

 下载:
下载:


-
With the development of urbanization and globalization, viruses previously confined to small, remote geographic areas is keeping spreading all over the world through global business[1]. Classical viral diseases are invading novel populations. For example, the transmission of Middle East respiratory syndrome (MERS) coronavirus from camel to human in Arabic countries, and to remote areas, as far as Asia[2-7], the transmission of yellow fever virus from Africa to Asia[8-12], and the transmission of Zika virus from South America to the whole world[1, 13-17]. Even in the area with inconvenient transportation, well-defined viruses can cause new headaches for public health, like the outbreak of Ebola in 2014-2016[18-25].
The identification of epidemic pathogens is therefore challenging. Traditionally, serological tests and PCR-based methods are used as first-line identification methods in outbreak of epidemic diseases[26]. However, these methods rely on pre-defined specific antibody, primer, probes, and do not work with new pathogens. The development of next generation sequencing (NGS), with unprecedented sequencing throughput, has promised to address these problems[27-31]. With the use of random oligo nucleic acids instead of predefined primers, unbiased sequencing is able to identify any virus sequence, known or unknown, local or imported, or even the pathogens that have never been reported[32].
However, unbiased sequencing or shotgun sequencing is susceptible to interference from environmental sequences. According to reported results as well as our own experience in processing traditional shotgun sequences, over 95% of success reads are commonly found to be host or environmental ribosomal RNAs[33]. Consequently, background depletion techniques are particularly useful for samples where viral load is much lower than environment sequences. Some studies reported different methods for background depletion, but virome enrichment techniques are usually needed to access a broader set of viruses[34].
In the present study, we aimed to develop a Viral Sequence Independent Targeted Amplification (VSITA) approach using a set of non-ribosomal and virus-enriched octamers (V8) and compare with traditionally used random hexamers (N6) in the reverse transcription step followed by library construction and NGS analysis, attempting to reduce the interference from unwanted RNA sequences and improve NGS-based virus identification in clinical samples.
-
First, we removed a total of 3, 985 hexamers that primed reverse-transcription of human or bacterial ribosomal RNA (Accession Number: NR_046235.3 of human 45S pre-ribosomal sequence, J01859.1 for Escherichia coli 16S ribosomal RNA, and NR_037007.2 for Staphylococcus aureus 16S ribosomal RNA) from a candidate dataset of 4, 096 (46) random hexamers. Second, we constructed a single FASTA sequence file with all the virus-related sequences available from NCBI (8, 584 viral genome sequences) (release data 8 July 2014) and corresponding complementary sequences (17, 168 virus-related sequences in total). Third, we aligned the remaining 111 hexamers with both virus-related sequences, and scored each of them according to the numbers of perfect matching to either sequence. Forth, we then obtained the first 30 hexamers with the highest scores in matching to virus-related sequences (sense and antisense). Finally, we intended to add 2 random nucleotides to the 5' tail of the hexame to increase flexibility and diversity of targeted priming. Thus these 6+2N octamers (V8) (listed in Table 1) were used in reverse-transcription step for VSITA.
Table 1. Octamer (V8) Sequences Used for VSITA (5'-3')
Motif Motif Motif Motif Motif Motif NNGGCAAT NNGATATC NNGCATTG NNGTCTAG NNAGTATA NNAAGTAT NNAATTGT NNAATCTA NNAATTGT NNACAACG NNACAATT NNACTATT NNATACTT NNATTGCC NNATTTTA NNCGTATG NNCGTTGT NNCAATGC NNCAATTG NNCATACG NNCCTAGA NNCTAGAC NNCTCGAG NNTAGATT NNTAAAAT NNTATAAA NNTATACT NNTATATA NNTCTAGG NNTTTATA -
Forty-five archived clinical samples of different types (serum, feces, and throat swabs) were included in this study. These samples were previously tested by quantitative RT-PCR assay (qRT-PCR) (Supplementary Table 1 available in www.besjournal.com) and found to be positive for either of 14 different virus types/subtypes [Japanese encephalitis virus, Rotavirus, Norovirus, Human Adenovirus (hADV), Dengue virus, Influenzavirus A H1N1, Influenzavirus A H3N2, Influenzavirus A swirl H1N1 (swlH1N1), Enterovirus 71 (EV71), Coxsackievirus A16 (CA16), Cytomegalovirus (CMV), Human immunodeficiency virus (HIV), Hepatitis B virus (HBV), Hepatitis C virus (HCV)]. These 45 samples were used in parallel to compare the V8 and N6 enrichment performance of viral sequences and removal performance of ribosomal sequences in the step of reverse transcription followed by quantitative PCR (qPCR). Additional two separate groups, one group containing 10 serum samples from patients with fever of unknown origin (F1-F10) and one containing 10 feces samples from patients with diarrhea of unknown origin (D1-D10), were also included in this study. These 20 samples were used in comparison of V8 and N6 enrichment performance following NGS analysis.
For all the 65 samples, total RNA was extracted with RNease Mini Plus Kit (QIAGEN) following manufacturer's instruction. To evaluate the performance of VSITA, both 4, 096 random hexamers (N6) and VSITA primer set (V8) were used in parallel to synthesize first strand of cDNA with Superscript Ⅲ (Invitrogen) for each sample following manufacture's instruction. For second strand synthesis of each sample, Klenow fragment was added to first strand synthesis system for 2-hour incubation at 37 ℃.
-
For the 45 previously validated positive samples, both N6-primed and V8 primed cDNAs were evaluated by qPCR assays of ribosomal RNA and viral target sequence in parallel according to the protocols described in previous reports (Supplementary Table 1). Previously reported sequences of primers and probes were used in this study to ensure reliability of real-time PCR results (as listed in Supplementary Table 1). For comparison, relative quantity of target sequence using V8-primed cDNA vs. N6 counterpart was calculated as follows:
$$ {\rm{R}} = {2^{({\rm{C}}{{\rm{t}}_{{\rm{N}}6}} - {\rm{C}}{{\rm{t}}_{{\rm{V}}8}})}} $$ (1) R: relative quantity of V8-primed target cDNA vs. N6-primed target cDNA; CtN6: Average Ct value of quantitative real-time PCR using N6-synthesized cDNA of four replicates; CtV8: Average Ct value of quantitative real-time PCR using V8-synthesized cDNA of four replicates.
-
For the 20 unidentified clinical samples, both V8 and N6 primed cDNAs were pre-enriched by multiple displacement amplification (MDA) using phi29 DNA polymerase (QIAGEN) according to manufacturer's instruction prior to library construction to guarantee enough amounts of templates for sequencing. Enriched cDNA products were evaluated with Agilent high sensitivity DNA assay on Agilent 2100 bioanalyzer. Fragmentation, end repair, adaptor ligation, size selection and purification were performed successively following instruction of Ion Torrent Hi-Q kit (Thermo Fisher Scientific). Adaptors with sequence barcodes were used to identify different libraries. After library evaluation with Agilent 2100 and real-time PCR, equal amount of each of the 40 libraries (20 samples for either V8 or N6 treatment) were grouped into four libraries and added to four 316v2 chips for sequencing. Sequencing was performed by 316 Chip Kit v2 on Ion Torrent PGM systems (Thermo Fisher Scientific) following instruction of Ion Torrent.
-
Raw data from sequencing was generated by software of Ion Torrent PGM system and downloaded in BAM format. The raw data was analyzed with our in-house software virus identification pipeline (VIP)[35].
-
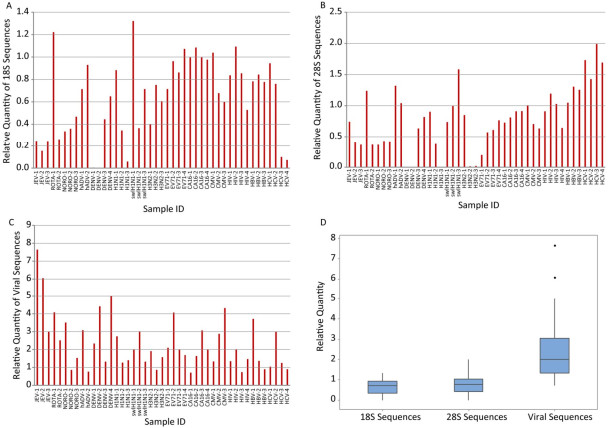
Relative quantity (the ratio) of V8-synthesized cDNA of target sequences vs. those N6-synthesized cDNA of the same sample was calculated to evaluate the ability of V8 primers in enriching target viral sequences and eliminating unwanted ribosomal sequences. Compared to N6, most V8-synthesized cDNA (39/45 for 18S, 31/45 for 28S) showed obvious decrease in the quantities of human 18S and 28S ribosomal RNA sequences (Figure 1A and 1B). The discrepancy occurred in partial samples infected with Rotavirus, hADV, swlH1N1, CMV, and HIV and all the samples infected with HBV and HCV. On the other hand, viral sequences were obviously enriched by V8 in most (38/45) samples (Figure 1C). The inconsistency occurred in partial samples infected with Norovirus, hADV, H3N2, coxsackievirus 16, HIV, HBV and HCV. In general, the relative quantities of 18S and 28S sequences were under 1 (Figure 1D), indicating the better performance of VSITA to inhibit the amplification of sequences from 18S and 28S. Meanwhile, the relative quantities of viral sequences were over 1 (Figure 1D), representing the better enrichment of viral genome sequences by VSITA.
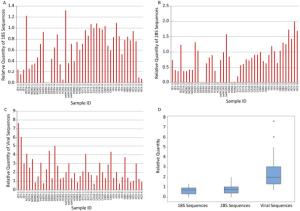
Figure 1. V8 versus N6 ratio of different RNA levels quantified by qPCR. (A) Human 18S ribosomal RNA level; (B) Human 28S ribosomal RNA level; (C) Viral RNA level; (D) Overview of relative quantity. The calculation of relative quantity of V8 versus N6 was showed in material and methods.
-
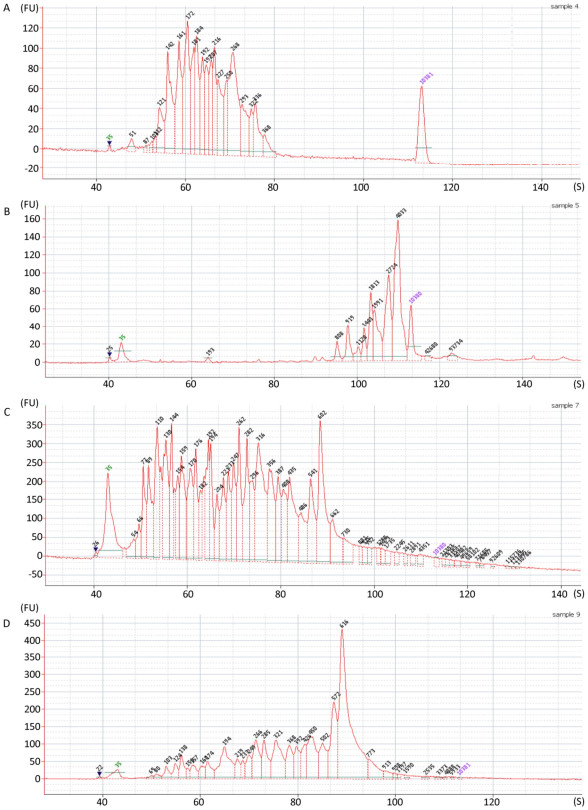
Fragment distribution of the enriched cDNA was evaluated by Agilent 2100 Bioanalyzer system. The obvious difference in the fragment distribution was observed between V8 and N6 priming. Taking the first-strand synthesis of 2 samples as examples for comparison (Figure 2A vs. 2B, 2C vs. 2D), the N6 priming showed higher diversity of the fragments, the fragments were more evenly distributed along the length axis (Figure 2A, 2C) while V8 priming tended to generate larger fragments more concentrated in some certain regions (Figure 2B, 2D). Results from other samples were showed in Supplementary Figure 1 (available in www.besjournal.com).
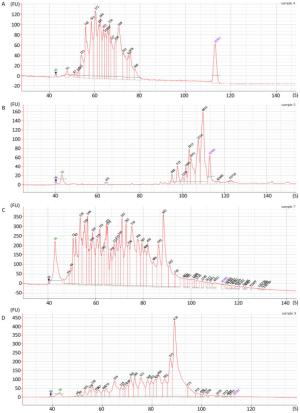
Figure 2. Fragment length analysis with Agilent 2100 bioanalyzer with: (A) N6 library from sample F1; (B) V8 library from sample F1; (C) N6 library from sample D1; (D) V8 library from sample D1.
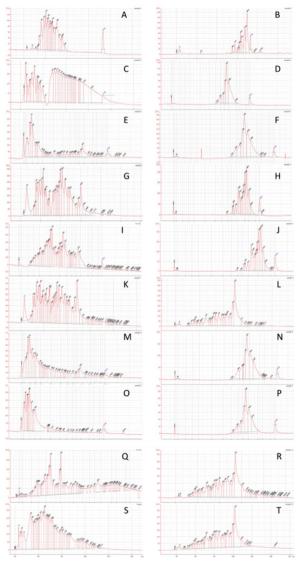
Figure Supplementary Figure 1. Fragment length analysis with Agilent 2100 bioanalyzerwith samples: (A) N6 library from sample F1; (B) V8 library from sample F1; (C) N6 library from sample F2; (D) V8 library from sample F2; (E) N6 library from sample F3; (F) V8 library from sample F3; (G) N6 library from sample F7; (H) V8 library from sample F7; (I) N6 library from sample F10; (J) V8 library from sample F10; (K) N6 library from sample D1; (L) V8 library from sample D1; (M) N6 library from sample D5; (N) V8 library from sample D5; (O) N6 library from sample D6; (P) V8 library from sample D6; (Q) N6 library from sample D8; (R) V8 library from sample D8; (S) N6 library from sample D10; (T) V8 library from sample D10.
-
Of all the 10 samples from patients with fever of unknown origin, N6 primers approach succeeded to identify 185 reads of dengue virus (DENV Ⅲ) in sample F10 and covered 25.1% of dengue virus genome, while V8 approach identified 2, 257 reads and covered 94.88% of the same dengue virus genome (Table 2). The presence of dengue virus genome in sample F10 was later validated by Real-time PCR (Supplementary Table 1). Of 4 samples (F1, F2, F3, and F7, Table 2), a few reads were generated by V8 but not by N6 approach. However, very few reads from F1, F2, and F3 were found to match with Hantavirus and Phlebovirus and real-time PCR assay failed to amplify any sequence from these samples. For sample F7, V8 approach was only able to identify 46 reads (22.31% of genome) due to low quantity of dengue virus genome (real-time PCR Ct value of 35.14). Both methods failed in the detection of the remaining 5 samples.
Table 2. Comparison of the NGS Results between the Two Methods by Clinical Samples
Sample Number Identified Virus Ct Valuea V8 N6 Reads Hit Coverage (%) Viral Reads Percentage (%) Reads Hit Coverage (%) Viral Reads Percentage (%) F1 HFRS neg 3 2.76 0 0 0 0 F2 HFRS neg 1 0.38 0 0 0 0 F3 SFTS neg 3 0.94 0 0 0 0 F7 DENV Ⅲ 35.14 46 22.31 0.08 0 0 0 F10 DENV Ⅲ 25.97 2, 257 94.88 0.61 185 25.10 0.19 D1 Echovirus 6 24.49 1, 134 92.33 8.11 817 90.13 4.42 D1 Norovirus 28.75 48, 344 86.53 8.11 25, 276 86.93 4.42 D5 Coxsackievirus A6 24.09 233 79.54 0.50 22 39.27 0.08 D6 Poliovirus 26.70 60 71.94 0.40 20 32.59 0.14 D8 Coxsackievirus A4 26.50 171 78.26 0.80 51 47.55 0.30 D10 Coxsackievirus B2 23.47 346 96.09 1.90 116 88.58 0.50 Note. aTests with reported real-time PCR methods to validate the identification of viruses. Virus sequences were identified from 5 out of 10 diarrhea samples of unknown origin (D1, D5, D6, D8, and D10) by both V8 and N6 approaches. Both methods found co-infection of Echovirus 6 and Norovirus in sample D1 with high possibility (with genome coverage of 92.33% and 90.13% for Echovirus 6, 86.53% and 86.93% for Norovirus, respectively). Of the other four samples, infection of Coxsackievirus A6, Poliovirus, Coxsackievirus A4, Coxsackievirus B2 was found by both methods and validated by real-time PCR. For virus identification with NGS, V8 showed more viral reads hit and higher genome coverage in all the samples tested (Table 2).
-
Shotgun NGS sequencing with random primer- based library construction has become the preferred choice for investigation of unknown infections in many laboratories[32]. However, clinical sample is always a combination of different genomes from the host, the pathogen, and environmental species. Under such circumstance, viral sequence is usually at disadvantage in random library construction, regardless of actual viral load[36]. Ribosomal RNA, above all, is predominantly amplified in random sequencing, and therefore became major interference in shotgun NGS-based pathogen identification, especially in viral infection cases[37]. Unlike prokaryotes and eukaryotes, there is little similarity between virus genomes. Accordingly, efforts have been made to enrich viral sequences in NGS investigation of complex clinical samples[34, 36]. Endoh et al. has reported a set of 96 non-ribosomal hexanucleotides, which preferentially primes the virus genome RNA[38], however, the viral enrichment abilities of both random hexamers and 96 non-ribosomal hexanucleotides primer sets were nearly comparative in model experiments as described in our recent report[36]. We speculate that the reduced diversity in non-ribosomal hexamers (from 46 = 4, 096 down to 96) might explain the limited coverage or sensitivity of viral pathogens found by 96 non-ribosomal hexamers.
To address this situation, we developed a novel VSITA approach by designing V8 primer set for virus-targeted selection in this study. After the removal of ribosomal RNA-targeted hexamers, we further narrowed virus-targeted hexamer sets down to an even smaller group containing only 30 virus-targeted hexamers. To expand the diversity of 30 primer set and improve detection sensitivity of viral pathogen, we intentionally added two random nucleotides (NN) to the 5' end of all the hexamers. As a result, the final V8 primer set was composed of 480 (30 × 42 = 480) non-ribosomal virus-targeted octamers. We infer that the virus-targeted specificity of V8 will not be obviously affected as the specificity of a primer is predominantly determined by its 3' end sequence.
Comparison of the virus-enrichment in cDNA synthesis between the V8 and random hexamers (N6) approaches was carried out by qPCR assays of 18S rRNA, 28S rRNA, and viral sequences. Our results showed V8 approach revealed improved virus enrichment and removal of human ribosomal RNA sequences in the majority of 45 archived clinical samples. Among a few samples (7/45), V8 approach showed inconsistent results (lower than N6) in virus (Norovirus, hADV, H3N2, coxsackievirus, HIV, HBV and HCV) enrichment. This phenomenon might be due to the difference in matrix constitution and targeted viral concentrations in extracted RNA of these clinical samples. Also, among some samples (6/45 for 18S, 14/45 for 28S), the performance of random hexamers was better in depletion of human ribosomal RNA than V8. Especially in the case of 28S sequences, N6 showed better performance than V8 in all of the 3 HBV-positive and 4 HCV-positive samples. The reason might be the nonspecific 28S amplification using cyber green-based qPCR analysis because subsequent melting curve analysis indicated they were nonspecific products. Though dsDNA viruses such as hADV and HBV were not primed in the step of reverse transcription by VSITA, they were involved in the virus-targeted enrichment in the PCR step due to the specificity of V8. More efforts will be needed to fine-tune the balance between virus-targeted sensitivity and specificity of V8 primer set in the future work. Nevertheless, given a large variety of virus-positive clinical samples of different types (serum, feces, and throat swabs) were tested, our data suggested VSITA is a worthwhile alternative to improve the enrichment of virus targets in the clinical samples.
Furthermore, 20 clinical samples with fever or diarrhea of unknown causes were used to evaluate the performance of V8 and N6 in parallel, the fragment length analysis of generated library was conducted with Agilent 2100 Bioanalyzer. The results showed that V8 produced longer fragments distribution and lower length diversity as compared to N6. This phenomenon might be attributed to the difference of V8 and N6 primer sets in the specificity and diversity. V8 has stronger specificity inclining to amplify limited viral genome regions, while N6 has higher diversity tending to generate fragments with a broad range. The sequencing results with ion torrent PGM system revealed an obviously superior performance using V8. By the use of V8 primers, one more case of dengue virus infection was found and validated using real-time PCR in serum sample tests. In the testing of fecal samples, both methods were able to report the causative viruses in several cases, including one case of multiple infections. However, V8 showed apparently higher genome coverage and more reads hit for all virus-positive samples in comparison with N6, except for one case with Norovirus infection, where N6 covered slightly larger region on the Norovirus genome (86.93% vs. 86.53%), although V8 achieved more reads hit. Overall, NGS data implied that V8 approach was superior to N6 in the identification of virus from clinical samples.
Other than virus-targeted amplification, efforts have also been made in pre-treatment of samples to improve virus identification with NGS. It is reported that ultracentrifugation of clinical samples improved virus discovery by removal of free RNA and DNA as well as bacteria[39]. Pre-degradation of free nucleic acid with cocktail enzyme system has also been reported[33]. The enrichment strategy in this study can be combined with those reported methods to provide an integrated and comprehensive enrichment solution.
In this study, only serum, swab, and feces samples were used to test the reliability of the VSITA approach followed by NGS. The performance of VSITA in other sample types, such as cerebrospinal fluid (CSF), urine, and other viral infection needs to be further validated in future study.
In conclusion, the VSITA approach designed in this study is demonstrated to possess higher sensitivity and broader genome coverage than traditionally used hexamers in NGS-based identification of viral pathogens directly from clinical samples.
-
All the authors approved the final manuscript and they have no conflict of interest to declare.
-
All aspects of the study were performed in accordance with national ethics regulations and approved by the Institutional Review Boards of National Institute for Viral Disease Control and Prevention, Center for Disease Control and Prevention of China.
-
We acknowledge the Pediatric Research Institute, Children's Hospital of Hebei Province, Shijiazhuang, Hebei, China for providing fecal clinical samples, the National Laboratory for Hemorrhagic Fever, National Institute for Viral Disease Control and Prevention (IVDC), Chinese Center for Disease Control and Prevention (CDC) for providing serum clinical samples.
-
Table Supplementary Table 1. Real-time PCR Methods of Each Viral Pathogen
Name Sequence (5'-3') Citation 18S rRNAa TACCACATCCAAGGAAGGGAGCA [1] TGGAATTACCGCGGCTGCTGGCA 28S rRNAa AACGAGATTCCCACTGTCCC [1] CTTCACCGTGCCAGACTAGAG JEV AGAGCACCAAGGGAATGAAATAGT [2] AATAGGTTGTAGTTGGGCACTCTG FAM-CCACGCCACTCGACCCATAGACTG-BHQ Rota virus ACCATCTACACATGACCCTC [3] GGTCACATAACGCCCC FAM-ATGAGCACAATAGTTAAAAGCTAACACTGTCAA-BHQ Noro virus GⅡ CARGARBCNATGTTYAGRTGGATGAG [4] TCGACGCCATCT TCATTCACA FAM-TGGGAGGGCGATCGCAATCT-BHQ ADV GATGGCCACCCCATCGATGMTGC [5] GCGAACTGCACCAGACCCGGAC VIC-TACATGCACATCGCCGGACAGGAMGCTTCGGAGT-TAMRA DENV GGAAGTAGAGCAATATGGTACATGTG [6] CCGGCTGTGTCATCAGCATAYAT FAM-TGTGCAGTCCTTCTCCTTCCACTCCACT-BHQ H1N1 GACCRATCCTGTCACCTCTGAC [7] GGGCATTYTGGACAAAKCGTCTACG HEX-TGCAGTCCTCGCTCACTGGGCACG-BHQ SWL-H1N1 GGGTAGCCCCATTGCAT [8] AGAGTGATTCACACTCTGGATTTC HEX-TGGGTAAATGTAACATTGCTGGCTGG-BHQ H3N2 ACCCTCAGTGTGATGGCTTCCAAA [9] TAAGGGAGGCATAATCCGGCACAT HEX-ACGCAGCAAAGCCTACAGCAACTGT-BHQ EV71 GAG AGT TCT ATA GGG GAC AGT [10] AGC TGT GCT ATG TGA ATT AGG AA FAM-ACT TAC CCA GGC CCT GCC AGC TCC-TAMRA CaV16 GGGAATTTCTTTAGCCGTGC [11] CCCATCAARTCAATGTCCC FAM-ACAATGCCCACCACGGGTACACA-BHQ CMV CATGAAGGTCTTTGCCCAGTAC [12] GGCCAAAGTGTAGGCTACAATAG FAM-TGGCCCGTAGGTCATCCACACTAGG-TAMRA HIV AGCATTATCAGAAGGAGCCA GCAGCCTCTTCATTGATGGT FAM-TGCATGGCTGCTTGATGTCCCC-TAMRA HBV CCGTCTGTGCCTTCTCATCTG [13] AGTCCAAGAGTYCTCTTATGYAAGACCTT FAM-CCGTGTGCACTTCGCTTCACCTCTGC-MGB HCV TGCGGAACCGGTGAGTACA [14] CTTAAGGTTTAGGATTCGTGCTCAT FAM-CACCCTATCAGGCAGTACCACAAGGCC-TAMRA Enterovirusb GGCTGCGYTGGCGGCC [11] CCAAAGTAGTCGGTTCCGC FAM-CTCCGGCCCCTGAATGCGG-BHQ Note. aTested with real-time PCR by cyber green. bEchovirus 6, Coxsackievirus A6, Coxsackievirus A4, Coxsackievirus B2 were all tested by HEV primer and probe, followed by sanger sequencing for genotyping. 1. Chuang TW, Lee KM, Lou YC, et al. A Point Mutation in the Exon Junction Complex Factor Y14 Disrupts Its Function in mRNA Cap Binding and Translation Enhancement. J Biol Chem, 2016; 291, 8565-74.
2. Kalia M, Khasa R, Sharma M, et al. Japanese encephalitis virus infects neuronal cells through a clathrin-independent endocytic mechanism. J Virol, 2013; 87, 148-62.
3. Verheyen J, Timmen-Wego M, Laudien R, et al. Detection of adenoviruses and rotaviruses in drinking water sources used in rural areas of Benin, West Africa. Appl Environ Microbiol, 2009; 75, 2798-801.
4. Kageyama T, Kojima S, Shinohara M, et al. Broadly reactive and highly sensitive assay for Norwalk-like viruses based on real-time quantitative reverse transcription-PCR. J Clin Microbiol, 2003; 41, 1548-57.
5. Krafft AE, Russell KL, Hawksworth AW, et al. Evaluation of PCR testing of ethanol-fixed nasal swab specimens as an augmented surveillance strategy for influenza virus and adenovirus identification. J Clin Microbiol, 2005; 43, 1768-75.
6. Pang Z, Li A, Li J, et al. Comprehensive multiplex one-step real-time TaqMan qRT-PCR assays for detection and quantification of hemorrhagic fever viruses. PLoS One, 2014; 9, e95635.
7. Zou S, Han J, Wen L, et al. Human influenza A virus (H5N1) detection by a novel multiplex PCR typing method. J Clin Microbiol, 2007; 45, 1889-92.
8. Poon LL, Chan KH, Smith GJ, et al. Molecular detection of a novel human influenza (H1N1) of pandemic potential by conventional and real-time quantitative RT-PCR assays. Clin Chem, 2009; 55, 1555-8.
9. Chen Y, Cui D, Zheng S, et al. Simultaneous detection of influenza A, influenza B, and respiratory syncytial viruses and subtyping of influenza A H3N2 virus and H1N1 (2009) virus by multiplex real-time PCR. J Clin Microbiol, 2011; 49, 1653-6.
10. Tan EL, Yong LL, Quak SH, et al. Rapid detection of enterovirus 71 by real-time TaqMan RT-PCR. J Clin Virol, 2008; 42, 203-6.
11. Cui A, Xu C, Tan X, et al. The development and application of the two real-time RT-PCR assays to detect the pathogen of HFMD. PLoS One, 2013; 8, e61451.
12. Sugita S, Shimizu N, Watanabe K, et al. Use of multiplex PCR and real-time PCR to detect human herpes virus genome in ocular fluids of patients with uveitis. Br J Ophthalmol, 2008; 92, 928-32.
13. Zhao JR, Bai YJ, Zhang QH, et al. Detection of hepatitis B virus DNA by real-time PCR using TaqMan-MGB probe technology. World J Gastroenterol, 2005; 11, 508-10.
14. Martell M, Gomez J, Esteban JI, et al. High-throughput real-time reverse transcription-PCR quantitation of hepatitis C virus RNA. J Clin Microbiol, 1999; 37, 327-32.
doi: 10.3967/bes2018.035
VSITA, an Improved Approach of Target Amplification in the Identification of Viral Pathogens
-
Abstract:
Objective Unbiased next generation sequencing (NGS) is susceptible to interference from host or environmental sequences. Consequently, background depletion and virome enrichment techniques are usually needed for clinical samples where viral load is much lower than background sequences. Methods A viral Sequence Independent Targeted Amplification (VSITA) approach using a set of non-ribosomal and virus-enriched octamers (V8) was developed and compared with traditionally used random hexamers (N6). Forty-five archived clinical samples of different types were used in parallel to compare the V8 and N6 enrichment performance of viral sequences and removal performance of ribosomal sequences in the step of reverse transcription followed by quantitative PCR (qPCR). Ten sera samples from patients with fever of unknown origin and 10 feces samples from patients with diarrhea of unknown origin were used in comparison of V8 and N6 enrichment performance following NGS analysis. Results A minimum 30 hexamers matching to viral reference sequences (sense and antisense) were selected from a dataset of random 4, 096 (46) hexamers (N6). Two random nucleotides were added to the 5' end of the selected hexamers, and 480 (30×42) octamers (V8) were obtained. In general, VSITA approach showed higher enrichment of virus-targeted cDNA and enhanced ability to remove unwanted ribosomal sequences in the majorities of 45 predefined clinical samples. Moreover, VSITA combined with NGS enabled to detect not only more viruses but also achieve more viral reads hit and higher viral genome coverage in 20 clinical samples with diarrhea or fever of unknown origin. Conclusion The VSITA approach designed in this study is demonstrated to possess higher sensitivity and broader genome coverage than traditionally used random hexamers in the NGS-based identification of viral pathogens directly from clinical samples. -
Key words:
- Virus /
- Next generation sequencing /
- Non-ribosomal /
- Virus targeted
-
Figure 1. V8 versus N6 ratio of different RNA levels quantified by qPCR. (A) Human 18S ribosomal RNA level; (B) Human 28S ribosomal RNA level; (C) Viral RNA level; (D) Overview of relative quantity. The calculation of relative quantity of V8 versus N6 was showed in material and methods.
Figure 2. Fragment length analysis with Agilent 2100 bioanalyzer with: (A) N6 library from sample F1; (B) V8 library from sample F1; (C) N6 library from sample D1; (D) V8 library from sample D1.
Supplementary Figure 1. Fragment length analysis with Agilent 2100 bioanalyzerwith samples: (A) N6 library from sample F1; (B) V8 library from sample F1; (C) N6 library from sample F2; (D) V8 library from sample F2; (E) N6 library from sample F3; (F) V8 library from sample F3; (G) N6 library from sample F7; (H) V8 library from sample F7; (I) N6 library from sample F10; (J) V8 library from sample F10; (K) N6 library from sample D1; (L) V8 library from sample D1; (M) N6 library from sample D5; (N) V8 library from sample D5; (O) N6 library from sample D6; (P) V8 library from sample D6; (Q) N6 library from sample D8; (R) V8 library from sample D8; (S) N6 library from sample D10; (T) V8 library from sample D10.
Table 1. Octamer (V8) Sequences Used for VSITA (5'-3')
Motif Motif Motif Motif Motif Motif NNGGCAAT NNGATATC NNGCATTG NNGTCTAG NNAGTATA NNAAGTAT NNAATTGT NNAATCTA NNAATTGT NNACAACG NNACAATT NNACTATT NNATACTT NNATTGCC NNATTTTA NNCGTATG NNCGTTGT NNCAATGC NNCAATTG NNCATACG NNCCTAGA NNCTAGAC NNCTCGAG NNTAGATT NNTAAAAT NNTATAAA NNTATACT NNTATATA NNTCTAGG NNTTTATA  下载: 导出CSV
下载: 导出CSV
Table 2. Comparison of the NGS Results between the Two Methods by Clinical Samples
Sample Number Identified Virus Ct Valuea V8 N6 Reads Hit Coverage (%) Viral Reads Percentage (%) Reads Hit Coverage (%) Viral Reads Percentage (%) F1 HFRS neg 3 2.76 0 0 0 0 F2 HFRS neg 1 0.38 0 0 0 0 F3 SFTS neg 3 0.94 0 0 0 0 F7 DENV Ⅲ 35.14 46 22.31 0.08 0 0 0 F10 DENV Ⅲ 25.97 2, 257 94.88 0.61 185 25.10 0.19 D1 Echovirus 6 24.49 1, 134 92.33 8.11 817 90.13 4.42 D1 Norovirus 28.75 48, 344 86.53 8.11 25, 276 86.93 4.42 D5 Coxsackievirus A6 24.09 233 79.54 0.50 22 39.27 0.08 D6 Poliovirus 26.70 60 71.94 0.40 20 32.59 0.14 D8 Coxsackievirus A4 26.50 171 78.26 0.80 51 47.55 0.30 D10 Coxsackievirus B2 23.47 346 96.09 1.90 116 88.58 0.50 Note. aTests with reported real-time PCR methods to validate the identification of viruses.
下载: 导出CSV
Supplementary Table 1. Real-time PCR Methods of Each Viral Pathogen
Name Sequence (5'-3') Citation 18S rRNAa TACCACATCCAAGGAAGGGAGCA [1] TGGAATTACCGCGGCTGCTGGCA 28S rRNAa AACGAGATTCCCACTGTCCC [1] CTTCACCGTGCCAGACTAGAG JEV AGAGCACCAAGGGAATGAAATAGT [2] AATAGGTTGTAGTTGGGCACTCTG FAM-CCACGCCACTCGACCCATAGACTG-BHQ Rota virus ACCATCTACACATGACCCTC [3] GGTCACATAACGCCCC FAM-ATGAGCACAATAGTTAAAAGCTAACACTGTCAA-BHQ Noro virus GⅡ CARGARBCNATGTTYAGRTGGATGAG [4] TCGACGCCATCT TCATTCACA FAM-TGGGAGGGCGATCGCAATCT-BHQ ADV GATGGCCACCCCATCGATGMTGC [5] GCGAACTGCACCAGACCCGGAC VIC-TACATGCACATCGCCGGACAGGAMGCTTCGGAGT-TAMRA DENV GGAAGTAGAGCAATATGGTACATGTG [6] CCGGCTGTGTCATCAGCATAYAT FAM-TGTGCAGTCCTTCTCCTTCCACTCCACT-BHQ H1N1 GACCRATCCTGTCACCTCTGAC [7] GGGCATTYTGGACAAAKCGTCTACG HEX-TGCAGTCCTCGCTCACTGGGCACG-BHQ SWL-H1N1 GGGTAGCCCCATTGCAT [8] AGAGTGATTCACACTCTGGATTTC HEX-TGGGTAAATGTAACATTGCTGGCTGG-BHQ H3N2 ACCCTCAGTGTGATGGCTTCCAAA [9] TAAGGGAGGCATAATCCGGCACAT HEX-ACGCAGCAAAGCCTACAGCAACTGT-BHQ EV71 GAG AGT TCT ATA GGG GAC AGT [10] AGC TGT GCT ATG TGA ATT AGG AA FAM-ACT TAC CCA GGC CCT GCC AGC TCC-TAMRA CaV16 GGGAATTTCTTTAGCCGTGC [11] CCCATCAARTCAATGTCCC FAM-ACAATGCCCACCACGGGTACACA-BHQ CMV CATGAAGGTCTTTGCCCAGTAC [12] GGCCAAAGTGTAGGCTACAATAG FAM-TGGCCCGTAGGTCATCCACACTAGG-TAMRA HIV AGCATTATCAGAAGGAGCCA GCAGCCTCTTCATTGATGGT FAM-TGCATGGCTGCTTGATGTCCCC-TAMRA HBV CCGTCTGTGCCTTCTCATCTG [13] AGTCCAAGAGTYCTCTTATGYAAGACCTT FAM-CCGTGTGCACTTCGCTTCACCTCTGC-MGB HCV TGCGGAACCGGTGAGTACA [14] CTTAAGGTTTAGGATTCGTGCTCAT FAM-CACCCTATCAGGCAGTACCACAAGGCC-TAMRA Enterovirusb GGCTGCGYTGGCGGCC [11] CCAAAGTAGTCGGTTCCGC FAM-CTCCGGCCCCTGAATGCGG-BHQ Note. aTested with real-time PCR by cyber green. bEchovirus 6, Coxsackievirus A6, Coxsackievirus A4, Coxsackievirus B2 were all tested by HEV primer and probe, followed by sanger sequencing for genotyping.
下载: 导出CSV
-
[1] Haug CJ, Kieny MP, Murgue B. The Zika Challenge. N Engl J Med, 2016; 374, 1801-3. doi: 10.1056/NEJMp1603734 [2] Su S, Wong G, Liu Y, et al. MERS in South Korea and China a potential outbreak threat? Lancet, 2015; 385, 2349-50. doi: 10.1016/S0140-6736(15)60859-5 [3] MERS-the latest threat to global health security. Lancet, 2015; 385, 2324. [4] Drosten C, Meyer B, Muller MA, et al. Transmission of MERS-coronavirus in household contacts. N Engl J Med, 2014; 371, 828-35. doi: 10.1056/NEJMoa1405858 [5] Azhar EI, El-Kafrawy SA, Farraj SA, et al. Evidence for camel-to-human transmission of MERS coronavirus. N Engl J Med, 2014; 370, 2499-505. doi: 10.1056/NEJMoa1401505 [6] Perlman S, McCray PB Jr. Person-to-person spread of the MERS coronavirus-an evolving picture. N Engl J Med, 2013; 369, 466-7. doi: 10.1056/NEJMe1308724 [7] Assiri A, McGeer A, Perl TM, et al. Hospital outbreak of Middle East respiratory syndrome coronavirus. N Engl J Med, 2013; 369, 407-16. doi: 10.1056/NEJMoa1306742 [8] Markoff L. Yellow fever outbreak in Sudan. N Engl J Med, 2013; 368, 689-91. doi: 10.1056/NEJMp1300772 [9] Chen Z, Liu L, Lv Y, et al. A fatal yellow fever virus infection in China: description and lessons. Emerg Microbes Infect, 2016; 5, e69. doi: 10.1038/emi.2016.89 [10] The L Yellow fever a global reckoning. Lancet, 2016; 387, 1348. [11] Ahmed QA, Memish ZA. Yellow fever from Angola and Congo: a storm gathers. Trop Doct, 2017; 47, 92-6. doi: 10.1177/0049475517699726 [12] Wilder-Smith A, Leong WY. Importation of yellow fever into China: assessing travel patterns. J Travel Med, 2017; 24. http://www.thelancet.com/journals/laninf/article/PⅡS1473-3099(17)30494-2/fulltext [13] Ferguson NM, Cucunuba ZM, Dorigatti I, et al. EPIDEMIOLOGY. Countering the Zika epidemic in Latin America. Science, 2016; 353, 353-4. doi: 10.1126/science.aag0219 [14] Jouannic JM, Friszer S, Leparc-Goffart I, et al. Zika virus infection in French Polynesia. Lancet, 2016; 387, 1051-2. http://www.thelancet.com/pdfs/journals/lancet/PⅡS0140-6736(16)00625-5.pdf [15] Lessler J, Chaisson LH, Kucirka LM, et al. Assessing the global threat from Zika virus. Science, 2016; 353, aaf8160. doi: 10.1126/science.aaf8160 [16] Petersen LR, Jamieson DJ, Powers AM, et al. Zika Virus. N Engl J Med, 2016; 374, 1552-63. doi: 10.1056/NEJMra1602113 [17] The L Zika's emerging threat for the Asia-Pacific region. Lancet, 2016; 388, 1026. [18] Gire SK, Goba A, Andersen KG, et al. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science, 2014; 345, 1369-72. doi: 10.1126/science.1259657 [19] Whitty CJ, Farrar J, Ferguson N, et al. Infectious disease: tough choices to reduce Ebola transmission. Nature, 2014; 515, 192-4. doi: 10.1038/515192a [20] Althaus CL. Ebola superspreading. Lancet Infect Dis, 2015; 15, 507-8. doi: 10.1016/S1473-3099(15)70135-0 [21] Tong YG, Shi WF, Liu D, et al. Genetic diversity and evolutionary dynamics of Ebola virus in Sierra Leone. Nature, 2015; 524, 93-6. doi: 10.1038/nature14490 [22] Zinszer K, Morrison K, Anema A, et al. The velocity of Ebola spread in parts of west Africa. Lancet Infect Dis, 2015; 15, 1005-7. http://europepmc.org/abstract/MED/26333328 [23] Currie J, Grenfell B, Farrar J. Infectious diseases. Beyond Ebola. Science, 2016; 351, 815-6. doi: 10.1126/science.aad8521 [24] Rico A, Brody D, Coronado F, et al. Epidemiology of Epidemic Ebola Virus Disease in Conakry and Surrounding Prefectures, Guinea, 2014-2015. Emerg Infect Dis, 2016; 22, 178-83. doi: 10.3201/eid2202.151304 [25] Wang Q, Zhang Y, Wang HY, et al. Detection and Analysis of Ebola Virus in Sierra Leone-China Friendship Biosafety Laboratory from March 11 to April 20, 2015. Biomed Environ Sci, 2016; 29, 443-7. http://www.besjournal.com/Articles/Archive/2016/No6/201607/t20160721_132800.html [26] Boonham N, Kreuze J, Winter S, et al. Methods in virus diagnostics: from ELISA to next generation sequencing. Virus Res, 2014; 186, 20-31. doi: 10.1016/j.virusres.2013.12.007 [27] Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet, 2016; 17, 333-51. http://europepmc.org/abstract/med/27184599 [28] Han Y, Gong L, Sheng J, et al. Prediction of virological response by pretreatment hepatitis B virus reverse transcriptase quasispecies heterogeneity: the advantage of using next-generation sequencing. Clin Microbiol Infect, 2015; 21, 797. e1-8. doi: 10.1016/j.cmi.2015.03.021 [29] Harrison A, Binder H, Buhot A, et al. Physico-chemical foundations underpinning microarray and next-generation sequencing experiments. Nucleic Acids Res, 2013; 41, 779-96. https://www.researchgate.net/profile/Peter_Noble2/publication/234105625_ChemInform_Abstract_Physico-Chemical_Foundations_Underpinning_Microarray_and_Next_Generation_Sequencing_Experiments/links/09e415102ad1930416000000/ChemInform-Abstract-Physico-Chemical-Foundations-Underpinning-Microarray-and-Next-Generation-Sequencing-Experiments.pdf [30] Marx V. Next-generation sequencing. The genome jigsaw. Nature, 2013; 501, 263-8. doi: 10.1038/501261a [31] Saliba AE, Westermann AJ, Gorski SA, et al. Single-cell RNA-seq: advances and future challenges. Nucleic Acids Res, 2014; 42, 8845-60. doi: 10.1093/nar/gku555 [32] Schlaberg R, Chiu CY, Miller S, et al. Validation of Metagenomic Next-Generation Sequencing Tests for Universal Pathogen Detection. Arch Pathol Lab Med, 2017; 141, 776-86. doi: 10.5858/arpa.2016-0539-RA [33] Neill JD, Bayles DO, Ridpath JF. Simultaneous rapid sequencing of multiple RNA virus genomes. J Virol Methods, 2014; 201, 68-72. doi: 10.1016/j.jviromet.2014.02.016 [34] Quince C, Walker AW, Simpson JT, et al. Shotgun metagenomics, from sampling to analysis. Nat Biotechnol, 2017; 35, 833-44. doi: 10.1038/nbt.3935 [35] Li Y, Wang H, Nie K, et al. VIP: an integrated pipeline for metagenomics of virus identification and discovery. Sci Rep, 2016; 6, 23774. doi: 10.1038/srep23774 [36] Endoh D, Mizutani T, Kirisawa R, et al. Species-independent detection of RNA virus by representational difference analysis using non-ribosomal hexanucleotides for reverse transcription. Nucleic Acids Res, 2005; 33, e65. doi: 10.1093/nar/gni064 [37] Armour CD, Castle JC, Chen R, et al. Digital transcriptome profiling using selective hexamer priming for cDNA synthesis. Nat Methods, 2009; 6, 647-9. doi: 10.1038/nmeth.1360 [38] Wang CH, Nie K, Zhang Y, et al. An Improved Barcoded Oligonucleotide Primers-based Next-generation Sequencing Approach for Direct Identification of Viral Pathogens in Clinical Specimens. Biomed Environ Sci, 2017; 30, 22-34. http://www.besjournal.com/Articles/Archive/2017/No1/201702/t20170224_138604.html [39] Kohl C, Brinkmann A, Dabrowski PW, et al. Protocol for metagenomic virus detection in clinical specimens. Emerg Infect Dis, 2015; 21, 48-57. http://cn.bing.com/academic/profile?id=20fce381488b41dd56b4f041f4fcb18d&encoded=0&v=paper_preview&mkt=zh-cn -

 点击查看大图
点击查看大图
计量
- 文章访问数: 1557
- HTML全文浏览量: 585
- PDF下载量: 47
- 被引次数: 0

 Quick Links
Quick Links