German)[5].
For the assay of osteogenic differentiation, hHF-MSCs were cultured in osteogenic differentiation medium which contained High Glucose DMEM (Gibco, USA) supplemented with 10% FBS (Gibco, USA), 0.1 mmol/L dexamethasone (Sigma-Aldrich, USA), 50 mmol/L ascorbate-2-phosphate (Sigma-Aldrich, USA), and 10 nmol/L β-glycerophosphate. The formation of mineralized nodules in hHF-MSCs stained by Alizarinred S (Sigma-Aldrich, USA) after a month culture was observed by inversion fluorescence microscope (Leica, German)[4].
-
Day 1, 6 × 106 293T cells were cultured in 8 mL High Glucose DMEM (Gibco by Life Technologies, USA) with 10% FBS in a 10 cm dish. Day 2, the lentivirus vector containing NANOG coding region 10 μg, transfection reagents (GeneCopoeia, USA) 22 μL, two packaging plasmids (7.5 μg of psPAX2- gag/pol/tat/rev and 2.5 μg of pMD2.G-VSVG), were added into 1 mL Opti-MEM (Reduced Serum Medium, Gibco by Life Technologies, USA). Six hours later, the medium was replaced for new High Glucose DMEM with 10% FBS for the sake of better growth and lower toxic effect. After 36 h the supernatant was harvested and added into hHF-MSCs supplemented with 10 μg/mL polybrene (Santa, USA) and 2 ng/mL bFGF (Sino Biological Inc, China). Seventy-two hours later, the transduction efficiency was observed by inversion fluorescence microscope (Leica, German) and also was verified by Western blot.
-
hHF-MSCs were seeded at 5 × 103 cells per well in a 96-well plate with culture medium containing 100 μL DMEM/F-12 and 10% FBS without bFGF and cultured for 5 days. Each day, 10 μL of 3-(4, 5-Dimethylthiazol-2-yl)-2, 5-Diphenyltetrazolium Bromide (MTT, 5 mg/mL, Dingguo, China) were added into culture medium and incubated at 37 ℃, 5% CO2 in dark for 4 h. After removing the medium, 150 μL dimethyl sulfoxide were added into each well. Five minutes later, the MTT absorbance could be measured by microplate reader (SYNERGY H1 microplate reader, BioTek, USA) at 490 nm.
-
CCK-8 was used to assess the cell survival rate of hHF-MSCs treated with H2O2. hHF-MSC cells were plated in a 96-well plate and when the confluence of the cells was 80%, the cells were cultured with the medium supplemented with 0.5% FBS for 24 h. 24 h later, the concentration of FBS in the medium was changed to 1% and the cells were treated with different concentrations of H2O2 (0, 100, 200, 400, 800 μmol/L) at 37 ℃ with 5% CO2 for 2 h. Two hours later, the culture media was removed and 100 μL fresh culture medium and 10 μL CCK8 (Fuyuanbio, China) were added to the cells and cultured at 37 ℃ for 2 h. The absorbance was determined by microplate reader (SYNERGY H1 microplate reader, BioTek, USA) at 450 nm. Cell survival rate = (OD H2O2 damage-OD blank) / (OD control-OD blank) × 100%. In addition, another experiment was performed to explore the damage of 400 μmol/L H2O2 to the cells at different time (0, 1, 2, 4, 6 h) according to the CCK-8 assay above.
-
CM-H2DCFDA (General Oxidative Stress Indicator, Invitrogen, USA) was used to measure intracellular ROS production through fluorescence intensity. hHF-MSC cells were plated in a 96-well plate and incubated with 10 μmol/L CM-H2DCFDA at 37 ℃ for 30 min in darkness. Then cells were treated with 400 μmol/L H2O2 for 2 h after being washed with DMEM/F-12 medium without FBS three times. Next, the fluorescence intensity was detected by Cytation 3 Cell Imaging Multi-Mode Reader (BioTek, USA). The excitation wavelength and emission wavelength were 488 nm and 525 nm respectively.
-
Annexin V-FITC Apoptosis Analysis Kit (Sungene, Tianjin, China) was used to detect the cell apoptosis. Cells were collected and cell concentration was adjusted to 1 × 106/ tube, rinsed by precool 4 ℃ PBS and resuspended by 100 μL binding buffer, 5 μL Annexin-V FITC was added to the tube and tube was shaken gently, then incubated at room temperature in darkness for 10 min, 10 min later, 5 μL PI solution was added to the cells and incubated for 5 min, finally 400 μL PBS was added to the tube and the cells were detected (BD, FACSCalibur, USA).
-
To measure relative protein level, western blots were performed according to previous study of our team[4]. The antibody information was AKT (pan) (C67E7) Rabbit mAb (CST, USA, 1:1, 000), phospho-AKT (Ser473) Rabbit mAb (CST, USA, 1:2, 000), p44/42 MAPK (Erk1/2) Rabbit mAb (CST, USA, 1:1, 000), Phospho-p44/42 MAPK (Erk1/2) Rabbit mAb (CST, USA, 1:1, 000), NANOG Rabbit mAb (CST, USA, 1:1, 000), p21Waf1/Cip1 (12D1) Rabbit mAb (CST, USA, 1:1, 000), GAPDH Mouse mAb (ProteinTech, USA, 1:10, 000), HRP-conjugated Affinipure Goat Anti-Rabbit IgG (H+L) (ProteinTech Group Inc, USA, 1:5, 000), and HRP-conjugated Affinipure Goat Anti-Mouse IgG (H+L) (ProteinTech Group Inc, USA, 1:5, 000). The images were obtained by Chemiluminescence imaging analysis system (ECL from Tanon 5, 200, Shanghai, China) and band intensity was analyzed by Tanon Gis analytical software (Shanghai, China).
-
For western blot, NANOG hHF-MSCs were seeded into a 10 cm plate while for cell survial rate and ROS, the cells were seed in a 96-well plate. When the confluence of the cells was 80%, inhibitor LY294002 was added to the cells at the final concentration of 40 μmol/L for 24 h. The inhibitor LY294002 (MedChem Express, USA) was used to inhibiting PI3K/AKT pathway or blocking AKT phosphorylation. The optimal treatment concentration and time for LY294002 were determined by western blotting (data not shown).
-
Software SPSS version 24.0 (SPSS, Chicago, IL) was used for statistical analysis and all quantitative data was presented as mean ± SD. All data are from at least three independent experiments. Comparisons between two groups were tested by Student's t-test. P < 0.05 was considered statistically significant.
Isolation, Culture, and Identification of Human Hair Follicle Stem Cells
Differentiation Potential of Human Hair Follicle Stem Cells
Lentivirus Packaging
Assessment of Cell Proliferation
Assessment of Cell Survival Rate
Assay of Intracellular ROS Production
Cell Apoptosis Determination by Flow Cytometry
Western Blot
The Inhibitor LY294002 to Block PI3K/AKT Pathway
Statistical Analysis
-
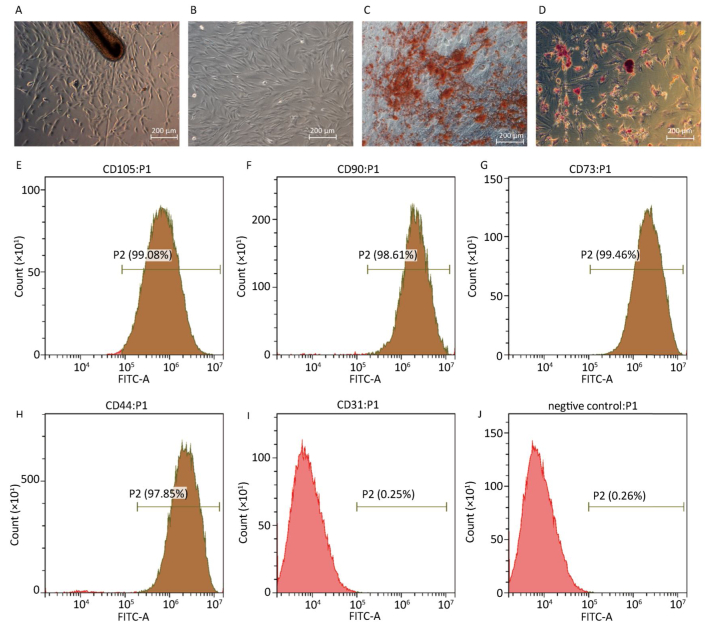
As shown in Figure 1A, when the hair follicle tissue was cultured for 1-2 weeks, hHF-MSCs migrated out of the hair follicles. The fourth passage of hHF-MSCs displayed uniform spindle-shape, fibroblastlike morphology shown in Figure 1B. After cultured in osteogenic differentiation medium for a month, the formation of mineralized nodules was observed by inversion fluorescence microscope in hHF-MSCs, as demonstrated in Figure 1C. After cultured in adipogenic differentiation medium for two weeks, the acellular lipid droplets were formed in hHF-MSCs demonstrating by Oil Red O staining. The acellular lipid droplets were stained red by Oil red O shown in Figure 1D. Flow cytometry revealed that hHF-MSCs had high expression of the surface markers for mesenchymal stem cells, CD44, CD73, CD90, and CD105; low expression of CD31 (Figure 1E-J).
Figure 1. Determination of human hair follicle stem cells and their differentiation. (A) Human hair follicle stem cells from human hair follicle; (B) The fourth passage of hHF-MSCs; (C) The mineralized nodules stained by Alizarinred S; (D) The acellular lipid droplets stained by Oil red O; (E-J) Expression of cell surface markers of CD105, CD90, CD73, CD44, CD31 and negative control determined by flow cytometry. Scale bar = 200 μm.
-
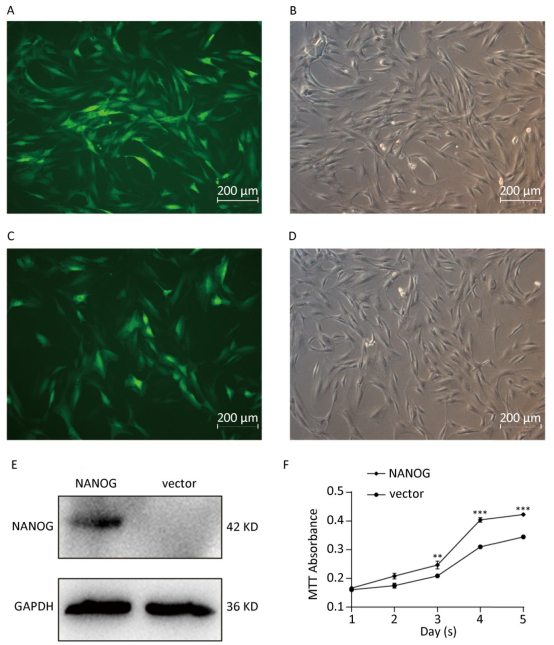
Green fluorescence exhibited that vector was transduced into the hHF-MSCs. Figure 2A-B showed vector hHF-MSCs and Figure 2C-D demonstrated NANOG hHF-MSCs. The expression of NANOG in NANOG hHF-MSCs was confirmed by Western blot analysis, which was shown in Figure 2E. Next, MTT results showed that NANOG promoted proliferation of hHF-MSCs compared with vector hHF-MSCs (Figure 2F).
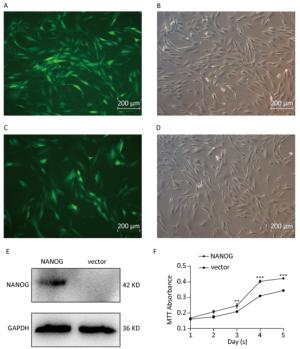
Figure 2. NANOG ectopic expression by lentivirus transduction. (A-B) The images of vector group with green fluorescence and bright light; (C-D) The images of NANOG group with green fluorescence and bright light; (E) Western blot measured the expression of NANOG in hHF-MSCs; (F) MTT assay measured cell growth for five days (**P < 0.01, ***P < 0.001). Scale bar = 200 μm.
-
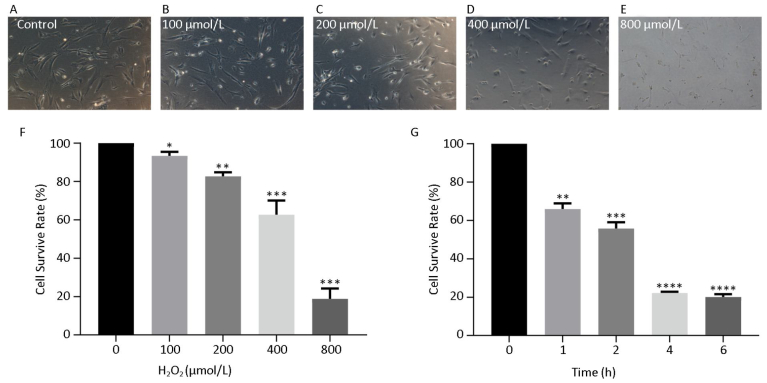
hHF-MSCs were treated with H2O2 at the concentration of 0, 100, 200, 400, and 800 μmol/L for 2 h, the cells became loss of normal morphology and enlarged, the number of viable cells decreased with the increasing of H2O2 concentration (Figure 3A-E). The cell survival rate determination by CCK-8 revealed that the cell survival rate decreased with the concentration increasing of H2O2. When H2O2 was at 400 μmol/L, the cell survival rate was around 50%, therefore the concentration of 400 μmol/L H2O2 was selected for the followed experiment (Figure 3F). The cells treated with 400 μmol/L H2O2 for different time (0, 1, 2, 4, 6 h) displayed decreased cell survival rate with the prolonged time of exposure to H2O2 (Figure 3G).
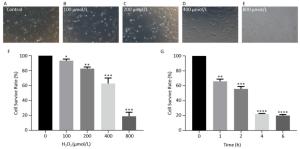
Figure 3. hHF-MSCs morphology treated with H2O2 and cell survival rate at different concentration of H2O2 and at different time of 400 μmol/L H2O2. (A) Cells without H2O2; (B) Cells with 100 μmol/L H2O2; (C) Cells with 200 μmol/L H2O2; (D) Cells with 400 μmol/L H2O2; (E) Cells with 800 μmol/L H2O2; (F) Cell survival rate at different concentration of H2O2; (G) Cell survival rate at different time of 400 μmol/L H2O2 (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
-
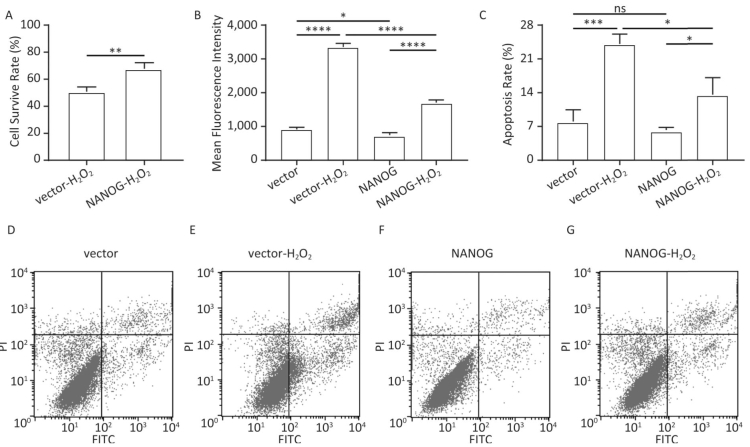
As shown in Figure 4A-B, NANOG hHF-MSCs showed higher cell survival rate and less ROS compared with vector hHF-MSCs after being treated with H2O2. The apoptosis of hHF-MSCs was also decreased in NANOG hHF-MSCs compared with vector hHF-MSCs after bening treated with H2O2 (Figure 4C-G).
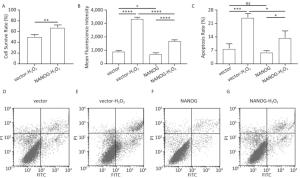
Figure 4. Cell survival rate, ROS production and apoptosis. (A) Cell survival rate comparison of NANOG hHF-MSCs and vector hHF-MSCs after being treated with H2O2; (B) Comparison of ROS production between NANOG hHF-MSCs and vector hHF-MSCs after being treated with H2O2; (C) Analysis results of apoptosis rate by flow cytometry; (D) Apoptosis of vector hHF-MSCs; (E) Apoptosis of vector hHF-MSCs after being treated with H2O2; (F) Apoptosis of NANOG hHF-MSCs; (G) Apoptosis of NANOG hHF-MSCs after being treated with H2O2 (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
-
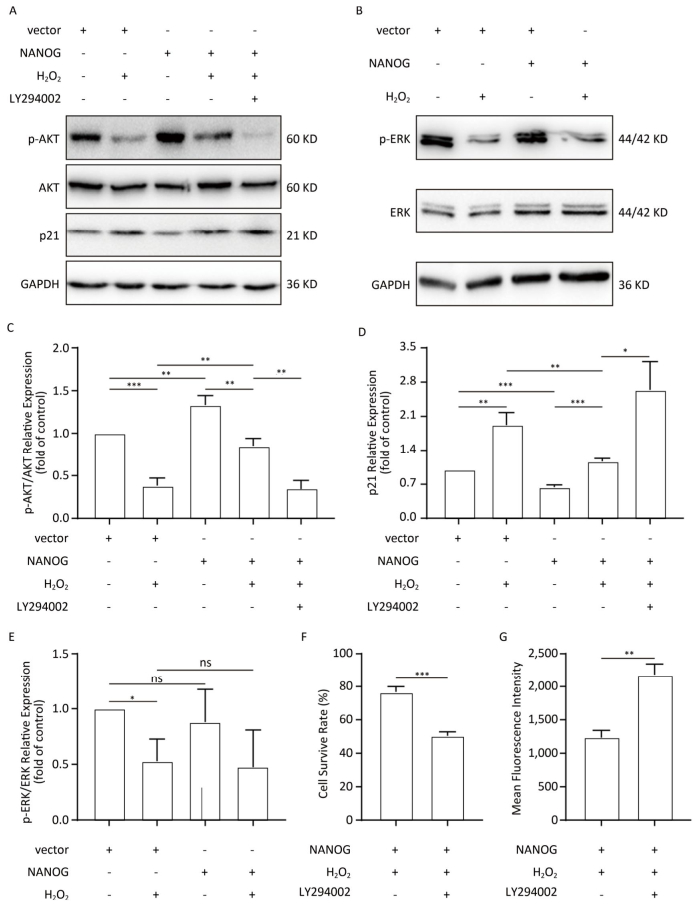
NANOG hHF-MSCs showed higher expression of p-AKT and ratio of p-AKT/AKT compared with vector hHF-MSCs, which indicates that NANOG could activate AKT. Treated with H2O2, both NANOG hHF-MSCs and vector hHF-MSCs also manifested decreased expression of p-AKT and ratio of p-AKT/AKT, which indicates that H2O2 could inhibit AKT. In addition, NANOG hHF-MSCs treated with H2O2 exhibited higher expression of p-AKT and ratio of p-AKT/AKT compared with vector hHF-MSCs treated with H2O2 (Figure 5A, 5C).
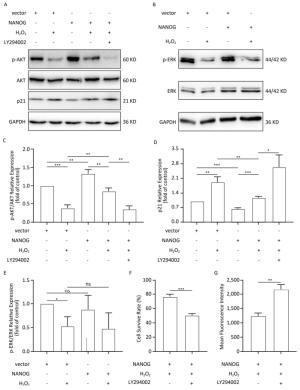
Figure 5. Protection of hHF-MSCs against H2O2 by NANOG through activation AKT signaling. (A) The expression of p-AKT, AKT, and p21 in hHF-MSCs; (B) The expression of p-ERK and ERK in hHF-MSCs; (C) The relative expression levels of p-AKT and AKT in hHF-MSCs were quantitatively analyzed with Tanon Gis analytical software; (D) The relative expression levels of p21 in hHF-MSCs were quantitatively analyzed with Tanon Gis analytical software; (E) The relative expression levels of p-ERK and ERK in hHF-MSCs were quantitatively analyzed with Tanon Gis analytical software; (F) Cell survival rate of NANOG hHF-MSCs with H2O2 at the presence of inhibitor LY294002; (G) ROS production of NANOG hHF-MSCs with H2O2 at the presence of inhibitor LY294002 (*P < 0.05, **P < 0.01, ***P < 0.001).
In addition, p21 is regarded as cell cycle inhibitor and downstream molecule of AKT which AKT can repress its expression. Our results showed that NANOG hHF-MSCs had lower expression of p21 compared with vector hHF-MSCs with or without H2O2 (Figure 5D).
After treated with H2O2, NANOG hHF-MSCs with LY294002 had lower survival rate and higher ROS production. These results indicated that NANOG protected hHF-MSCs through activating AKT pathway (Figure 5F, 5G).
There was no significant difference in the expression of p-ERK between NANOG hHF-MSCs and vector hHF-MSCs, and after being treated with H2O2, the expression of p-ERK was decreased in both NANOG hHF-MSCs and vector hHF-MSCs. Moreover, there was no significant different between NANOG hHF-MSCs and vector hHF-MSCs (Figure 5B, 5E).
hHF-MSCs Determination and Their Potential of Differentiation
Generation of NANOG Ectopic Expression
hHF-MSC Cell Survival Rate Treated with H2O2
Protection of hHF-MSCs by NANOG Against H2O2
Protection of hHF-MSCs by NANOG through Activation of AKT Pathway
-
Hair follicle is one of the mammalian skin appendages. Hair follicle growth is periodic, and hair follicle can support and promote hair growth, which is the basis of hair survival. Hair follicle is abundant of stem cells[3], which can be affected by outside factors and result in changes of hair structure and prolonged growth cycle[6]. It is revealed that hHF-MSCs are involved in the hair follicle formation, self-renewal and cycle formation, and play an important role in hair growth, regeneration and periodic growth[17]. hHF-MSCs are damaged, hair withering or even hair loss will develop.
It is suggested that oxidative stress induces hair greying and inhibits hair follicle cell cycle progression[18]. Exposure to harmful environmental factors such as ultraviolet radiation causes the injury of hair follicle stem cells by oxidative stress[18, 19]. It is well established that H2O2 is regarded as inducer to establish oxidative stress model in vitro for cell research[20]. H2O2 is a strong oxidant and will cause the damage of cells through the production of ROS, even apoptosis[21]. It is worthy to note that the concentration of H2O2 varies greatly from different cell types[22]. In our study, we chose 400 μmol/L H2O2 to treat hHF-MSCs for 2 h and found out that cell morphology, cell survival rate, ROS production and apoptosis changed significantly.
There is no doubt that NANOG is a crucial transcription factor in maintaining stem cells property. However, there is few study of NANOG against cell damage from oxidative stress. Our previous study shows that NANOG is able to promote proliferation and to delay the senescence of hHF-MSCs through activation of AKT pathway. Thus, we hypothesized that NANOG would contribute to protecting hHF-MSCs against the damage of oxidative stress caused by H2O2.
In our study, NANOG hHF-MSCs displayed higher cell survival rate, higher expression of p-AKT and lower ROS production than those were absent of NANOG when they were treated with H2O2. Moreover, AKT could be activated by NANOG as shown in the higher expression of p-AKT when NANOG was transduced into hHF-MSCs. p21 is a downstream molecule of AKT. When AKT was upregulated, the expression of p21 would be downregulated. In our study, NANOG could activate and upregulate AKT, p21 was downregulated, which further confirms that NANOG can function through AKT/p21 pathway.
ERK signal pathway is also implicated in the process of cell survival and repairing[23]. However, the findings of our study show that NANOG could not activate ERK pathway, thus our result suggests that NANOG attenuates cell damage by activation AKT but not ERK.
In conclusion, the present study established a cell damage model using H2O2 to investigate the protective effects produced by NANOG against H2O2. The findings of our study show that NANOG ectopic expression can increase the cell survival rate, decrease ROS production and apoptosis of hHF-MSCs treated with H2O2 through activation of AKT pathway. NANOG could be applied in agents to protect hair follicle cells which needs future study.
-
SHI Jia Hong performed the experiment and wrote the manuscript. ZUO Kui Yang, ZHANG Ying Yao, and other persons were involved in collecting data and analyzing the results. LIU Jin Yu and SHI Jia Hong contributed to the study design.
-
All the authors declare that there is no conflict of interest.
the Jilin Province Science and Technology Development Plan 20190304044YY
the China Natural National Science Foundation 81573067
the Joint construction project between Jilin province and provincial colleges SXGJQY2017-12
the Open Research Project of the State Key Laboratory of Industrial Control Technology, Zhejiang University, China ICT1800381
the Innovative special industry fund project in Jilin province 2018C049-2

 Quick Links
Quick Links
 DownLoad:
DownLoad: