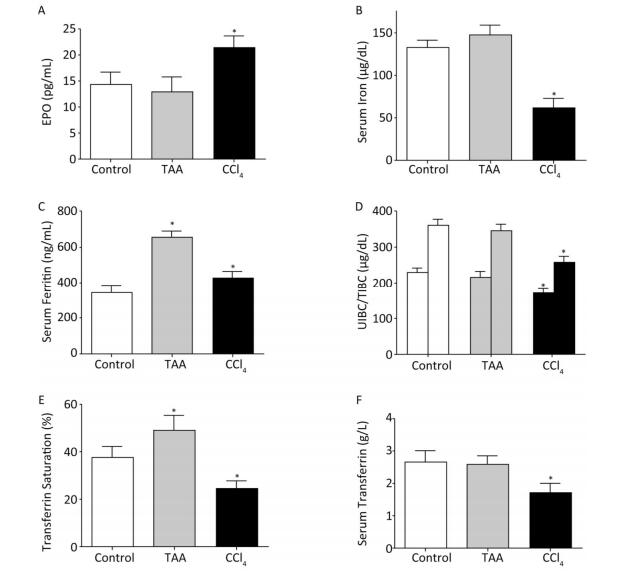
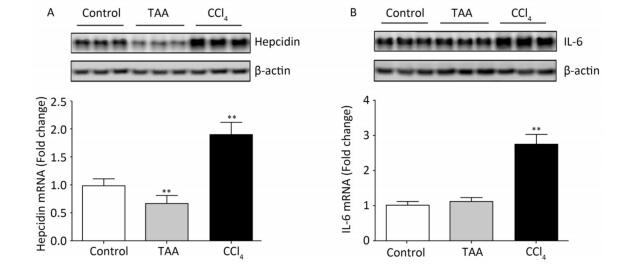
Objective This study was designed to evaluate hematological disorders and the orchestrating roles of hepcidin and IL-6 in rat models of thioacetamide (TAA) and carbon tetrachloride (CCl4) hepatotoxicity.Methods Rats were intraperitoneally injected with TAA (10 mg/100 g rat weight dissolved in isosaline) or CCl4 (100 μL/100 g rat weight diluted as 1:4 in corn oil) twice weekly for eight consecutive weeks to induce subchronic liver fibrosis. Blood and tissue samples were collected and analyzed.Results CCl4 but not TAA significantly decreased the RBCs, Hb, PCV, and MCV values with minimal alterations in other erythrocytic indices. Both hepatotoxins showed leukocytosis, granulocytosis, and thrombocytopenia. By the end of the experiment, the erythropoietin level increased in the CCl4 model. The serum iron, UIBC, TIBC, transferrin saturation%, and serum transferrin concentration values significantly decreased, whereas that of ferritin increased in the CCl4 model. TAA increased the iron parameters toward iron overload. RT-PCR analysis revealed increased expression of hepatic hepcidin and IL-6 mRNAs in the CCl4 model and suppressed hepcidin expression without significant effect on IL-6 in the TAA model.Conclusion These data suggest differences driven by hepcidin and IL-6 expression between CCl4 and TAA liver fibrosis models and are of clinical importance for diagnosis and therapeutics of liver diseases.
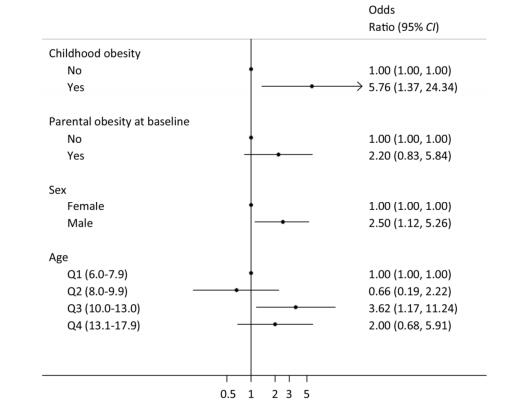
Objective Obesity is recognized as a significant risk factor for diabetes and hypertension. The present study aimed to examine the associations between adults' obesity risk and childhood and parental obesity.Methods A total of 204 children aged 6-17 years were recruited in 2002 with an average follow-up period of 13.2 years. Height and body weight were measured by trained staffs. Overweight and obesity were defined based on the Chinese standard for children and adults. T-test, analysis of variance, and Chi-square analysis were used for single factor analysis. Multiple linear and logistic regression analyses were used to perform multifactor analysis.Results The percentage of non-obese children who grew up to be non-obese adults was 62.6%, and that of obese children who grew up to be obese adults was 80.0%. There was a significant association between childhood body mass index (BMI) and adulthood BMI with a β regression coefficient of 3.76[95% confidence interval (CI):1.36-6.16], and between childhood obesity and adulthood obesity with an odds ratio of 5.76 (95% CI:1.37-24.34). There was no statistical difference between parental obesity at baseline and children's adulthood obesity, after adjustment of confounders. Male participants and those aged 10.0-13.0 years had a higher risk of adulthood obesity with odds ratios of 2.50 (95% CI:1.12-5.26) and 3.62 (95% CI:1.17-11.24), respectively.Conclusion Childhood obesity is an important predictor of adulthood obesity.
Objective We aimed to evaluate the combined effects of a high body shape index (ABSI) and a high serum C-reactive protein (CRP) level on the incidence of ischemic stroke in a Mongolian population in China.Methods A prospective cohort study was conducted among 2, 589 participants from June 2002 to July 2012 in Inner Mongolia, China. The participants were categorized into 4 groups according to their level of ABSI and CRP. Cox proportional hazards models were used to assess the hazard ratios (HRs) and 95% confidence intervals (CIs) for ischemic stroke among all groups.Results The multivariate adjusted HRs (95% CI) of ischemic stroke for high ABSI and high CRP level were 1.46 (0.89-2.39) and 1.63 (0.95-2.79), respectively. Compared with the low ABSI/low CRP level group, the multivariate adjusted HRs (95% CI) of ischemic stroke in the low ABSI/high CRP, high ABSI/low CRP, and high ABSI/high CRP groups were 1.04 (0.46-2.35), 1.06 (0.58-1.95) and 2.52 (1.27-5.00), respectively. The HR of ischemic stroke for the high ABSI/high CRP level group was the highest and most statistically significant.Conclusion We found that participants with simultaneously high ABSI and high CRP levels had the highest risk of ischemic stroke in the Mongolian population. Our findings suggest that the combination of high ABSI and high CRP levels may increase the risk of ischemic stroke.
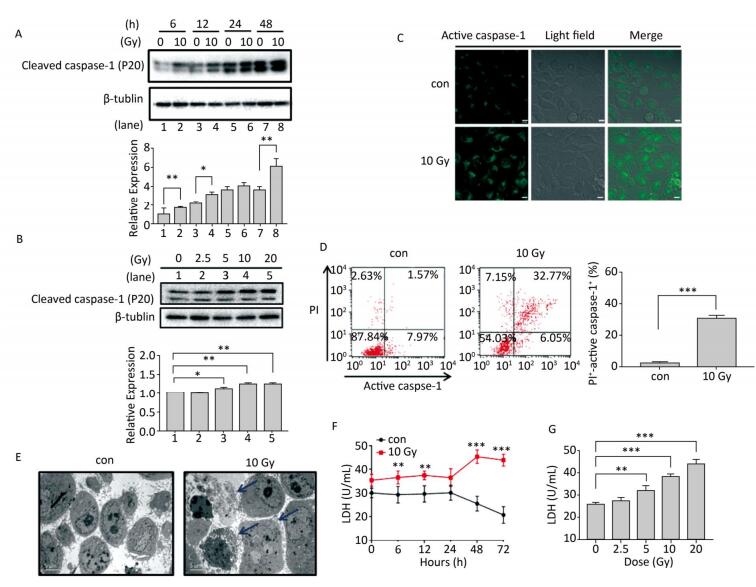
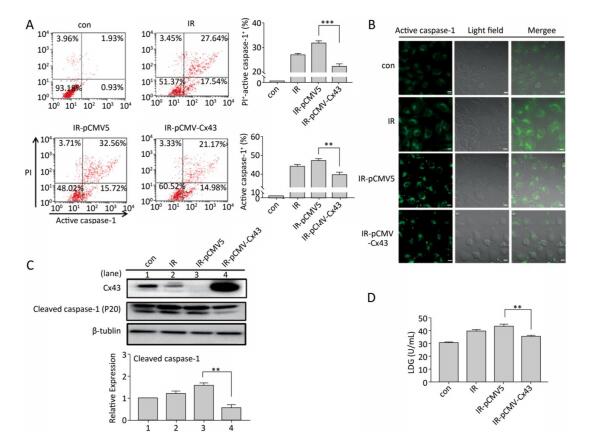
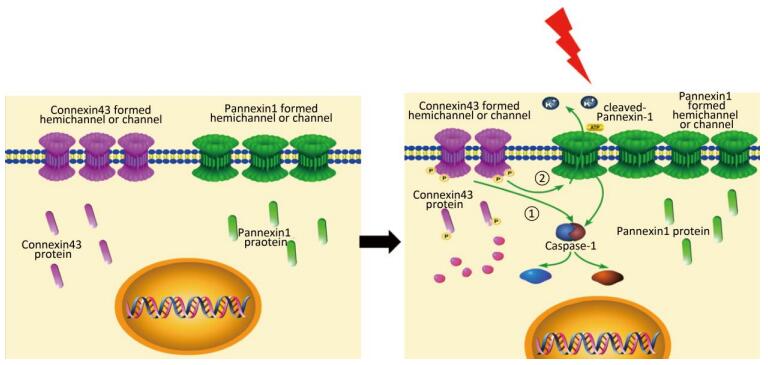
Objective Pyroptosis is an inflammatory form of programmed cell death. This phenomenon has been recently reported to play an important role in radiation-induced normal tissue injury. Connexin43 (Cx43) is a gap junction protein that regulates cell growth and apoptosis. In this study, we investigated the effect of Cx43 on X-ray-induced pyroptosis in the human umbilical vein endothelial cells (HUVECs).Methods HUVECs, Cx43 overexpression, and Cx43 knockdown strains were irradiated with 10 Gy. Proteins were detected using western blot analysis. Cell pyroptosis was evaluated using the fluorescence-labeled inhibitor of caspase assay (FLICA) and propidium iodide staining through flow cytometry and confocal microscopy. Cell morphology and cytotoxicity were detected by scanning electron microscopy and lactate dehydrogenase release assay, respectively.Results Irradiation with 10 Gy X-ray induced pyroptosis in the HUVECs and reduced Cx43 expression. The pyroptosis in the HUVECs was significantly attenuated by overexpression of Cx43 as it decreased the level of active caspase-1. However, interference of Cx43 expression with siRNA significantly promoted pyroptosis by increasing the active caspase-1 level. Pannexin1 (Panx1), a gap junction protein regulates pyroptosis, and its cleaved form is used to evaluate channel opening and active state. The level of cleaved Panx1 in the HUVECs and Cx43 knockdown strains increased in the presence of X-ray, but decreased in the Cx43 overexpression strains. Furthermore, interference of Panx1 with siRNA alleviated the upregulation of pyroptosis caused by Cx43 knockdown.Conclusion Results suggest that single high-dose X-ray irradiation induces pyroptosis in the HUVECs. In addition, Cx43 regulates pyroptosis directly by activating caspase-1 or indirectly by cleaving Panx1.
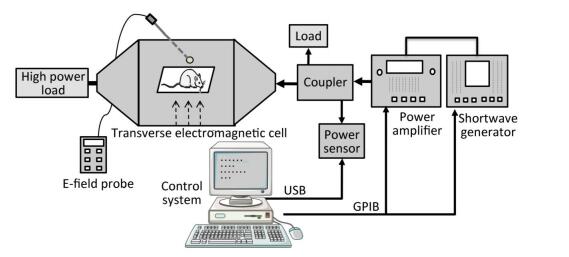
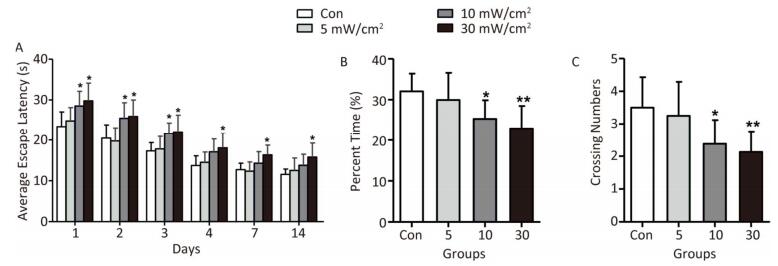
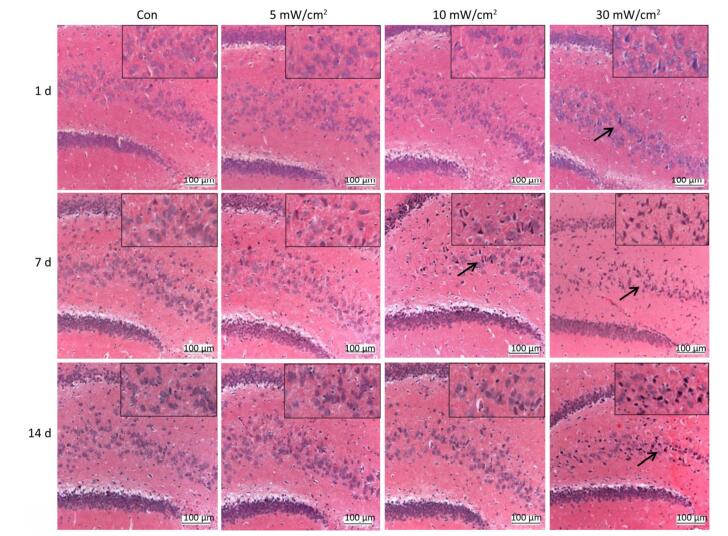
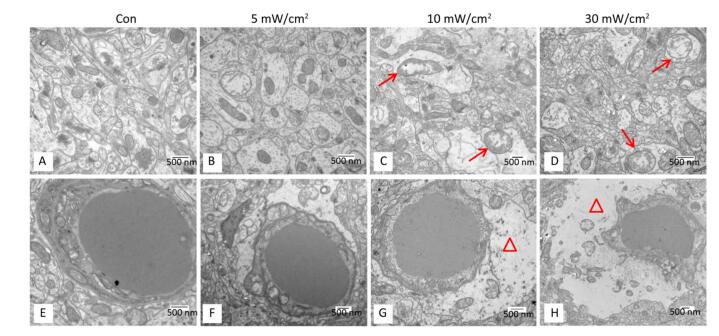
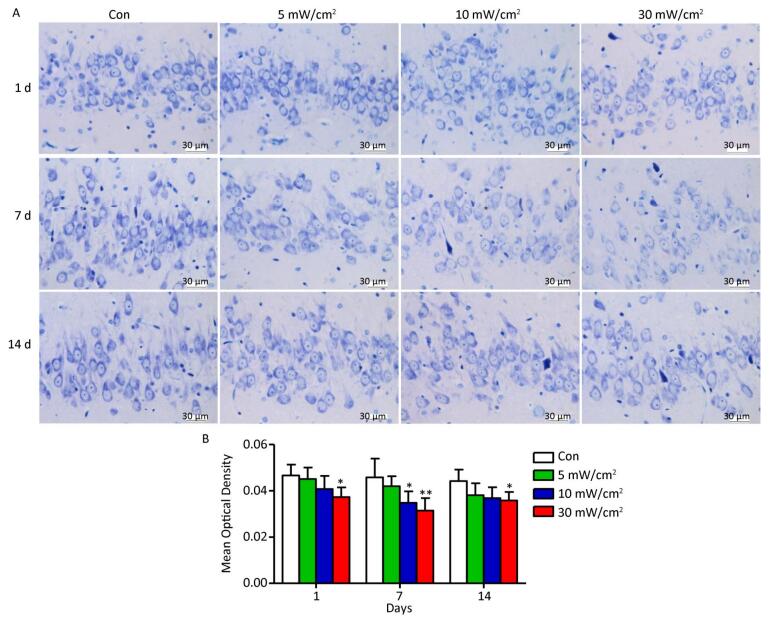
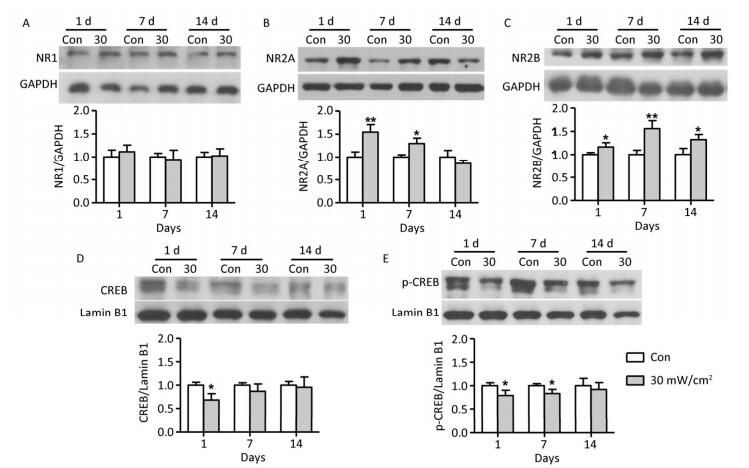
Objective To estimate the detrimental effects of shortwave exposure on rat hippocampal structure and function and explore the underlying mechanisms.Methods One hundred Wistar rats were randomly divided into four groups (25 rats per group) and exposed to 27 MHz continuous shortwave at a power density of 5, 10, or 30 mW/cm2 for 6 min once only or underwent sham exposure for the control. The spatial learning and memory, electroencephalogram (EEG), hippocampal structure and Nissl bodies were analysed. Furthermore, the expressions of N-methyl-D-aspartate receptor (NMDAR) subunits (NR1, NR2A, and NR2B), cAMP responsive element-binding protein (CREB) and phosphorylated CREB (p-CREB) in hippocampal tissue were analysed on 1, 7, and 14 days after exposure.Results The rats in the 10 and 30 mW/cm2 groups had poor learning and memory, disrupted EEG oscillations, and injured hippocampal structures, including hippocampal neurons degeneration, mitochondria cavitation and blood capillaries swelling. The Nissl body content was also reduced in the exposure groups. Moreover, the hippocampal tissue in the 30 mW/cm2 group had increased expressions of NR2A and NR2B and decreased levels of CREB and p-CREB.Conclusion Shortwave exposure (27 MHz, with an average power density of 10 and 30 mW/cm2) impaired rats' spatial learning and memory and caused a series of dose-dependent pathophysiological changes. Moreover, NMDAR-related CREB pathway suppression might be involved in shortwave-induced structural and functional impairments in the rat hippocampus.
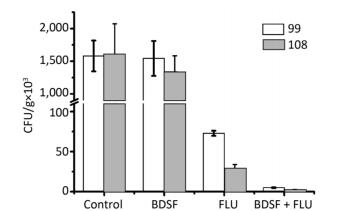
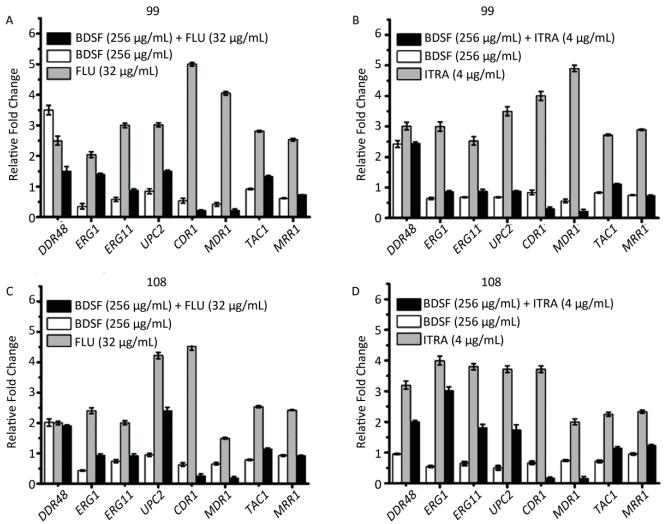
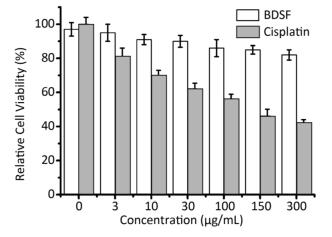
Objective To evaluate the synergy of the Burkholderia signaling molecule cis-2-dodecenoic acid (BDSF) and fluconazole (FLU) or itraconazole (ITRA) against two azole-resistant C. albicans clinical isolates in vitro and in vivo.Methods Minimum inhibitory concentrations (MICs) of antibiotics against two azole-resistant C. albicans were measured by the checkerboard technique, E-test, and time-kill assay. In vivo antifungal synergy testing was performed on mice. Analysis of the relative gene expression levels of the strains was conducted by quantitative reverse-transcription polymerase chain reaction (qRT-PCR).Results BDSF showed highly synergistic effects in combination with FLU or ITRA with a fractional inhibitory concentration index of ≤ 0.08. BDSF was not cytotoxic to normal human foreskin fibroblast cells at concentrations of up to 300 μg/mL. The qRT-PCR results showed that the combination of BDSF and FLU/ITRA significantly inhibits the expression of the efflux pump genes CDR1 and MDR1via suppression of the transcription factors TAC1 and MRR1, respectively, when compared with FLU or ITRA alone. No dramatic difference in the mRNA expression levels of ERG1, ERG11, and UPC2 was found, which indicates that the drug combinations do not significantly interfere with UPC2-mediated ergosterol levels. In vivo experiments revealed that combination therapy can be an effective therapeutic approach to treat candidiasis.Conclusion The synergistic effects of BDSF and azoles may be useful as an alternative approach to control azole-resistant Candida infections.































 Quick Links
Quick Links