
-
Poyang Lake is one of the most important wintering sites for waterfowl along the migration route from East Asia to Australia. This area is a Ramsar site, a wetland site designated internationally important under the Ramsar Convention[1]. Hundreds of thousands of migratory birds, including rare and endangered species, from Siberia, Mongolia, Japan, and northeastern and northwestern China overwinter at Poyang Lake annually[2]. Bird monitoring showed that over 98% of the global population of Siberian cranes, 50% of swan geese, and 50% of white-napped cranes overwinter at Poyang Lake. In addition, 80% of the global population of oriental white storks is also found here in winter[3,4].
Wild waterfowl hosts a vast diversity of well-known and many potential pathogens[5,6]. Farmers raise poultry in the water system of the Poyang Lake area[7]. Thus, migratory birds may share food, water, and even habitats with domestic poultry, creating opportunities for pathogen transmission[7]. Specifically, the effective habitat area for migratory birds to forage in dry seasons (autumn and winter) is greatly reduced[8,9], resulting in significantly more waterfowl foraging on farmland around the lake than in wet seasons, exacerbating the spread of pathogens between domestic and wild birds[7].
The emergence of new infections poses a threat to both animal and human health. To gain a better understanding of newly emerging pathogens at Poyang Lake, we characterized the bacterial microbiome and virome diversity of samples through metagenomic analysis in the Duchang Tangkou Wildlife Rescue Station and identified a complete genome of new pigeon mesivirus-like virus.
-
A total of ten specimens, one surface-smearing swab per cage, were collected at the Duchang Tangkou Wildlife Rescue Station (29.209982 N, 116.463861 E) on March 10, 2019, named S01–S10. The caged bird species of S01–S08 are unknown. The species of S09 and S10 were a skylark and a red turtle dove, respectively. The swabs were immediately placed in sterile tubes at 4 °C until transported to the laboratory for storage at −75 °C.
-
Total RNA was extracted from each sample using the MagMAX CORE Nucleic Acid Purification Kit (Applied Biosystems, Shanghai, China, Cat# A32702) according to the manufacturer’s instructions. RNA was eluted in a final volume of 85 µL.
-
An RNA reverse transcription kit (MatriDx Biotech Corp., Hangzhou, China, Cat# MD017) was used for library preparation. For each sample, 14 µL of RNA was mixed with 2 µL of the enzyme mixture in buffer in a 0.2 mL tube of as the first step. The total reaction volume was 20 µL. PCR using Bio-Rad T-100 cycler (Hercules, CA, USA) was conducted. The reaction conditions were as follows: 25 °C for 10 min, 50 °C for 30 min, 75 °C for 10 min, and held at 4 °C. The chain synthesis products (20 µL) were added to 2 µL of the enzyme mixture in the buffer and 18 µL nuclease-free ddH2O in the second step. A total volume of 40 µL was used in the PCR reaction. The reaction conditions were as follows: 16 °C for 15 min and stored at 4 °C. DNA purification was performed using a DNA purification kit (magnetic bead method) (Matridx Biotech Corp., Hangzhou, China, Cat# MD012T). The 35 µL purification solution was prepared for the follow-up experiments. The cDNA concentrations were determined using a Qubit X-Green II dsDNA Quantitation Kit (Yuheng Biotech Corp., Suzhou, China, Cat# Q2038). All cDNA was diluted 1:200 with dsDNA HS Buffer.
-
Libraries were constructed according to the manufacturer’s protocol using the Metagenomic DNA Library Preparation Kit (MatriDx Biotech Corp., Hangzhou, China, Cat# MD001T). The DNA was fragmented using an enzyme. The reaction (50 µL) was incubated at 37 °C for 10 min, 75 °C for 10 min, and held at 4 °C. The adaptors were added to the fragmented DNA solution. The reaction conditions were set at 20 °C for 15 min and 75 °C for 5 min. The solutions containing adaptors were purified again. DNA was quantified using a Magic dsDNA HS Assay Kit (magic-bio, Cat #VG00537). The thermocycling conditions were 98 °C for 45 s, followed by 13 cycles of denaturation (98 °C for 15 s), annealing (60 °C for 30 s), and extension (72 °C for 30 s). Equal concentrations of the samples were added to the final pool. Libraries were pooled and sequenced (75 bp single end) using a NextSeq500 sequencer. The clean raw reads were retained by removing low-quality and low-complexity reads. All sequencing was performed by MatriDx Biotech (Hangzhou, China).
-
For each library, sequencing reads were trimmed using Trimomatic V0.33[10]. The obtained reads were used to perform taxonomic analysis using the Kraken2 program with default parameter settings using the Standard PlusPF database (
https://benlangmead.github.io/aws-indexes/k2 , 5/17/2021)[11]. Species abundance after classification with Kraken2 was re-estimated using Bracken[12]. No filtering of the host/bacterial reads was performed before taxonomic analysis. For sample comparison, principal coordinate analysis (PCoA) based on Jaccard distance was performed on the bacterial microbiome or virome counts of the Bracken outputs.The trimmed reads of each sample were individually assembled using MEGAHIT v1.2.9 (default parameter)[13]. Prodigal V2.6.3 was used to perform gene prediction on the de novo results and extract the complete genes (“partial=00”)[14]. The predicted complete genes were searched against the nucleotide sequence database from the National Center for Biotechnology Information (NCBI) with e-values of 1e−5 using BLAST [
https://blast.ncbi.nlm.nih.gov/Blast.cgi ]. The hypothetical cleavage map of the picornavirus polyprotein was derived from alignment with the closest picornaviruses.The RNA-dependent RNA polymerase (RdRp) region of the viral gene and closely related amino acid sequences were aligned using MAFFT v7.222[15]. A phylogenetic tree was constructed using MEGA 7 based on the maximum-likelihood method, and the bootstrap value was tested with 1,000 replications[16]. The tree was edited and visualized using Interactive Tree of Life (iToL)[17].
-
Clean data and bio-samples can be found in NCBI BioProject PRJNA862611. The complete genome sequence of NC-BM-233 was submitted to GenBank and assigned the accession number OP169446.
-
Ten libraries were constructed, and 120,085,728 clean sequence reads were generated. Using the Standard PlusPF database, 7,507 reads (0.06‰ of the total reads) were assigned to Viruses by Kraken2 and Bracken, and 45,871,237 reads (38% of the total reads) were assigned to Bacteria (Table 1).
Sample Reads count Viruses reads (‰) Bacteria reads (%) Unclassified reads (%) S01 14,189,681 638 (0.04) 5,592,952 (39.42) 8,594,075 (60.57) S02 12,396,722 139 (0.01) 5,895,415 (47.56) 6,499,171 (52.43) S03 12,665,698 891 (0.07) 5,171,548 (40.83) 7,487,318 (59.11) S04 12,730,271 696 (0.05) 4,067,597 (31.95) 8,634,041 (67.82) S05 12,628,298 2,237 (0.18) 5,446,785 (43.13) 7,162,035 (56.71) S06 12,671,688 612 (0.05) 4,943,276 (39.01) 7,692,390 (60.71) S07 12,451,945 168 (0.01) 5,156,039 (41.41) 7,288,344 (58.53) S08 11,379,174 754 (0.07) 4,527,355 (39.79) 6,836,391 (60.08) S09 10,539,865 420 (0.04) 4,189,283 (39.75) 6,348,867 (60.24) S10 8,432,386 952 (0.11) 880,987 (10.45) 7,473,206 (88.63) Table 1. Overview of reads of samples
-
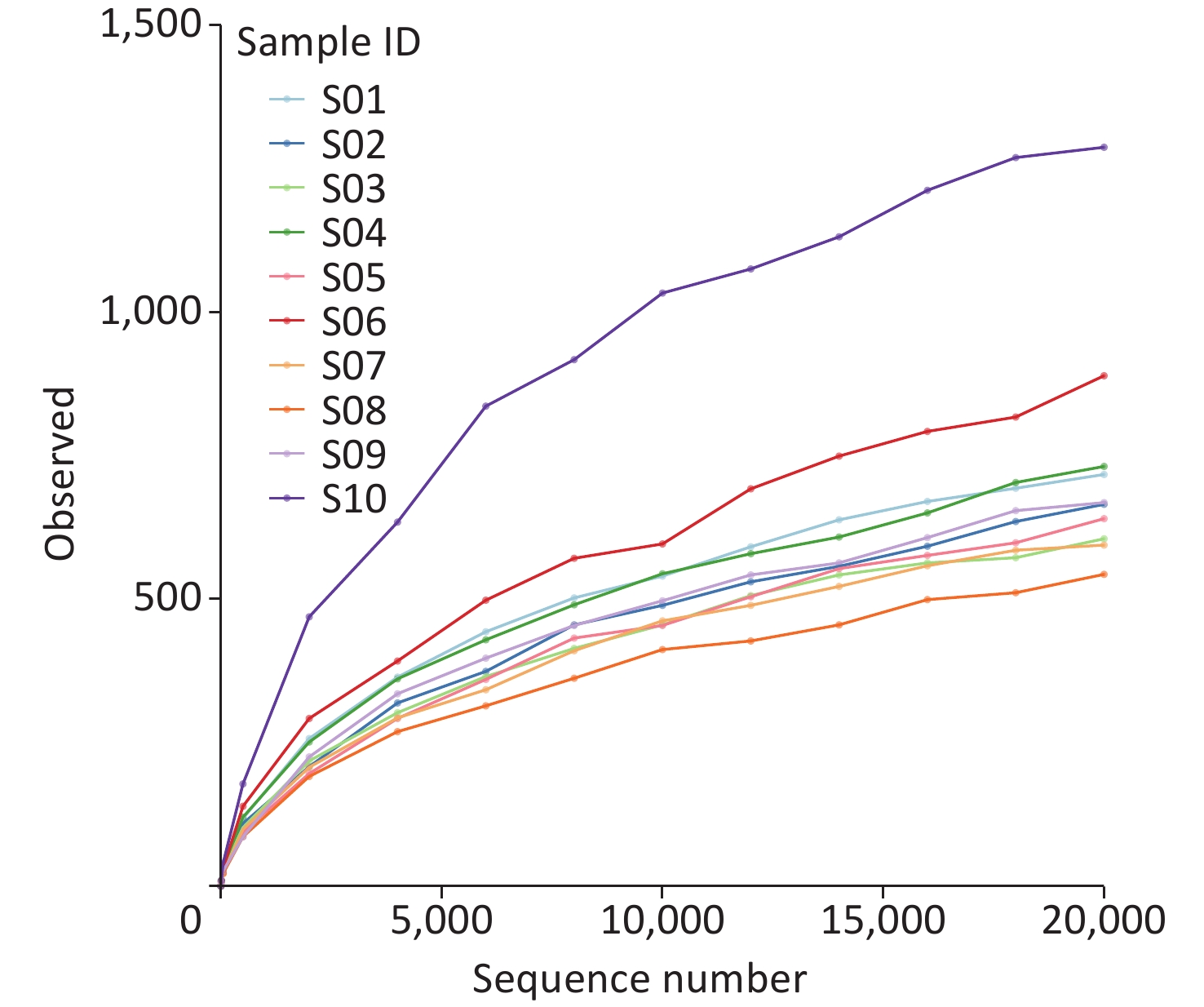
We examined the species richness of the identified taxa in ten samples. The number of observed species in S10 was the highest, followed by that in S06 and S04. In contrast, we identified a relatively low species abundance in S08 (Figure 1).
Figure 1. Rarefaction curve analysis of observed species on each sample.
-
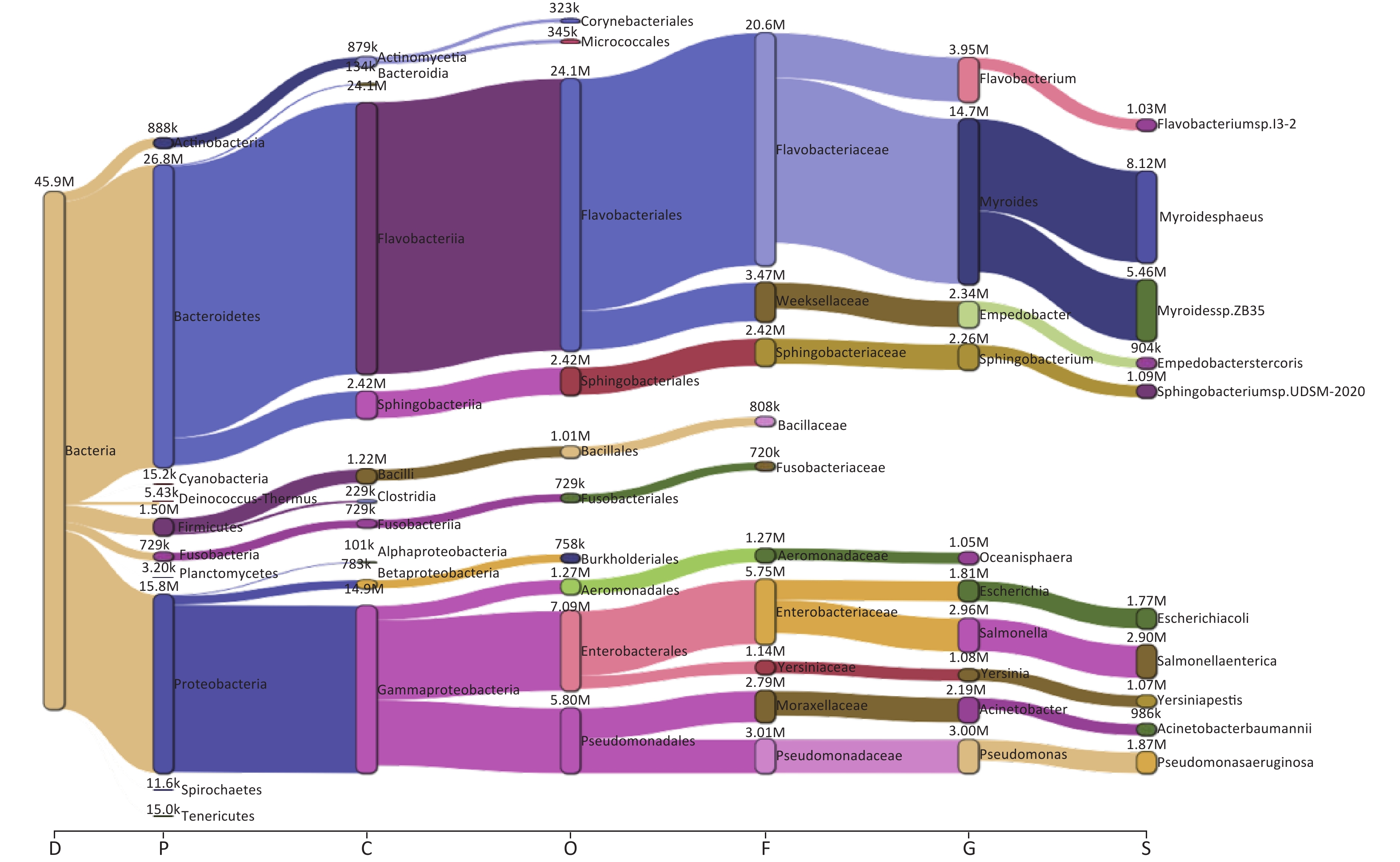
Taxonomic analysis of the bacterial microbiome in ten samples was performed using Kraken2 and Bracken, and the visualization of classification results is shown as Sankey diagrams (Supplementary Figure S1, available in www.besjournal.com). A total of 1,223 bacterial genera from 34 phyla, 71 classes, 162 orders, and 363 families were identified. The Bacteroidetes phylum comprised most of the bacterial microbiome, with an abundance of 58.52%, followed by Proteobacteria (34.53%), Firmicutes (3.28%), Actinobacteria (1.94%), and Fusobacteria (1.59%). The class Flavobacteriia was the most abundant (52.59%), followed by Gammaproteobacteria (32.40%), Sphingobacteriia (5.28%), Bacilli (2.66%), Actinomycetia (1.92%), Betaproteobacteria (1.71%), Fusobacteriia (1.59%), Clostridia (0.50%), Bacteroidia (0.29%), and Alphaproteobacteria (0.22%) (Figure 2A,
Supplementary Table S1 , available in www.besjournal.com).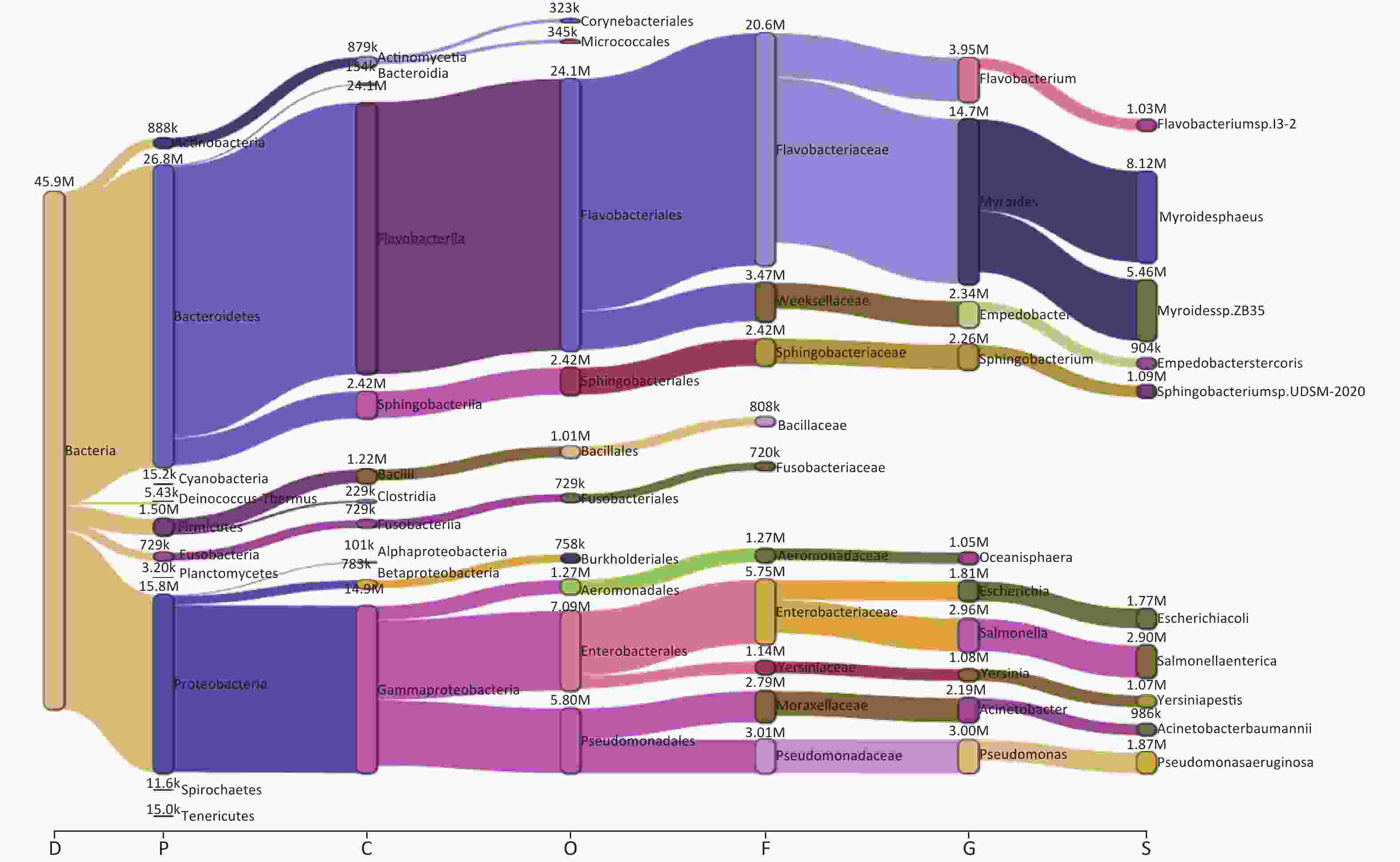
Figure S1. The Sankey plot indicating the bacterial microbiome taxon abundance of the 10 samples.
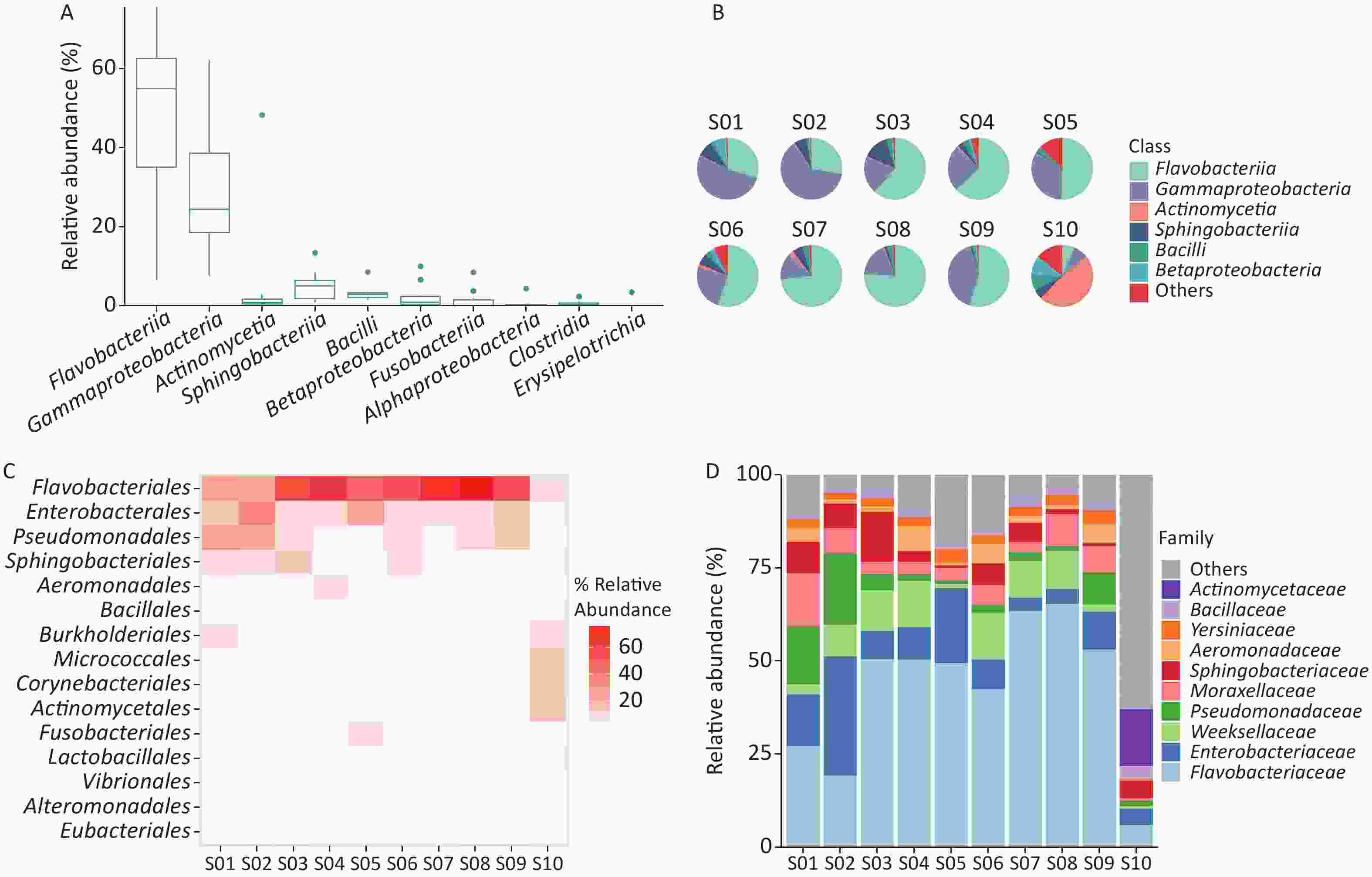
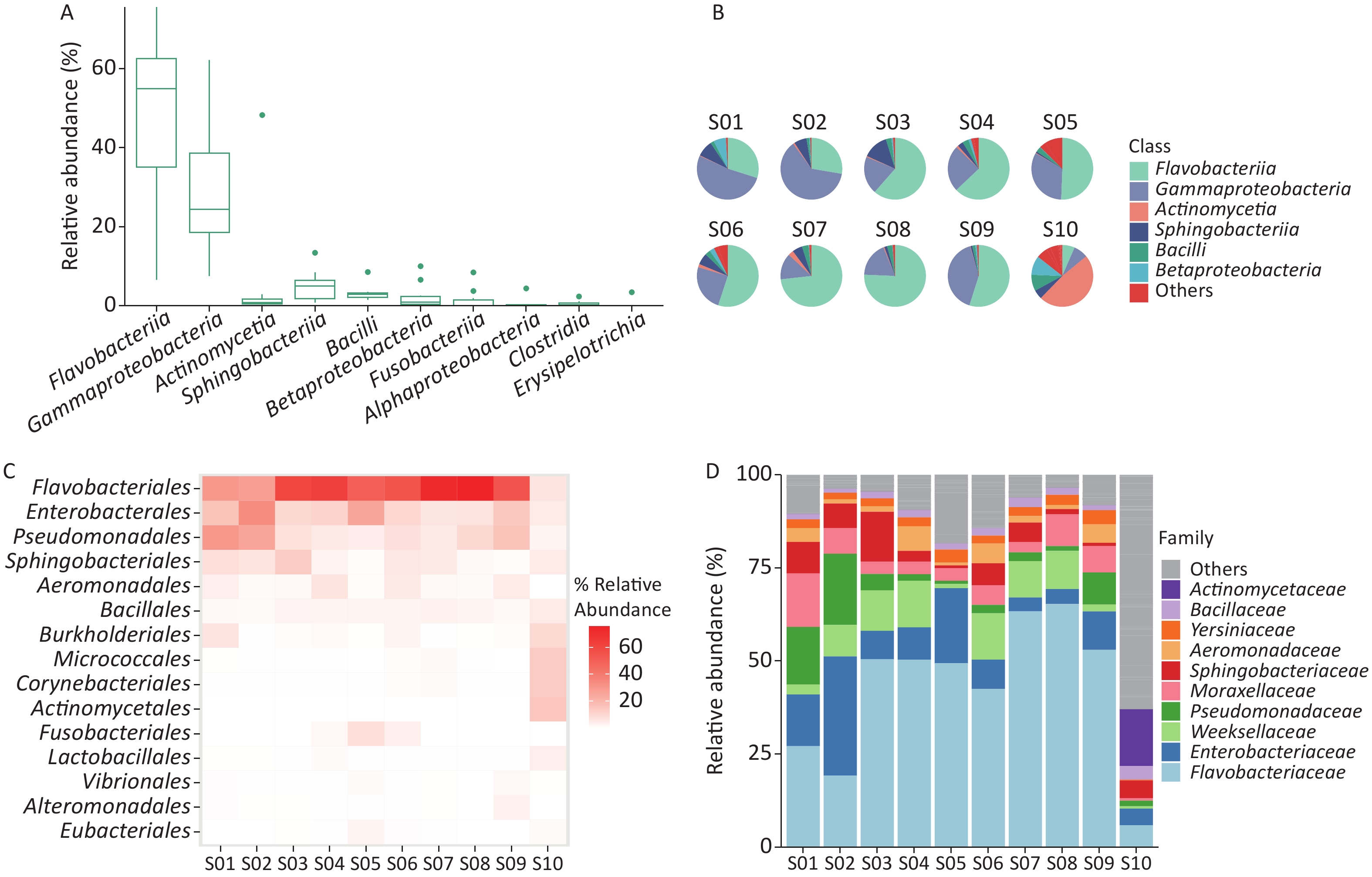
Figure 2. Identification of bacterial microbiome in environmental samples taken at Poyang Lake Animal Rescue Center using Kraken2 and Bracken. (A) The top 10 taxa at the class level of total relative abundance of identified bacterial sequence reads. The composition and diversity of bacterial microbiome identified in ten samples of (B) the group-mean pie plot of the top 6 taxa at the class level, (C) heatmap of the top 15 taxa at the order level and (D) bar plot of the top 10 taxa at the family level.
We examined the bacterial composition of each sample at the class, order, and family levels. Among S01 to S09, Flavobacteriia and Gammaproteobacteria classes were the most abundant, while Actinomycetia accounted for the largest proportion in S10 (Figure 2B). Among S01–S09, orders Flavobacteriales, Enterobacterales, and Pseudomonadales were the most abundant, and at the family level, Flavobacteriaceae and Enterobacteriaceae were the most abundant (Figure 2A, Supplementary Figure S1). In S10, Actinomycetales and Micrococcales accounted for the largest proportion, and at the family level, Actinomycetaceae and Microbacteriaceae accounted for the largest proportion (Figure 2C–D, Supplementary Figure S1).
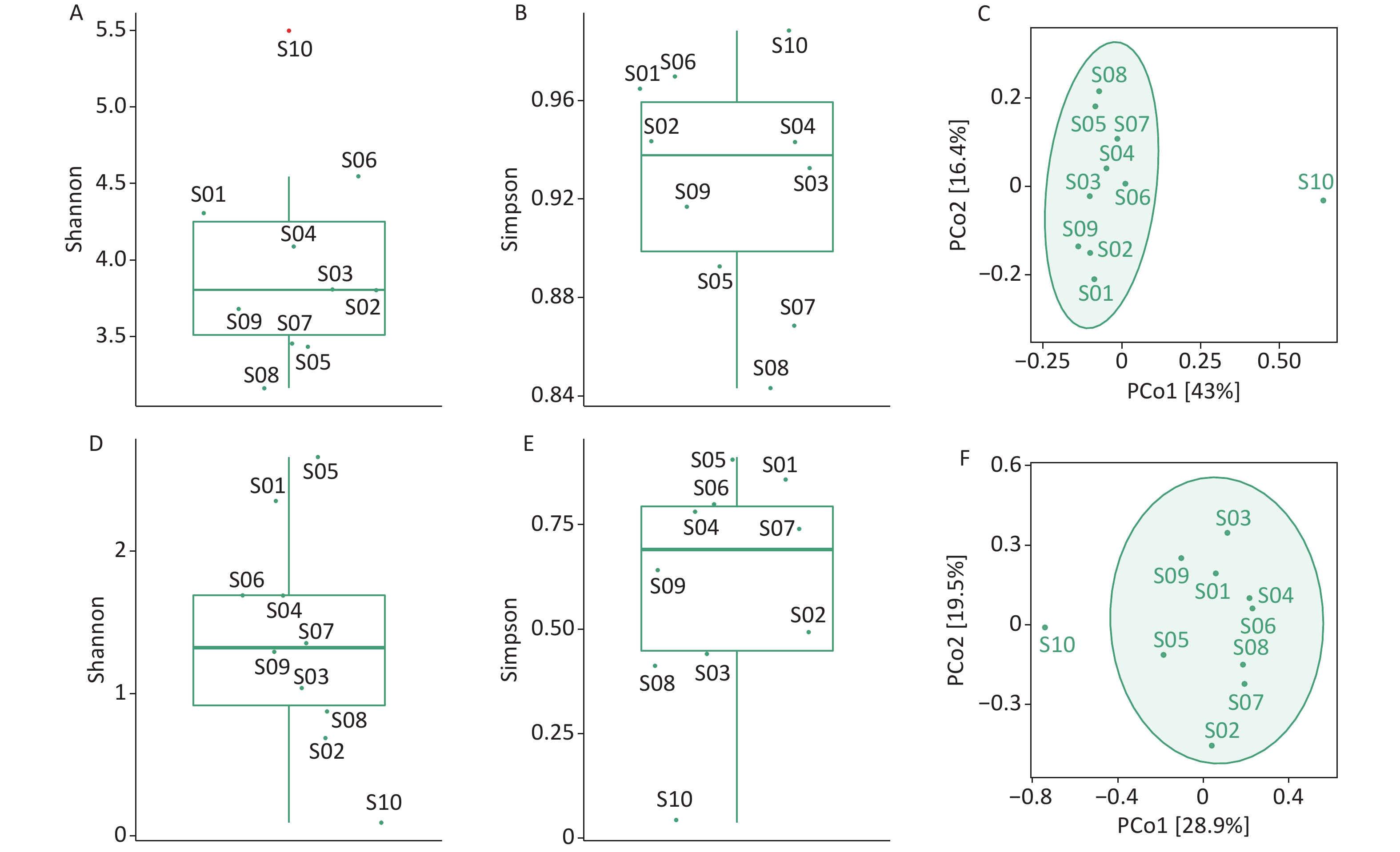
We used the Shannon and Simpson indices to illustrate bacterial microbiome diversity based on the Bracken output (Supplementary Figure S2A–B, available in www.besjournal.com). S10 was the only outlier of the bacterial microbiome Shannon index and had the highest Simpson index value among all samples. To further assess the differences between samples, PCoA results revealed that S10 differed from the other samples in the bacterial microbiome component (Supplementary Figure S2C).
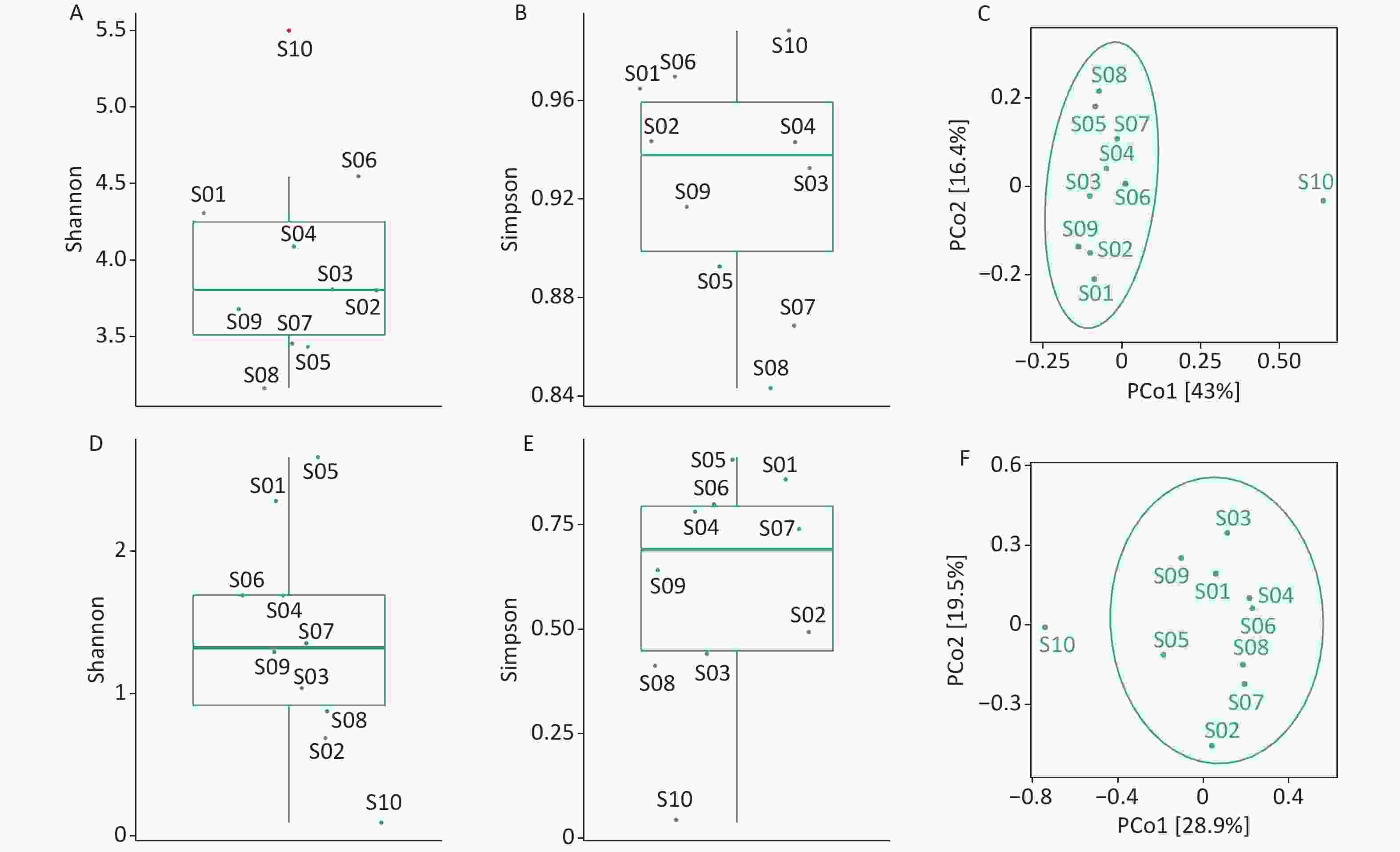
Figure S2. Diversity analysis of the bacterial microbiome and virome. (A) Box plot of Shannon index of the bacterial microbiome; (B) Box plot of Simpson index of the bacterial microbiome; (C) PCoA analysis of the bacterial microbiome based on Jaccard distance measures; (D) Box plot of Shannon index of the virus; (E) Boxplot of Simpson index of the virus; (F) PCoA analysis of virus based on Jaccard distance measures. Outliers are shown in red in the boxplot.
-
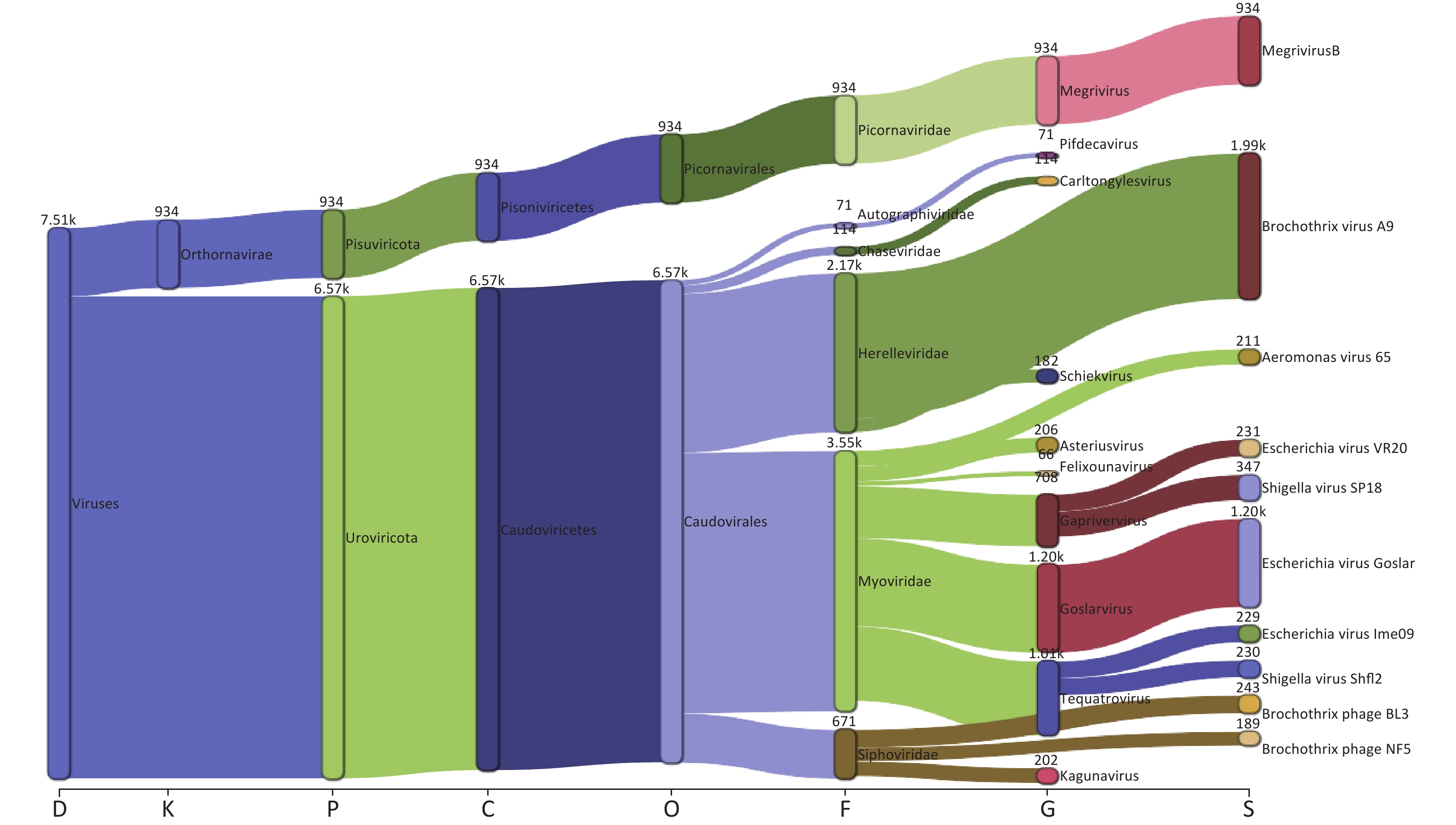
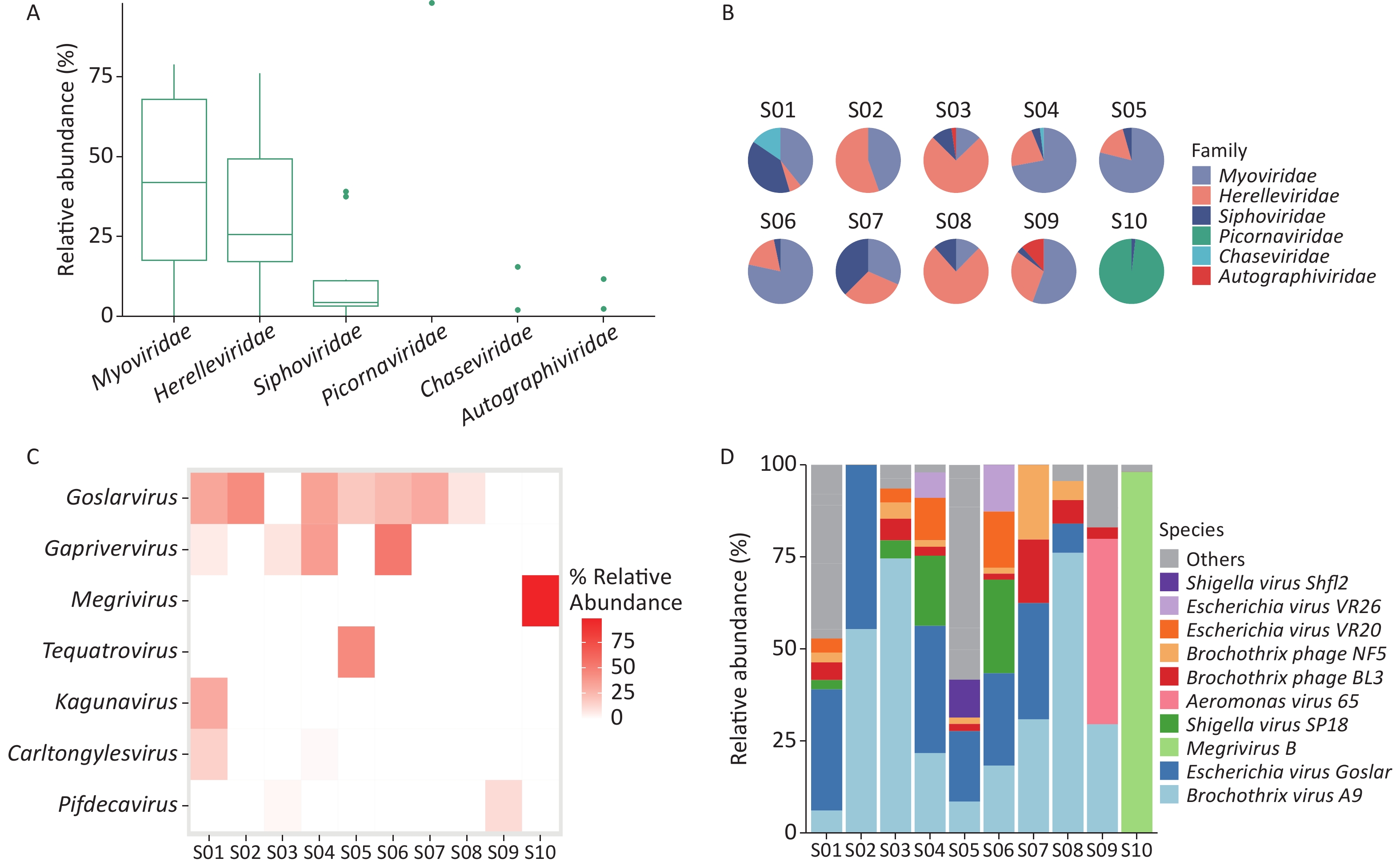
Taxonomic analysis of the virus results is shown in Sankey diagrams (Supplementary Figure S3, available in www.besjournal.com). A total of 38 virus species from 2 orders, 6 families, and 13 genera were identified. These two orders, Picornavirales and Caudovirales were from the phyla Pisuviricota (only in S10) and Uroviricota (S1–S10), respectively (Figure 3A,
Supplementary Table S1 ). Six virus families, Autographiviridae, Chaseviridae, Herelleviridae, Myoviridae, Picornaviridae, and Siphoviridae, were involved; only the family Picornaviridae belonged to the order Picornavirales. Myoviridae was the most abundant family (47.29 %), followed by Herelleviridae (28.87%) according to the taxonomic analysis results. (Figure 3A and Supplementary Figure S3).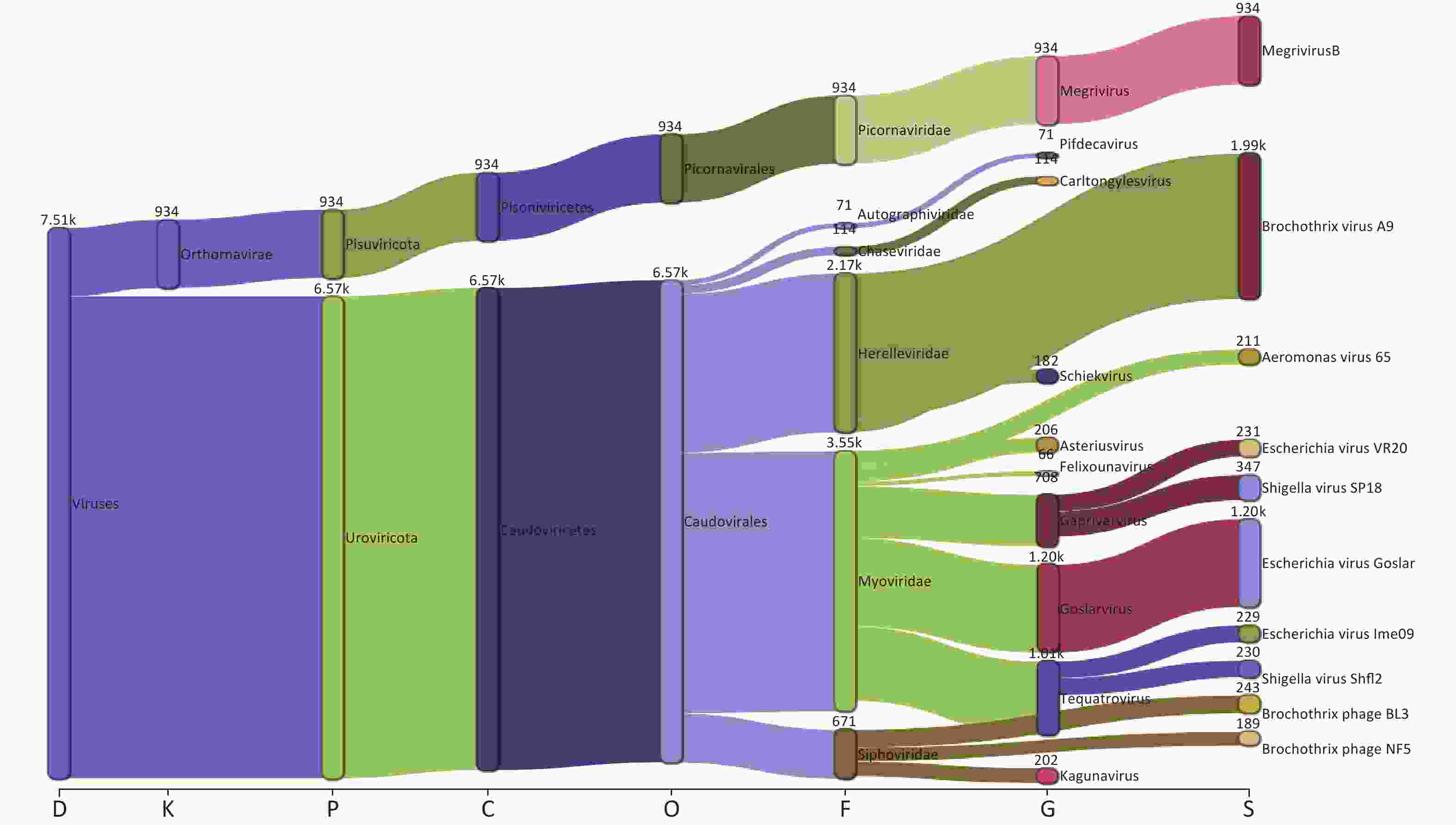
Figure S3. The Sankey plot indicating the virus taxon abundance of the 10 samples.
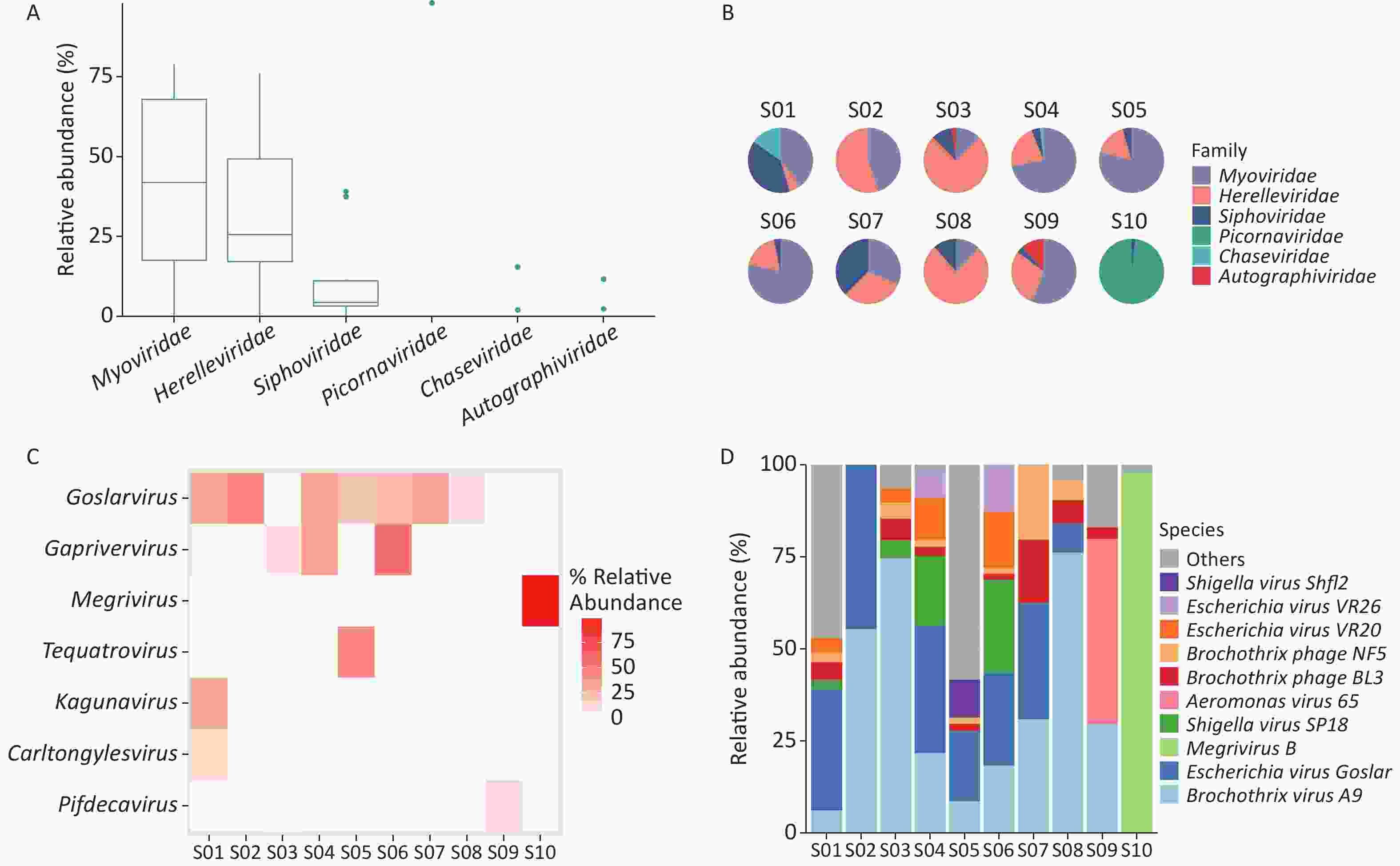
Figure 3. Identification of viruses in environmental samples taken at Poyang Lake Duchang Tangkou Wildlife Rescue Station using Kraken2 and Bracken. (A) The top 6 taxa at the family level of total relative abundance of identified virus sequence reads. The composition and diversity of viruses identified in the ten samples of (B) the group-mean pie plot of the top 6 taxa at the family level, (C) heatmap of the top 6 taxa at the genus level, and (D) bar plot of the top 10 taxa at the species level.
The proportion of Picornaviridae in S10’s virome was 98.11%, while there were no Picornaviridae in the other samples (Figure 3B). Megrivurus was the most abundant genus in S10, and was only found in S10 (Figure 3C). Unclassified species were observed in S01, S05, and S09 (Figure 3D).
We used the Shannon and Simpson indices to illustrate viral diversity based on the Bracken output (Supplementary Figure S2D–F). S10 had the lowest Shannon and Simpson index values of all samples. PCoA analysis showed that S10 were far removed from the other samples in the viral component.
-
Picornaviridae is a small, non-enveloped, single-stranded RNA virus that infects a wide range of hosts. We found Megrivirus in S10 and assembled a full-length genome of 9,184 nucleotides.
After mapping of the trimmed reads, 99,110 reads were mapped with 100% coverage and an average depth of 809.37-fold. This genome includes an open reading frame (ORF) of 8,124 nt and encodes 2,707 amino acids. The 5’UTR contained a highly conserved nucleotide motif ‘TGGTGCTGAAATATTGCAAG’ (with unknown function), which was also observed in Picornaviridae of avian origin, including turkey hepatitis virus (genus Megrivirus), duck hepatitis A virus-1 (genus Avihepatovirus), quail picornavirus (unassigned genus), pigeon picornavirus B (unassigned genus), and Anativirus (genus Anativirus)[18-21]. The BLAST result of the ORF showed the best hit to the polyprotein of pigeon mesivirus 2 (AGS15016.1), which belongs to Mesivirus with 84.10% amino acid identity. This virus was named NC-BM-233 pigeon mesivirus-like virus (NC-BM-233). The hypothetical cleavage map of the polyproteins of Mesiviruses was derived from alignments with other known picornaviruses. However, we could not determine the potential cleavage site of VP1/2A1 (Table 2).
Putative protein Proteinase Location Length (aa) P1 VP0 M1-Q390 390 VP3 T391-Q558 168 VP1 G559-D8071 249 P2 2A1 E8081-S1097 290 2A2 R1098-E1290 193 2B A1291-E1480 190 2C A1481-E1826 346 P3 3A S1827-E2008 182 3B A2009-E2036 28 3C G2037-Q2233 197 3D G2234-L2707 474 Note. 1The cleavage site of VP1/2A1 has not been fully predicted. Table 2. Coding potential/putative proteins of the genome of NC-BM-233_pigeon_mesivirus_like
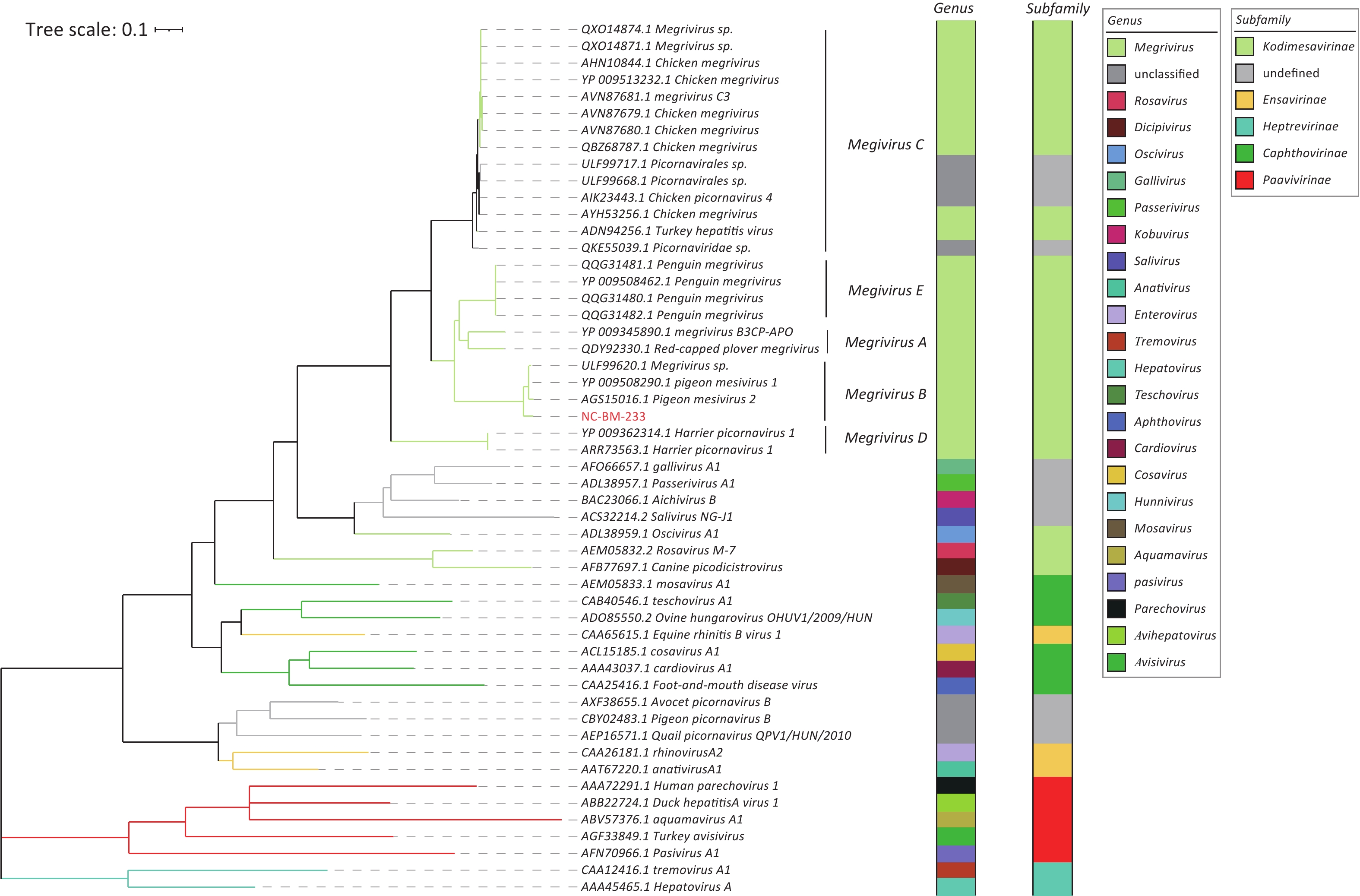
Based on the amino acid sequence of RdRp, phylogenetic analysis showed that NC-BM-233 clustered with viruses from pigeons (Figure 4). Sequences of NC-BM-233 and pigeon mesivirus clustered into Megrivirus B.

Figure 4. Phylogenetic analysis of viruses RdRps belonging to family Picornaviridae. The phylogenetic tree was constructed using the maximum-likelihood method with 1,000 bootstrap replicates. The branch widths are calculated from the bootstrap values. The virus sequence identified in this study is showed in red. Taxon information of genus and subfamily is shown as colored strips. The branch color was consistent with the subfamily information.
In addition, we found that 36 of the 39 host contigs in S10 belonged to the family Columbidae. This result was consistent with that of the caged turtle dove host.
-
The ecology of waterfowl is rich in microorganisms and viruses, which may cause cross-species transmission[22]. Poyang Lake is an important relay station for migratory birds, with many migratory birds stopping during the annual migration season. A few birds were injured for various reasons and could not survive independently. Migratory birds in need of rescue in the Poyang Lake District were rescued by volunteers and taken to the rescue station for treatment. There were many injured migratory birds in the limited space of the rescue station, which could have increased the risk of pathogen transmission. Therefore, it is particularly important to understand pathogenic pathogens by analyzing the diversity of bacteria and viruses in the living environment of birds at animal rescue stations. In this study, we used ten surface-smearing samples from rescue station cages to perform a metagenomic analysis of the high diversity of microorganisms and viruses.
Among the ten samples, the bacterial microbiome analysis results showed that the class Flavobacteriia was the most abundant, followed by Gammaproteobacteria and Actinomycetia. The proportion of class Actinomycetia in S10 was much higher than that in samples S01–S09, which mainly comprised Gammaproteobacteria and Flavobacteriia. Bacterial microbiome analysis results showed that the abundance of bacteria in cage-smearing samples was inconsistent with that of the Poyang Lake water body at the class level[22].
The class Actinomycetia is one of the largest lineages in the domain Bacteria. Actinomycetaceae, Microbacteriacea, Corynebacteriaceae, and Mycobacteriaceae were the top four of the most abundant families of Actinomycetia in S10. They were also the common families in avian-associated metagenomes[23,24]. A greater abundance of the family Actinomycetaceae was associated with obesity in humans[25]. However, the samples in this study were collected from the surface of the cages, and no host weight data were available. Microbacteriacea is a gram-positive bacteria common in bird droppings and soil environments[24,26]. Corynebacteriaceae and Mycobacteriaceae are collectively known as Corynebacterium-Mycobacterium-Nocardia (CMN) bacteria. The cell walls of CMN bacteria contain mycolic acid, which renders them less susceptible to a wide variety of antimicrobials[23].
The class Flavobacteria widely exists in fresh water, seawater, soil, and plants[22,27]. Gammaproteobacteria in S01–S09 included Salmonella enterica, Pseudomonas aeruginosa, Yersinia pestis and Acinetobacter baumannii. Salmonella enterica is a type of intestinal bacteria, which is often caused by ingesting unclean food, leading to severe diarrhea in infected individuals, and is one of the main pathogens of human food poisoning[28]. Salmonella pullorum mainly affects young chickens and causes septicemia and mass death. Salmonella pullorum causes brooding nest inflammation in adult hens and infected adult hens, carrying bacteria in the yolk and passing it to the chicks[29]. Pseudomonas aeruginosa is widely distributed in natural and normal skin, intestinal tract, and respiratory tract and is a common opportunistic pathogen in clinical practice[30]. Yersinia pestis is a vector of bubonic, pneumonic, and septicemic plague. Acinetobacter baumannii is a common nosocomial infection and pathogen in aquaculture animals, and it usually leads to bacteremia, pneumonia, meningitis, peritonitis, endocarditis, urinary tract, and skin infections[31].
The virus analysis results showed that Myoviridae, Herelleviridae, and Siphoviridae families were the dominant virus families of S01–S09, which is similar to the virome components of a freshwater Amazonian lake at the family level[32]. Myoviridae and Siphoviridae can infect Cyanobacteria, which are also known as cyanophages and are well-known phage families prevalent in multiple poultry feces[33]. An imbalance in phage diversity and abundance can lead to changes in poultry ecosystems.
The dominant virus family in S10 was Picornaviridae. Picornaviridae is a single-stranded RNA virus that is common in avians and can infect many types of avian-causing duck viral hepatitis, turkey viral hepatitis, and avian encephalomyelitis[18,34-39]. In S10, Megrivirus is the main component of the family Picornaviridae, which is widespread in healthy and diseased chickens and could lead to infectious viral gastritis in chickens[36]. We identified a new virus belonging to Megrivirus B, named NC-BM-233. A highly conserved 20 nucleotide motif was common in small RNA viruses of avian origin[18-20]. The genus Megrivirus can infect many avian species and cause diseases, indicating a potential threat to wild birds. Moreover, wild birds at Poyang Lake migrate hundreds of kilometers from Siberia to Australia, visiting many water bodies along the migration route, which enhances the chance of infection and transmission of multiple regions and hosts.
We must admit that the data results based on these reads have limitations. It is easy to misjudge the reads of low-abundance species in a taxon analysis, which may lead to inaccurate results. As metagenomics is developing rapidly, new classification technologies may provide new knowledge from our data in the future. The virome and bacterial microbiomes of S10 were different from those of the rest of the samples in this study, which might be caused by the presence of the virus NC-BM-233. Whether there is a correlation between the virome and the bacterial microbiome is worthy of further study. The hydrolysis site of VP1/2A1 in NC-BM-233 also needs further investigation.
In summary, these results indicate diverse bacterial microbiomes and viral communities in the Poyang Lake wildlife rescue station and provide a fresh perspective on the bacterial microbiome and virome diversity related to birds, and a new viral genome was discovered. Continuing to investigate bird-relative samples at Poyang Lake will provide more information about the microbiome and virome, especially for discovering potential pathogens.
-
The datasets presented in this study can be found in the following online repositories:
https://www.ncbi.nlm.nih.gov/ ;https://www.ncbi.nlm.nih.gov/sra/PRJNA862611 ;https://www.ncbi.nlm.nih.gov/nuccore/OP169446 .
HTML
Sample Collection
Nucleic Acid Extraction
Library Preparation
Metagenome Sequencing
Bioinformatic Analysis and Phylogenetic Analysis
Nucleotide Sequence Accession Numbers
Metagenomic Sequencing Overview
Rarefaction Analysis
Bacterial Microbiome Diversity
Viral Diversity
Novel Positive-Sense Single-Strand RNA Viruses of Megrivirus
CONFLICTS OF INTEREST The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
 22413+Supplementary Materials.pdf
22413+Supplementary Materials.pdf
|

|

 Quick Links
Quick Links
 DownLoad:
DownLoad: