
-
Hypobaric hypoxic environment in high-altitude areas can lead to various neurological impairments, and high altitude has a substantial impact on learning and memory (L&M)[1]. Increasing numbers of people have recently traveled to high-altitude regions, leading to an increase in the prevalence of altitude illnesses in various countries[2-3]. The human brain is highly susceptible to oxygen deprivation, and under hypoxic conditions, neurons respond to a lack of oxygen, undergo adaptive adjustments in morphology, and function to ensure the continuity of brain functions[4-5]. However, acute hypoxia can disrupt the delicate balance between adaptive neuroplasticity and pathological changes. Acute hypoxia can increase the neuronal oxidative stress response, resulting in damage of the brain function[6]. The hippocampus, which plays a crucial role in long-term spatial memory, is particularly vulnerable to oxygen deprivation[7].
Nogo-A is a myelin-enriched protein that inhibits axonal regeneration in the central nervous system after injury[8-10]. Nogo-A is encoded by Reticulon 4 (RTN4)[11]. In adulthood, Nogo-A is predominantly expressed in the glial cells and neurons located in brain areas with strong plasticity, such as the hippocampus and olfactory bulb[12-13]. Nevertheless, Nogo-A overexpression is involved in several cognitive impairment disorders and exacerbates oxidative stress[14-17]. Evidence indicates that Nogo-A levels increase in the hippocampal and neocortical neurons of patients with Alzheimer’s disease and increase with age[18]. In addition, suppressing Nogo-A can enhance synaptic plasticity and improve memory and cognitive functions[12]. Moreover, the expression of Nogo-A and its receptor NgR1 is upregulated in the ischemic cortex of neonatal rats, and the use of a Nogo-A-specific antibody could improve long-term neurological outcomes after stroke[19-20]. However, despite increasing evidence linking Nogo-A, NgR1, and hypoxia with cognitive impairment, it remains unclear whether Nogo-A is associated with L&M under high-altitude hypoxia (HH) conditions.
Based on previous studies, we hypothesized that HH impairs L&M via Nogo-A/NgR1. To verify this hypothesis, we used a low-pressure oxygen chamber to simulate high-altitude conditions and evaluated the L&M of rats using contextual conditioning fear and Morris water maze tests. Genetic and pharmaceutical approaches were used to block the Nogo-A/NgR1 pathway, and oxidative stress and dendritic spine density in the hippocampal region were measured. This study systematically validated the involvement of the Nogo-A/NgR1 pathway in L&M impairment following high-altitude exposure, providing a new theoretical guidance for the treatment of high-altitude L&M disorders.
-
Male Sprague-Dawley rats (200–250 g, 8-week-old) obtained from Peking University were used in this study. Female rats were excluded to avoid the influence of estrogen. Nogo-A-/- rats were donated by Professor Wang Jun of the School of Basic Medical Sciences at Peking University. Rats were housed in plastic cages at 23 ± 2 °C, 50%–60% humidity, with a 12-h light/dark cycle. They were provided with food and water ad libitum. All experiments were approved by the Animal Care and Utilization Committee of the Peking University Health Science Center. All experimental procedures and treatments were performed in accordance with the national guidelines stated in the “Guidelines for the Ethical Treatment of Experimental Animals” issued by the Ministry of Science and Technology of the People’s Republic of China.
-
Rats were placed in a hypobaric chamber (ProOx-810, Tawang Technology) and exposed to high-altitude hypoxia environment (20.8% O2, 54.02 kPa, equal to 5,000 m high altitude) for 3 or 7 days. Throughout the experiments, every effort was exerted to minimize the number of animals used and alleviate their suffering.
-
On the training day, the rats were placed in fear conditioning chambers for a 2-min acclimation period. Seven intermittent inescapable electric foot shocks (0.7 mA, 2 and 120 s interval) were delivered. After the final foot shock, the rats were left in a fear box for an additional 3 min. Twenty-four hours after training, during the contextual fear test phase, the fear-conditioned rats were reintroduced to the same fear-conditioning chamber for 5 min of free exploration each day. Spontaneous activity was recorded and analyzed using a Super Fear Conditioning Analysis System (SuperFcs) (XinRuan, Shanghai, China).
-
Rats were trained in a circular water tank with a diameter of 160 cm. Colorful and differently shaped cards were pasted onto the walls of the pool surrounding the water maze to provide extra maze cues. Each subject was tested in four trials/day for 4 consecutive days with a 15-min intertrial interval. If the participants did not reach the platform hidden 1 cm underwater within 60 s, they were manually guided. The platform was removed during the probe trial. All trials were analyzed using the Morris software (Mobile Datum).
-
For stereotaxic injection, the rats were anesthetized with isoflurane and placed in a stereotaxic instrument (68028, RWD). Adeno-associated viruses (AAVs) (108 VG/mL, 2 μL) for Nogo-A knockdown (the Nogo-A-shRNA sequence was determined as: GGAAGCATGTGAAAGTGAACT), and control viruses were simultaneously injected into both sides of the hippocampus (Anteroposterior: −3.8 mm; Mediolateral: ± 3.0 mm; Dorsoventral: −3.2 mm from bregma) according to the atlas of Paxinos and Watson (2007). AAVs were purchased from WZ Biosciences Inc. Twenty days after virus injection, modeling and behavioral tests were conducted. The effect of Nogo-A protein knockdown in the hippocampal tissue was validated using Western blotting after completion of the tests.
A cannula system was used for chronic drug infusion into the hippocampus with repeated injections for 3 or 7 days. NEP1-40 (10 μg/μL, 2 μL/day, MedChemExpress) or JTE-013 (10 μg/μL, 2 μL/day, MedChemExpress) was microinjected at a rate of 0.1 μL/min. The control group received vehicle. Intracranial drug delivery continued throughout the modeling period at a fixed time every day. We adapted the drug concentration and delivery method from that described by Fang PC et al. and Sancho-Alonso M et al.[21,22].
-
Total RNA was extracted from the tissues using a Total RNA Extraction Kit (Vazyme, RC101-01) according to the manufacturer’s protocol. The RNA was immediately used to generate cDNA using a cDNA Synthesis Kit (Vazyme, R223-01). The relative expression of mRNAs was determined using Taq Pro Universal SYBR qPCR Master Mix (Vazyme, R323-01). All results were normalized to β-actin gene expression using the comparative threshold cycle (2−ΔΔCT) method, and control samples were used to calculate the relative values. Each reaction was performed in triplicate and, 3–4 rats per condition were analyzed. All experiments were performed using a Quant Studio 1 Real-Time PCR System (Thermo, USA) according to the manufacturer’s protocol. β-actin forward primer: 5′-ACGGACCAGAGCGAAAGCAT-3′, β-actin reverse primer: 5′-TGTCAATCCTGTCCGTGTCC-3′. Shank1 (Qiagen, QT00183351), Shank2 (Qiagen, QT02350509), Shank3 (Qiagen, QT01568812), PSD95 forward primer: 5′-TAGGGCCCTGTTTGATTACG-3′, PSD95 reverse primer: 5′-TGGCCTTTAACCTTGACCAC-3′.
-
A Golgi apparatus staining kit PK401 (FD Rapid GolgiStainTM Kit) was used to analyze the density of neuronal dendritic spines within the hippocampal region. Brain tissues were selected and soaked in Golgi staining solution at 25 °C or 2 weeks. The slides were prepared according to the manufacturer’s instructions. To quantify the dendritic spine density, we reconstructed the 3D model of the spines using Laser Scanning Confocal Microscopy then analyzed straight terminal branches meeting a length exceeding 10 μm. Two or three brain slices were used from each group of rats, and two or three dendritic spines were sampled from each brain slice for statistical analysis. ImageJ software (National Institutes of Health) was used to analyze the number of 10-μm length dendritic spines.
-
Brain tissue samples were prepared in RIPA lysis buffer (P0013B; Beyotime). Protein concentrations were determined using bicinchoninic acid assay (Beyotime, P0012). Proteins were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes. The nitrocellulose membrane was incubated with the primary antibody solution at 4 °C overnight. Secondary antibodies conjugated to horseradish peroxidase were then incubated for 2 h at 25 °C. The protein levels were detected using an Enhanced Chemiluminescence solution. Densitometry analysis was performed using ImageJ software. The primary antibodies used in this study were as follows: Nogo-A (CST, 13401S), NgR1 (Abcam, ab184556), S1PR2 (Proteintech, 21180-1-AP), and β-actin (Bioss, bsm-33036M).
-
To detect the total antioxidant and oxidant capacity, the levels of malondialdehyde (MDA), reactive oxygen species (ROS), glutathione (GSH)/oxidized glutathione (GSSG) ratio, and superoxide dismutase (SOD) activity were measured according to the manufacturer’s protocol. The MDA (Beyotime, S0131S), ROS (YaJi Biological, YS470515), SOD (Beyotime, S0101S), and GSH/GSSH assay kits (Beyotime, S0053) were used.
-
Human neuroblastoma SH-SY5Y cell line was obtained from the Institute of Biophysics of the Chinese Academy of Sciences. The cells were cultured in DMEM/F12 (Gibco™, 11320033) supplemented with 10% fetal bovine serum (Gibco™, A5669701) in a humidified incubator containing 5% CO2 at 37 °C.
-
Cells were incubated in glucose-free DMEM (Gibco™, 11966025) in a hypoxic incubator (Ox-101C, Shanghai Tawang Technology, China) with a gas composition of 1% O2, 5% CO2, and 94% N2 at 37 °C for 6 h. Thereafter, glucose-free DMEM was replaced with complete medium. The cells were then returned to an incubator with 5% CO2 for 24 h to initiate reoxygenation.
-
Cells were cultured until they reached a confluency of 50%–60% prior to transfection. Transfection was performed using LV-Nogo-A-targeting shRNA (CAAATCCTAGGGAAGAAATAATTCAAGAGATTATTTCTTCCCTAGGATTTGTTTTTT) or scrambled shRNA (WZ Biosciences Inc., Shandong, China), according to the manufacturer’s instructions. The efficacy of shRNAi was confirmed by western blotting.
-
Cells treated with shRTN4 were plated in 24-well plates then treated with Nogo-P4 (ADI, cat#Nogo-P4) or Rtn-P4 (ADI, cat# Nogo-RTN4) at concentrations of 100 μg/mL. The cells were subjected to OGD/R modeling after 12 h.
-
Cells were fixed with 4% paraformaldehyde for 10 min and blocked by incubation with 10% goat serum for 20 min. This was followed by overnight incubation at 4 °C with the primary antibody (Nogo-A, Abcam, ab62024). The cells were then incubated with the secondary antibody (Abcam, ab150077) for 2 h at 25 °C. Nuclei were stained with DAPI (Abcam, ab104139).
-
After treatment, the cells were lysed in RIPA buffer containing a protease inhibitor cocktail, and the procedure was performed as described for tissue preparation in vivo. Antibodies against Nogo-A (Cell Signal Technology, 13401S), NgR1 (Abcam, ab184556), and β-Actin (Bioss, bsm-33036M) were utilized in Western blot.
-
The mitochondria of the cells were examined by TEM. The cells were first fixed with 2.5% glutaraldehyde solution for 2 h then post-fixed with 1% osmium tetroxide solution for 1 h. After dehydration with a series of alcohols, the samples were embedded in Epon resin and stained with lead citrate and uranyl acetate. Electron micrographs were obtained (JEM-1400, Japan).
-
According to the instruction manual of the JC-1 detection kit (C2006, Beyotime), the cells were supplemented with JC-1 staining working solution and cultured for 20 min at 37 °C. After centrifugation for 3 min, the mitochondrial membrane potential was measured using flow cytometry (Cytoflex, Beckman).
-
Following the instructions of the ROS detection kit (S0063, Beyotime), cells were treated with 5 µmol/L dihydroethidium solution, incubated at 37 °C for 30 min, then centrifuged to remove excess solution. Cells were resuspended in phosphate-buffered saline and analyzed using a flow cytometer to measure fluorescence intensity.
-
All data were expressed as mean ± standard error of the mean (sem). All statistical analyses were performed using GraphPad Prism version 10.0 software. Student’s t-test or one-way ANOVA was used to compare data. Pairwise comparisons between groups were conducted using Tukey’s post-hoc test. Each result contained at least three replicates of reproducibility and variation.
-
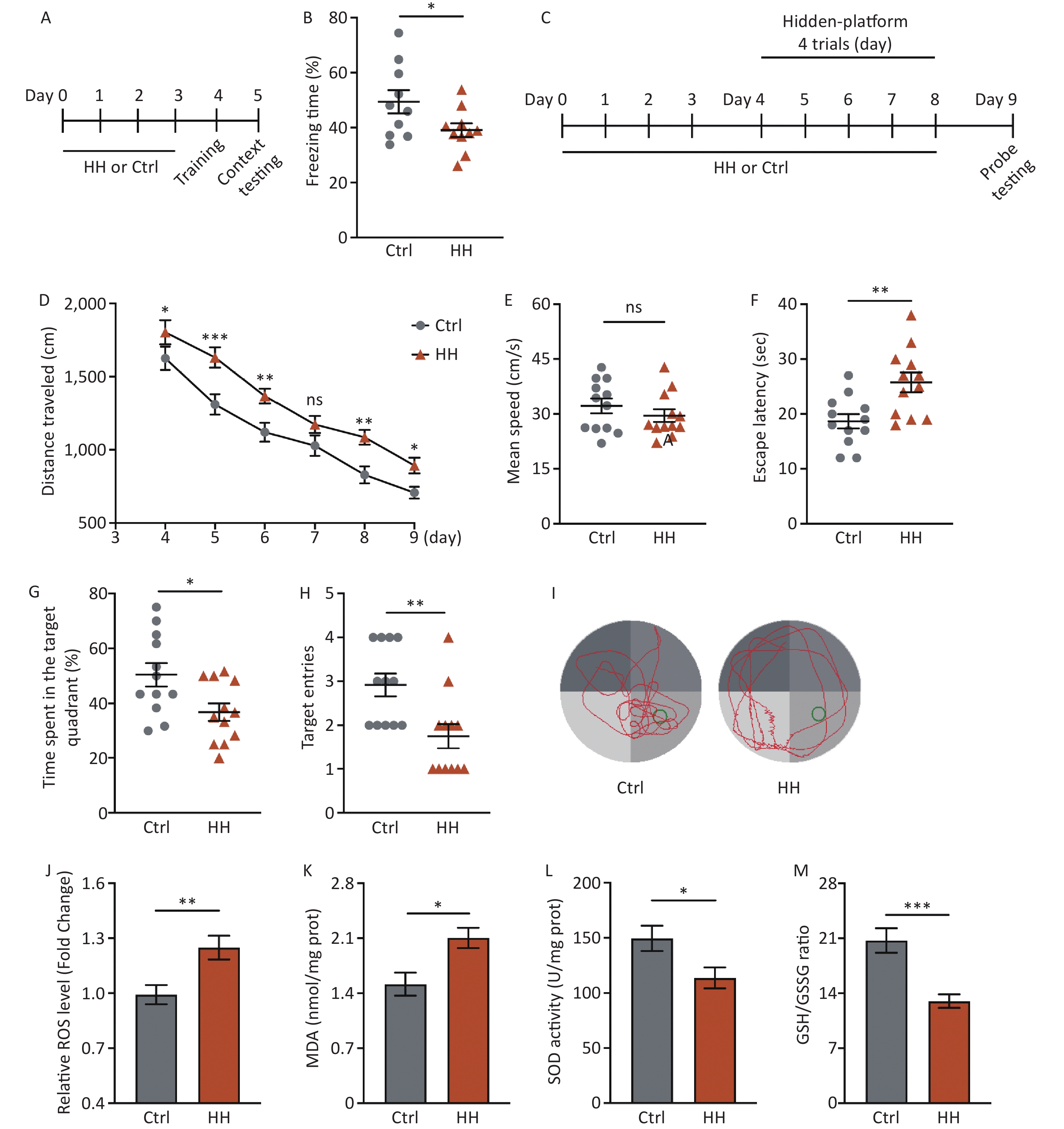
After acute HH exposure, the freezing time in the contextual fear memory test was significantly reduced (P < 0.05) (Figure 1A, B), suggesting that HH exposure detrimentally affects the ability of rats to acquire contextual fear. To further study the damaging effects of the HH environment on hippocampus-dependent L&M, we conducted an MWM test (Figure 1C). The results showed prolonged swimming distances during the training and testing days (P < 0.05, P < 0.001, P < 0.01, ns, P < 0.01, and P < 0.05, respectively) (Figure 1D); however, there was no change on motor ability (ns) (Figure 1E). The subsequent spatial probe test showed that the primary escape latency (the time taken by the rats to reach the original platform location for the first time) was noticeably extended, and the percentage of the target quadrant time and number of target quadrant crossings in the HH group were significantly decreased compared to those in the control group (P < 0.01, P < 0.05, and P < 0.01, respectively) (Figure 1F–I).
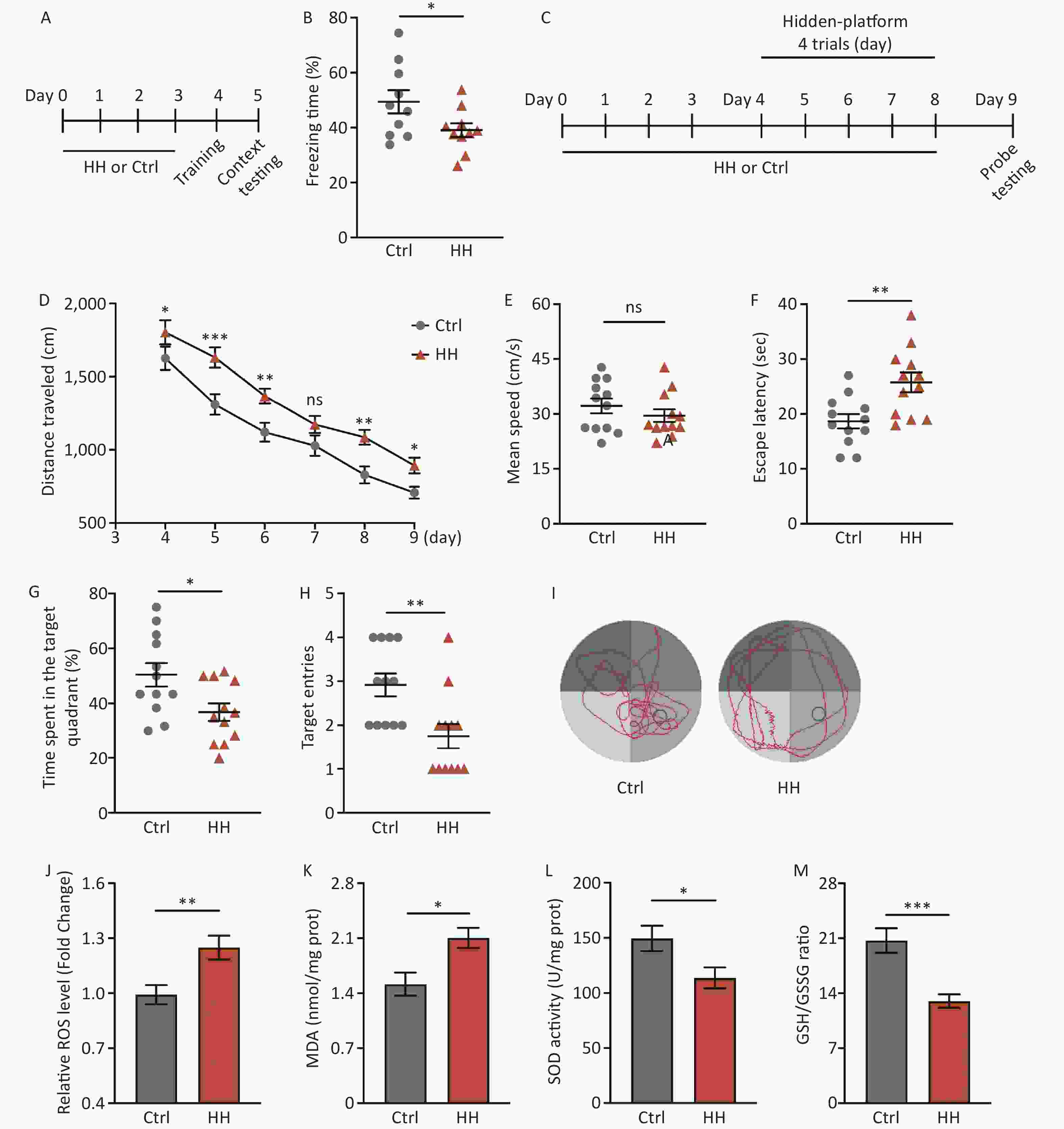
Figure 1. The effects of HH exposure on contextual fear conditioning test and Morris Water Maze (MWM) behavior in rats. (A) Schematic illustration of the strategy for a contextual fear conditioning test. (B) Freezing time in context test was measured after electric shock training. (Bars represent mean ± sem. Student’s t-test, *P < 0.05, N = 10). (C) Schematic illustration of the strategy for the MWM test pattern. (D) Escape distance to the platform, (E) mean swimming speed of the rats in the training phase, (F) primary escape latency, (G) time spent in the target quadrant, and (H) number of target entries of rats in the test phase of the MWM. (I) Representative picture of a rat track plot in the MWM test phase (data are shown as mean ± sem, N = 12, Student’s t-test, *P < 0.05, **P < 0.01, ***P < 0.001, ns, not significant). (J) Quantification of ROS levels, (K) MDA levels, (L) SOD activity, and (M) ratio of GSH-to-GSSG in hippocampus homogenates after HH exposure. SOD, superoxide dismutase; MDA, activity and malondialdehyde; ROS, reactive oxygen species; Ctrl, control; HH, high-altitude hypoxia. (Bars represent mean ± sem. Statistical significance was assessed using Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001, N = 6).
HH exposure can induce oxidative stress reactions[21]. Because the hippocampus plays an important role in regulating the contextual fear response and spatial L&M, we selected this brain region for further testing. Compared with the control group, ROS and MDA levels in the hippocampal homogenates of rats in the HH group were significantly increased (P < 0.01 and P < 0.05, respectively) (Figure 1J, K), whereas SOD activity and the GSH/GSSG ratio were significantly decreased (P < 0.05 and P < 0.001, respectively) (Figure 1L, M).
-
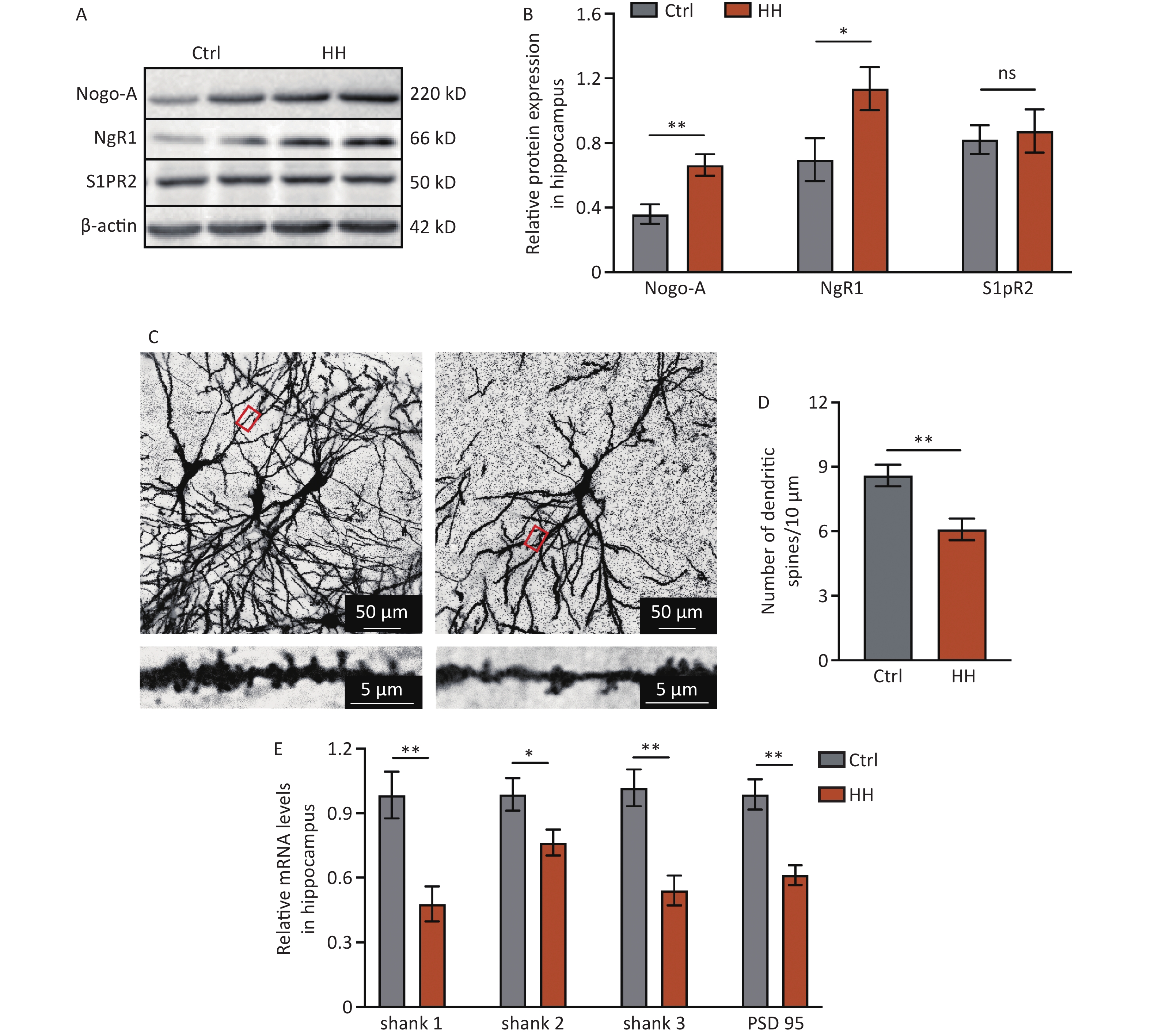
To determine the involvement of Nogo-A and its receptor in L&M impairment under HH conditions, immunoblotting was performed on rat hippocampal tissues. The results showed a significant increase in the protein expression of Nogo-A and NgR1, the receptor of the Nogo66 domain, in the rat hippocampal tissues after 3 days of exposure to HH, whereas the expression of S1PR2, the receptor of NogoΔ20, remained unchanged (P < 0.01, P < 0.05, and ns, respectively) (Figure 2A and B). Nogo-A is expressed in neurons in highly plastic central nervous system regions, such as the hippocampus and cortex, and has a substantial impact on neuronal morphology and function[22]. Therefore, we explored the dendritic spine density of hippocampal neurons and the mRNA levels of postsynaptic density proteins. Golgi staining revealed a significant decrease in dendritic spine density in hippocampal pyramidal neurons after HH exposure (P < 0.01) (Figure 2C, D). PSD95 is instrumental in the maintenance of synaptic plasticity and long-term potentiation (LTP)[23]. Similarly, Shank protein is a scaffolding protein predominantly located in the postsynaptic sites of glutamatergic synapses in the central nervous system[24]. In our study, qPCR revealed significant decreases in Shank1, Shank2, Shank3, and PSD95 mRNA levels in the hippocampal tissues of rats after HH exposure (P < 0.01, P < 0.05, P < 0.01, and P < 0.01, respectively) (Figure 2E).
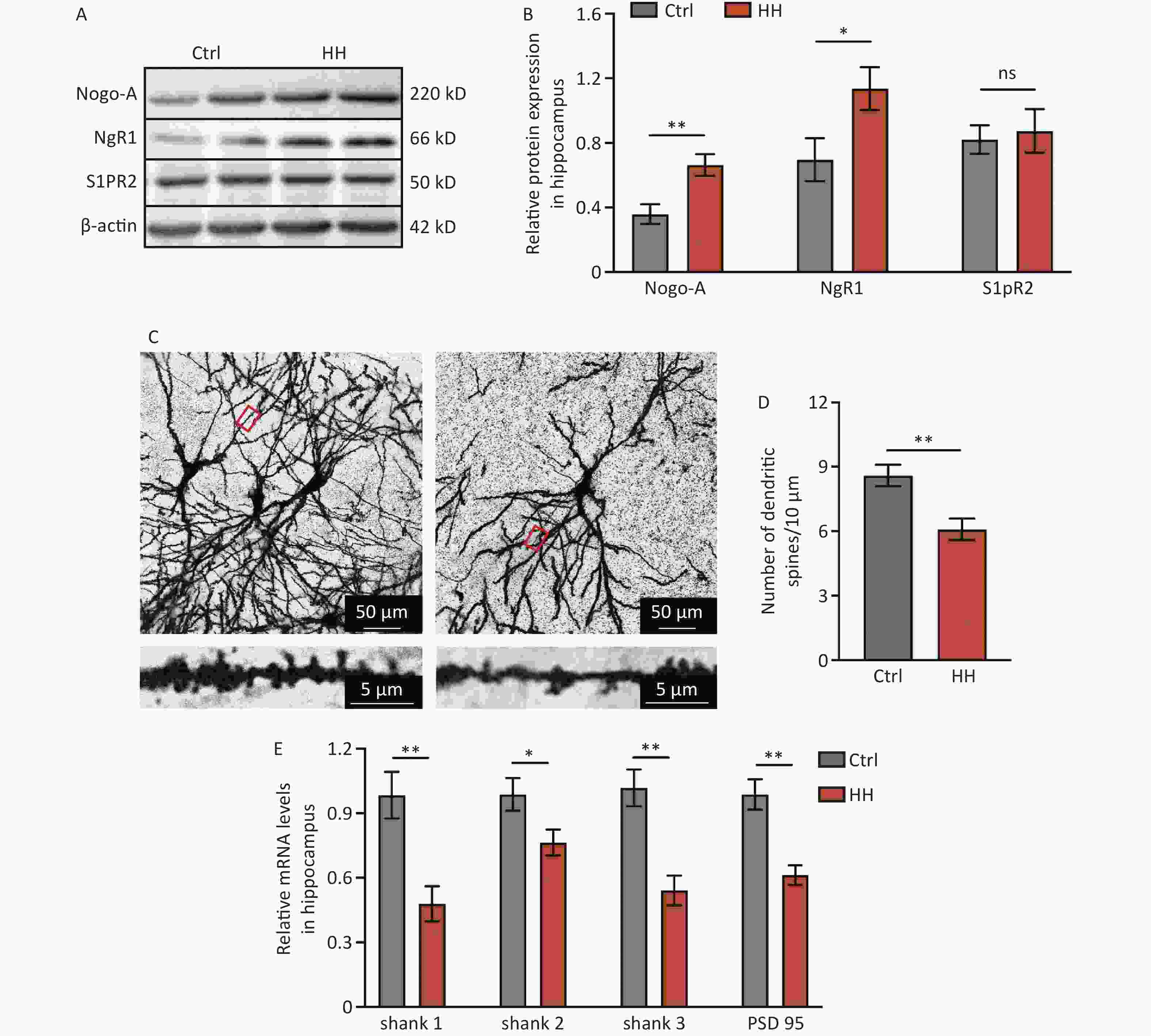
Figure 2. The effects of HH exposure on the expression of Nogo-A and NgR1 proteins in the hippocampal region and synaptic damage in hippocampal neurons. (A) Representative Western blot of Nogo-A, NgR1, S1PR2, and β-actin in rat hippocampal lysates. (B) The quantification of Nogo-A, NgR1, and S1PR2 protein levels normalized to β-actin. Bars represent mean ± sem. Statistical significance was assessed using Student’s t-test. *P < 0.05, **P < 0.01, ns, not significant (N = 5). (C) The dendritic spines of hippocampal neurons were visualized by Golgi staining. Scale bar, 50 μm and 5 μm. (D) Dendritic spine density was quantified by ImageJ and assessed using Student’s t-test. **P < 0.01 (N = 7). (E) The mRNA levels of Shank1, Shank2, Shank3, and PSD-95 were quantified and normalized to β-actin. Ctrl, control; HH, high-altitude hypoxia. Bars represent mean ± sem. Statistical significance was assessed using Student’s t-test. *P < 0.05, **P < 0.01 (N = 6).
-
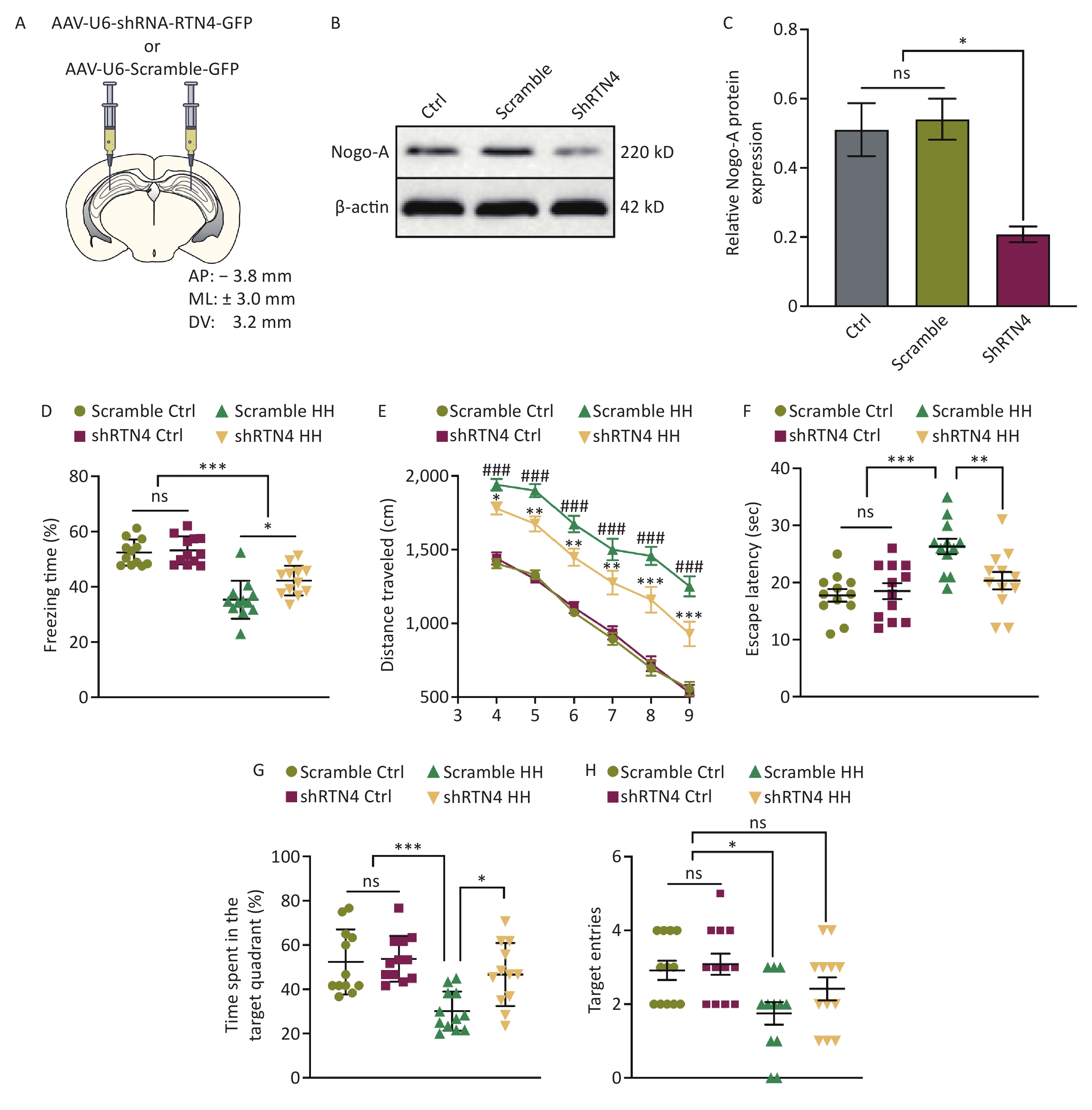
To precisely determine the function of Nogo-A in the hippocampal region of the HH model, stereotaxic injection of AAV expressing a specific targeted rat RTN4 shRNA was performed at designated rat hippocampal coordinates (Figure 3A). The knockdown efficiency of AAV-shRTN4 was validated using Western blotting, and the results showed that the knockout efficiency of AAV-shRTN4 at the protein level exceeded 50% (P < 0.05) (Figure 3B, C). Compared to that of the scrambled/HH group, the freezing time of rats in the shRTN4/HH group was markedly increased in the contextual fear test (P < 0.05) (Figure 3D). In the MWM experiment, shRTN4/HH rats exhibited improved L&M, as evidenced by a reduction in swimming distances during training and testing days (P < 0.05, P < 0.01, P< 0.01, P < 0.01, P < 0.001, and P < 0.001, respectively) (Figure 3E), reduction in the primary escape latency (P < 0.01) (Figure 3F), higher percentage of time spent in the target quadrant (P < 0.05) (Figure 3G), and compared to that of the control group, the number of entries into the target quadrant of rats in shRTN4/HH group exhibited no significant difference (P < 0.05) (Figure 3H).

Figure 3. The influence of injecting AAV-shRTN4 into the rat hippocampus on the learning and memory behavior of rats exposed to HH. (A) Schematic for stereotaxic injection. AAV-shRTN4 or AAV-Scramble solution was injected into both sides of the hippocampus. The stereotaxic coordinates used were: Anteroposterior (AP) = −3.8 mm, Mediolateral (ML) = ± 3.0 mm, Dorsoventral (DV) = 3.2 mm relative to the bregma. (B) Representative Western blot of Nogo-A and β-actin in rat hippocampus lysates. (C) The quantification of Nogo-A protein levels normalized to β-actin. Bars represent mean ± sem. Statistical significance was assessed using Student’s t-test. *P < 0.05, ns, not significant (N = 3). (D) Freezing time (%) in context test was measured after electric shock training and the freezing time (%) of Scramble HH and shRTN4 HH groups were analyzed. Statistical significance was assessed using one-way ANOVA followed by Tukey post hoc test. *P < 0.05, ***P < 0.001, ns, not significant (N = 12). (E) Swimming distance to the platform. Statistical significance was assessed using one-way ANOVA followed by Tukey post hoc test. *P < 0.05 vs. Scramble HH, **P < 0.01 vs. Scramble HH, ***P < 0.001 vs. Scramble HH, ###P < 0.001 vs. Scramble Ctrl (N = 10). (F) primary escape latency, (G) time spent in the target quadrant, and (H) number of target entries in the probe testing phase. Ctrl, control; HH, high-altitude hypoxia. Bars represent mean ± sem. Statistical significance was assessed using one-way ANOVA followed by Tukey post hoc test. *P < 0.05,
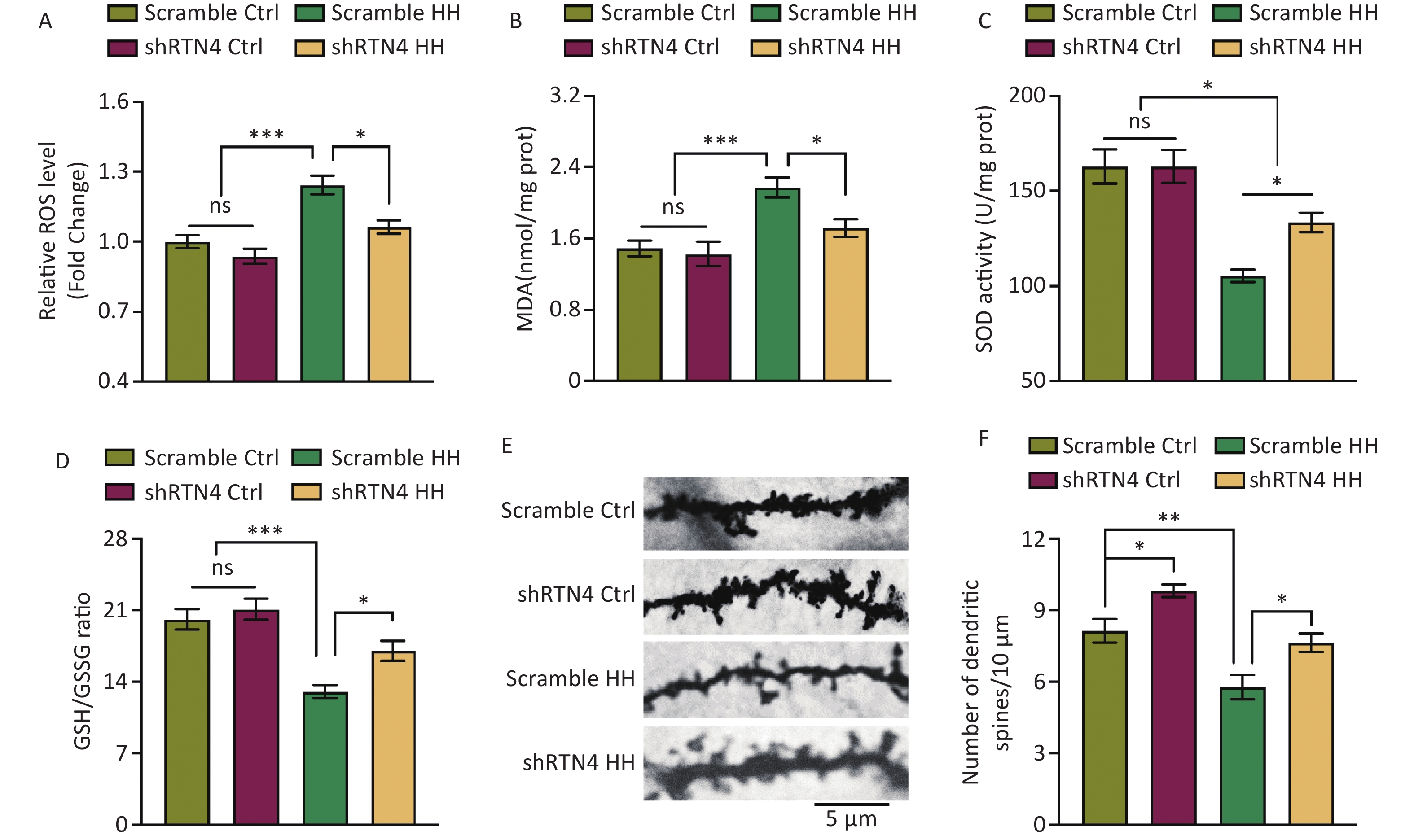
Injections of AAV-shRTN4 significantly reduced the increase in ROS and MDA levels in the hippocampal homogenates of HH-induced rats (P < 0.05 and P < 0.05, respectively) and significantly increased SOD activity and the GSH/GSSG ratio (P < 0.05 and P < 0.05, respectively) (Figure 4A and D). In addition, after exposure to HH, the density of dendritic spines in hippocampal pyramidal neurons in rats with shRTN4 interference significantly increased (P < 0.05) (Figure 4E, F).
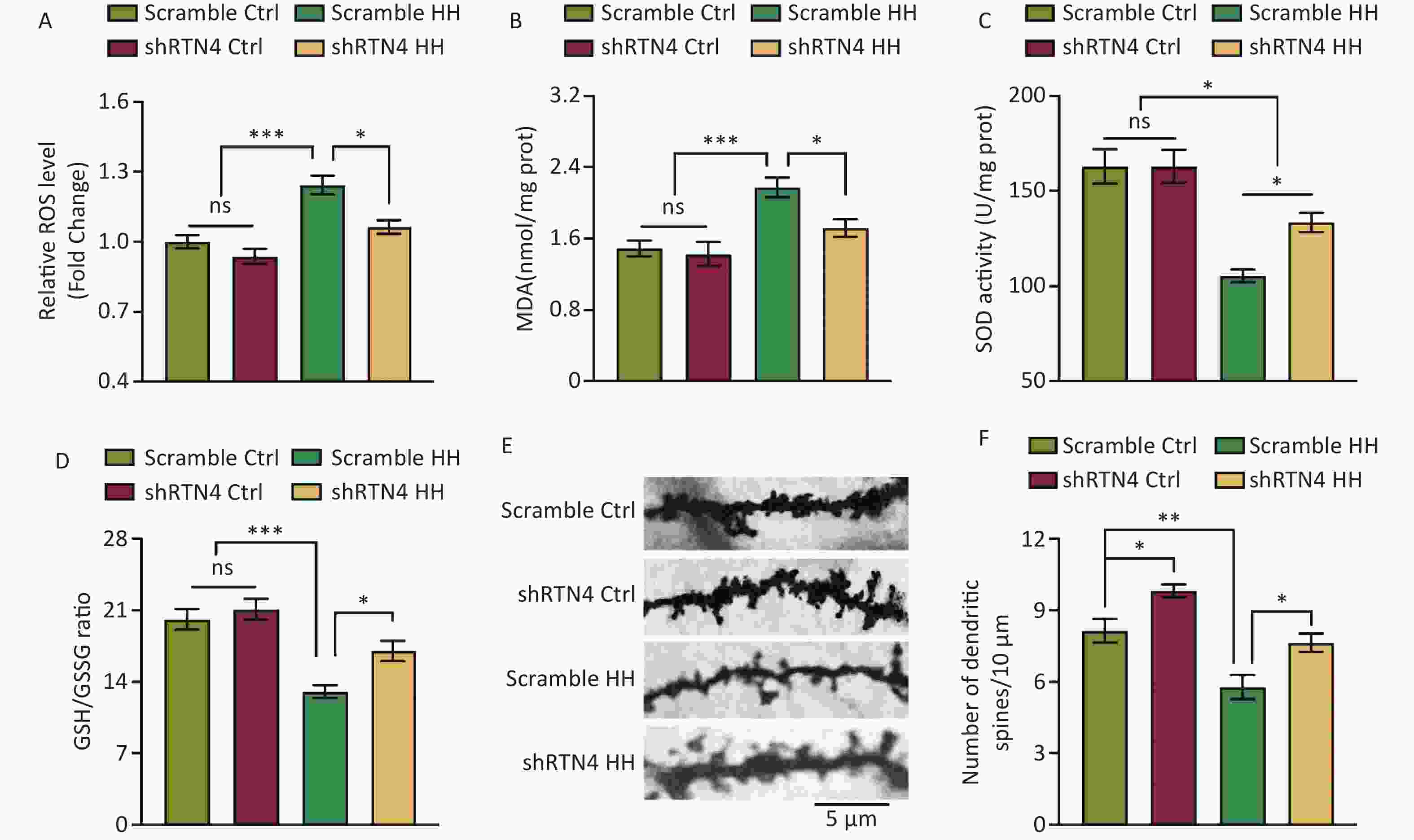
Figure 4. The influence of injecting AAV-shRTN4 into the rat hippocampus on hippocampal oxidative stress and dendritic spine density following exposure to HH. (A) Quantification of ROS levels, (B) MDA levels, (C) SOD activity, and (D) GSH/GSSG ratio in hippocampus homogenates after HH exposure. Bars represent mean ± sem. Statistical significance was assessed using one-way ANOVA followed by Tukey post hoc test. *P < 0.05, ***P < 0.001, ns, not significant (N = 6). (E–F) The dendritic spines of neurons in hippocampus were visualized and quantified. Ctrl, control; HH, high-altitude hypoxia. Scale bar, 5 μm. Statistical significance was assessed using one-way ANOVA followed by Tukey post hoc tests. **P < 0.05 (N = 7).
-
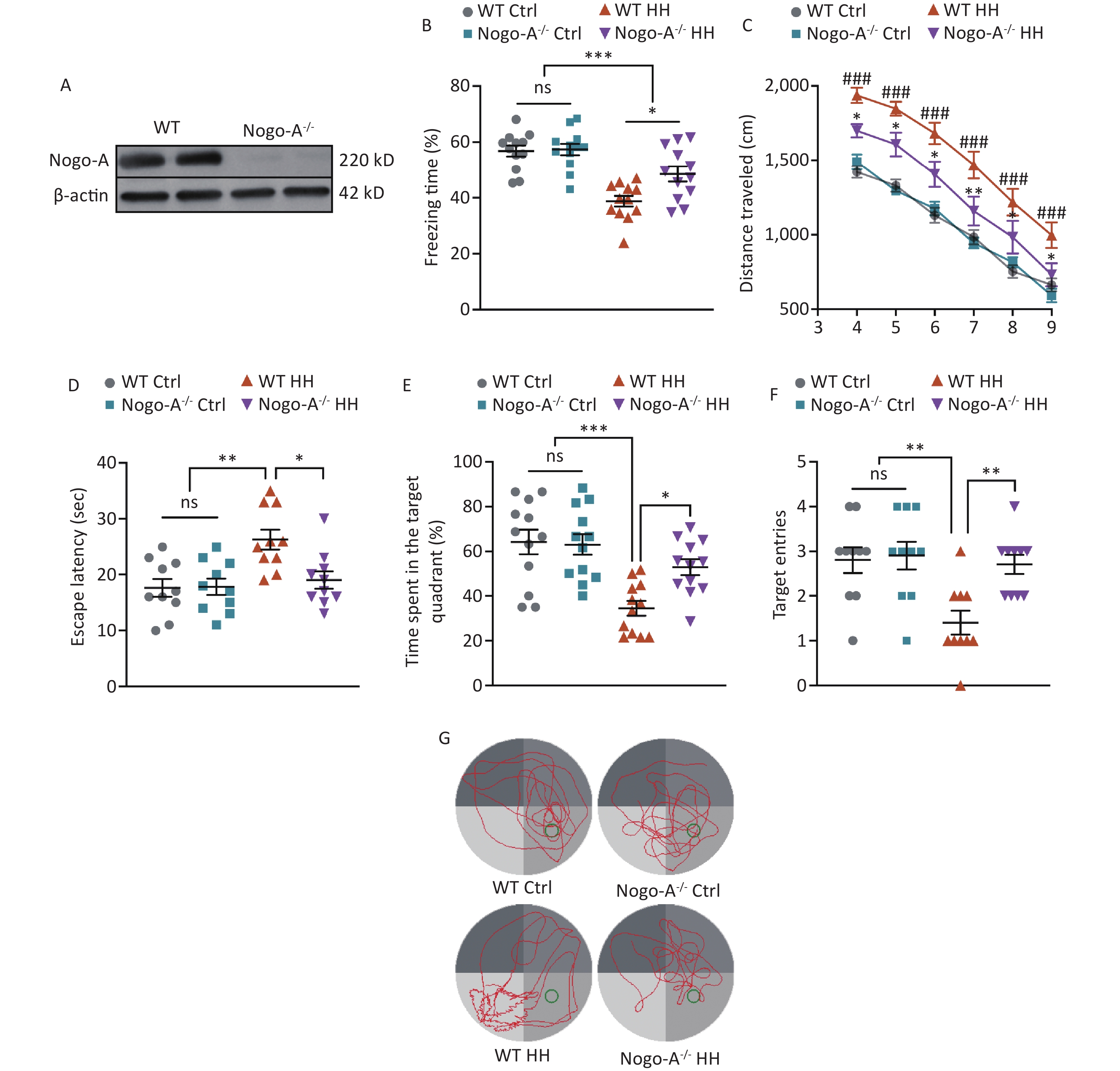
To further investigate the role of Nogo-A in the L&M impairment induced by HH exposure, we used Nogo-A-/- rats (Figure 5A). Under normal conditions (normoxic and normobaric), Nogo-A-/- rats did not show any changes in contextual fear or spatial memory learning in the MWM test compared to those of WT rats, indicating that Nogo-A knockout did not impair fear memory acquisition or spatial memory learning. After exposure to HH, Nogo-A-/- rats exhibited enhanced L&M in the contextual fear test compared with that of WT rats (P < 0.05) (Figure 5B). In the MWM tests, we found that the KO rats showed significantly better L&M performance. In the probe trial, KO rats displayed reduced swimming distance, shortened time to find the original platform location, increased time spent in the target quadrant, and improved accuracy (P < 0.05, P < 0.05, P < 0.05, and P < 0.01, respectively) (Figure 5C–G). The results of behavioral tests suggest that Nogo-A knockout mitigated the HH-induced impairment of L&M caused by HH.
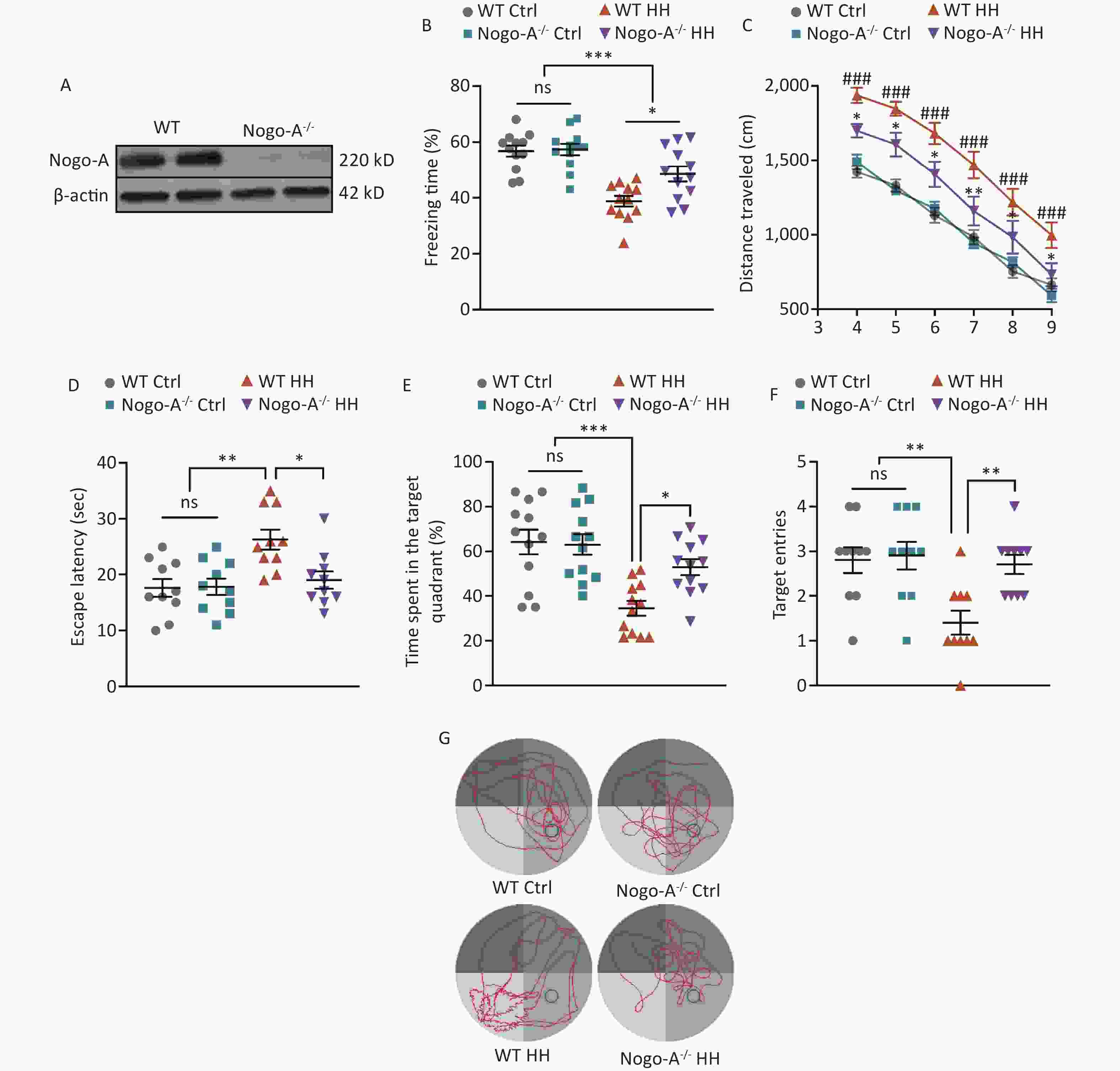
Figure 5. Nogo-A knockout improved HH-induced L&M damage. (A) Representative western blot images of Nogo-A and β-actin in the hippocampal homogenates of WT or Nogo-A-/- rats. (B) Freezing time (%) in context test was measured after electric shock training. Statistical significance was assessed using one-way ANOVA followed by Tukey post hoc tests. *P < 0.05, ***P < 0.001, ns, not significant (N = 12). (C) Escape distance to the platform. Data were analyzed using one-way ANOVA followed by Tukey post hoc tests. *P < 0.05 vs. WT HH, **P < 0.01 vs. WT HH, ###P < 0.001 vs. WT Ctrl (N = 10). (D) primary escape latency, (E) time spent in the target quadrant, and (F) number of target entries in the probe testing phase. (G) Representative swimming tracks in the MWM test. Ctrl, control; HH, high-altitude hypoxia. Data were analyzed using one-way ANOVA followed by Tukey post hoc tests. *P < 0.05, **P < 0.01, ***P < 0.001, ns, not significant (N = 10).
-
Oxidative stress was induced in SH-SY5Y cells in vitro using the OGD/R model. We confirmed Nogo-A expression in these cells (Figure 6A) using LV-Nogo-A shRNA to reduce Nogo-A levels, which was validated by immunoblotting (Figure 6B). Flow cytometry revealed that although the ROS levels in the OGD/R+/shRTN4+ group were higher than those in the OGD/R- group (P < 0.001) (Figures 6C, D), a significant decrease in ROS levels was observed in the OGD/R+/shRTN4+ group compared to those in the shRTN4- group following OGD/R modeling (P < 0.001) (Figure 6C, D). Cellular oxidative stress can lead to iron-mediated mitochondrial death, which is characterized by reduced mitochondrial volume, increased double-membrane density, and decreased or absent mitochondrial cristae[25]. The morphology of mitochondria was observed using TEM to investigate the impact of Nogo-A knockdown on OGD/R-induced mitochondrial ferroptosis in SH-SY5Y cells. After OGD/R, the mitochondria in the control group showed smaller size, increased membrane density, and thicker cristae, whereas the shRNA group exhibited improvement in mitochondrial volume and membrane density (Figure 6E). The mitochondrial inner membrane has a negative potential difference known as the mitochondrial membrane potential (Δψm), which is commonly monitored to assess mitochondrial function[26]. JC-1 was used to evaluate the Δψm by measuring the percentage of JC-1 monomers. Compared to that of the shRTN4- group, the shRTN4+ group exhibited a significantly decreased percentage of JC-1 monomers in the OGD/R model (P < 0.001) (Figure 6F). These findings are consistent with in vivo results, in which AAV-shRNA interference was used to suppress Nogo-A expression in the hippocampus.

Figure 6. The influence of the knockdown of Nogo-A in SH-SY5Y cells on OGD/R-induced oxidative stress response. (A) Representative immunofluorescence images of Nogo-A in SH-SY5Y cells. (B) Representative western blot images of Nogo-A and β-actin in SH-SY5Y cells after ShRNA treatment. The protein levels were normalized to β-actin. Bars represent mean ± sem. Statistical significance was assessed using Student’s t-test. ***P < 0.001 (N = 4). (C) Representative flow cytometry images of Dihydroethidium (DHE) staining, and (D) quantification of intracellular ROS in SH-SY5Y cells after OGD/R with administration of LV-scramble or LV-ShRTN4. Bars represent mean ± sem. Statistical significance was assessed using one-way ANOVA followed by Tukey post hoc tests. ***P < 0.001 ns, not significant (N = 4). (E) Representative TEM images of the mitochondria in SH-SY5Y cells after OGD/R with administration of LV-scramble or LV-ShRTN4. Scale bars (500 nm) are indicated in images. (F) Quantification of the ratio of depolarization of the cell mitochondrial membrane potential. Bars represent mean ± sem. Statistical significance was assessed using one-way ANOVA followed by Tukey post hoc tests. *P < 0.05, ***P < 0.001, ns, not significant (N = 4).
-
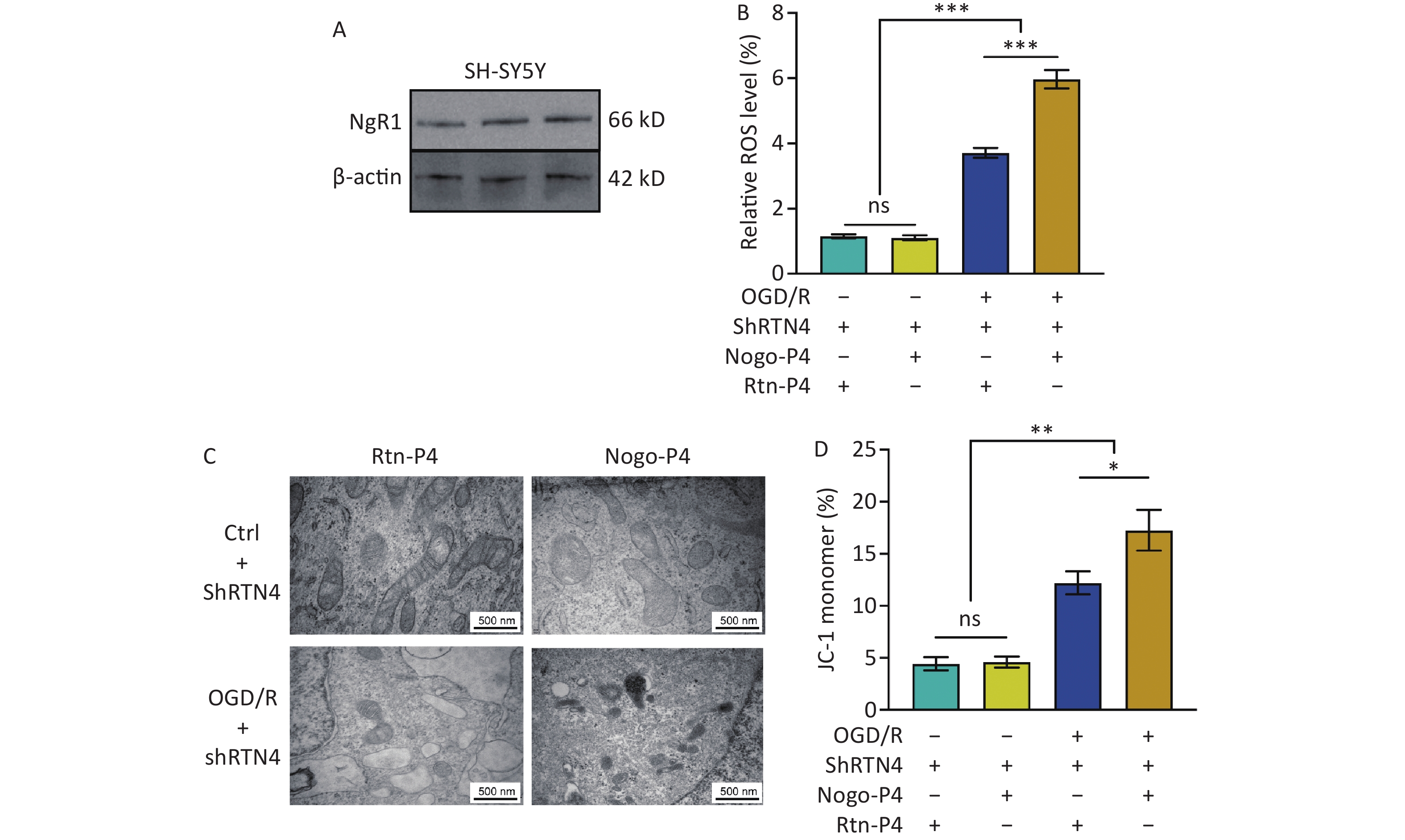
Nogo-P4 is an inhibitory peptide sequence composed of 25 amino acids (residues 31–55 of Nogo-66) and is the active fragment of Nogo-66[27]. The corresponding non-inhibitory peptide, Rtn-P4, was used as a control. We confirmed the expression of NgR1 in SH-SY5Y cells (Figure 7A). We found that in SH-SY5Y cells treated with shRTN4, supplementation with Nogo-P4 significantly increased cellular ROS levels after OGD/R (P < 0.001) (Figure 7B). Furthermore, we observed a smaller mitochondrial volume, increased membrane density, and an increased percentage of JC-1 monomers (P < 0.05) (Figure 7C, D).
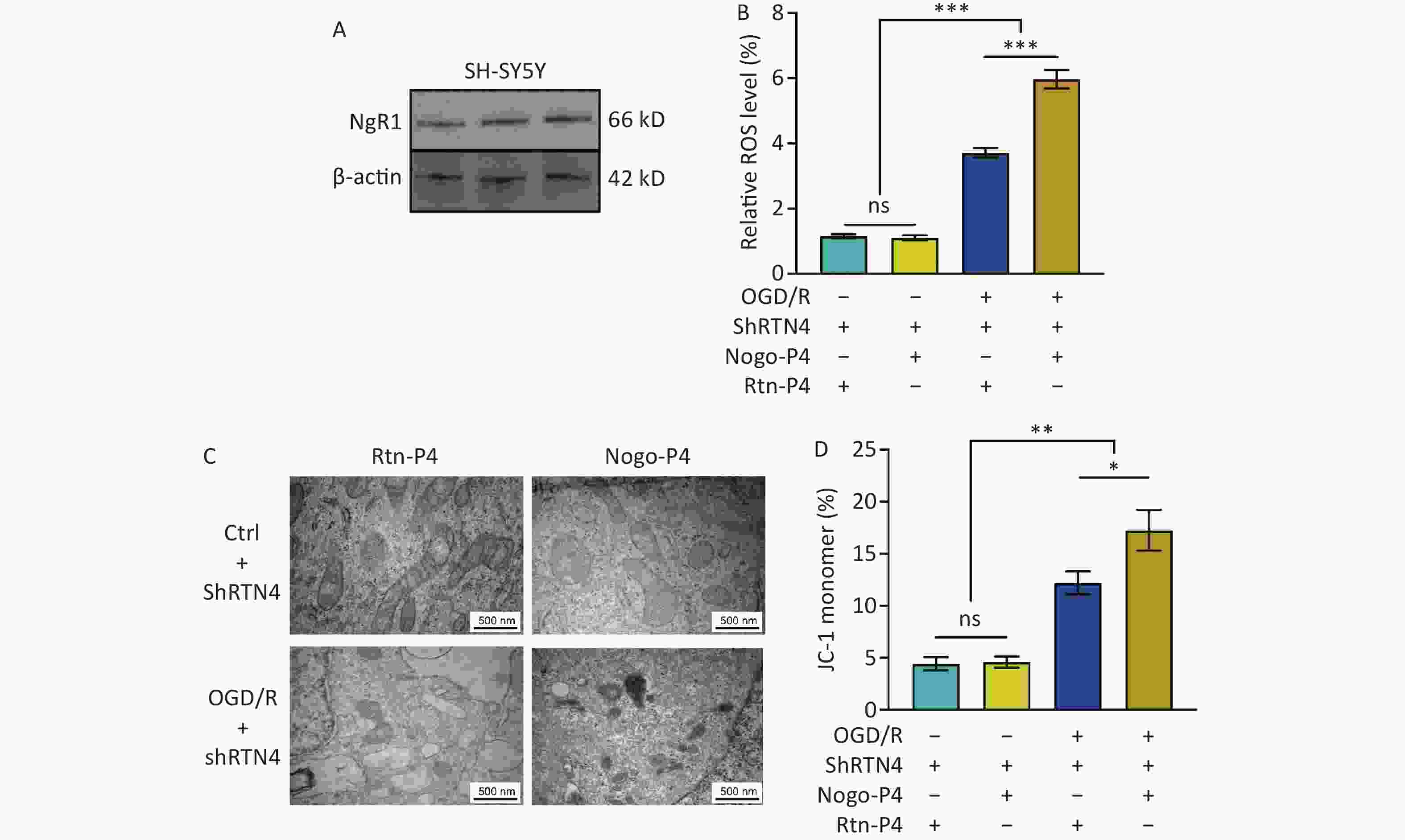
Figure 7. The effects of NgR1 agonists on mitochondrial morphology and function in SH-SY5Y cells treated with shRTN4 under the OGD/R modeling. (A) Expression of NgR1 in SH-SY5Y cells was determined using western blot. Representative western blot images of NgR1 and β-actin in SH-SY5Y cells. (B) Quantification of intracellular ROS in Nogo-A Knockdown cells after OGD/R with the treatment of Nogo-P4 or Rtn-P4. Bars represent mean ± sem. Statistical significance was assessed using one-way ANOVA followed by Tukey post hoc tests. ***P < 0.001, ns, not significant (N = 4). (C) Representative Transmission Electron Microscope (TEM) images of the mitochondria in SH-SY5Y cells treated with Nogo-P4 or Rtn-P4. Scale bars (500 nm) are indicated in the images. (D) Quantification of the ratio of depolarization of cell mitochondrial membrane potential. Bars represent mean ± sem. Scale bars assessed using one-way ANOVA followed by Tukey post hoc tests. *P < 0.05, **P < 0.01, ns, not significant (N = 4).
-
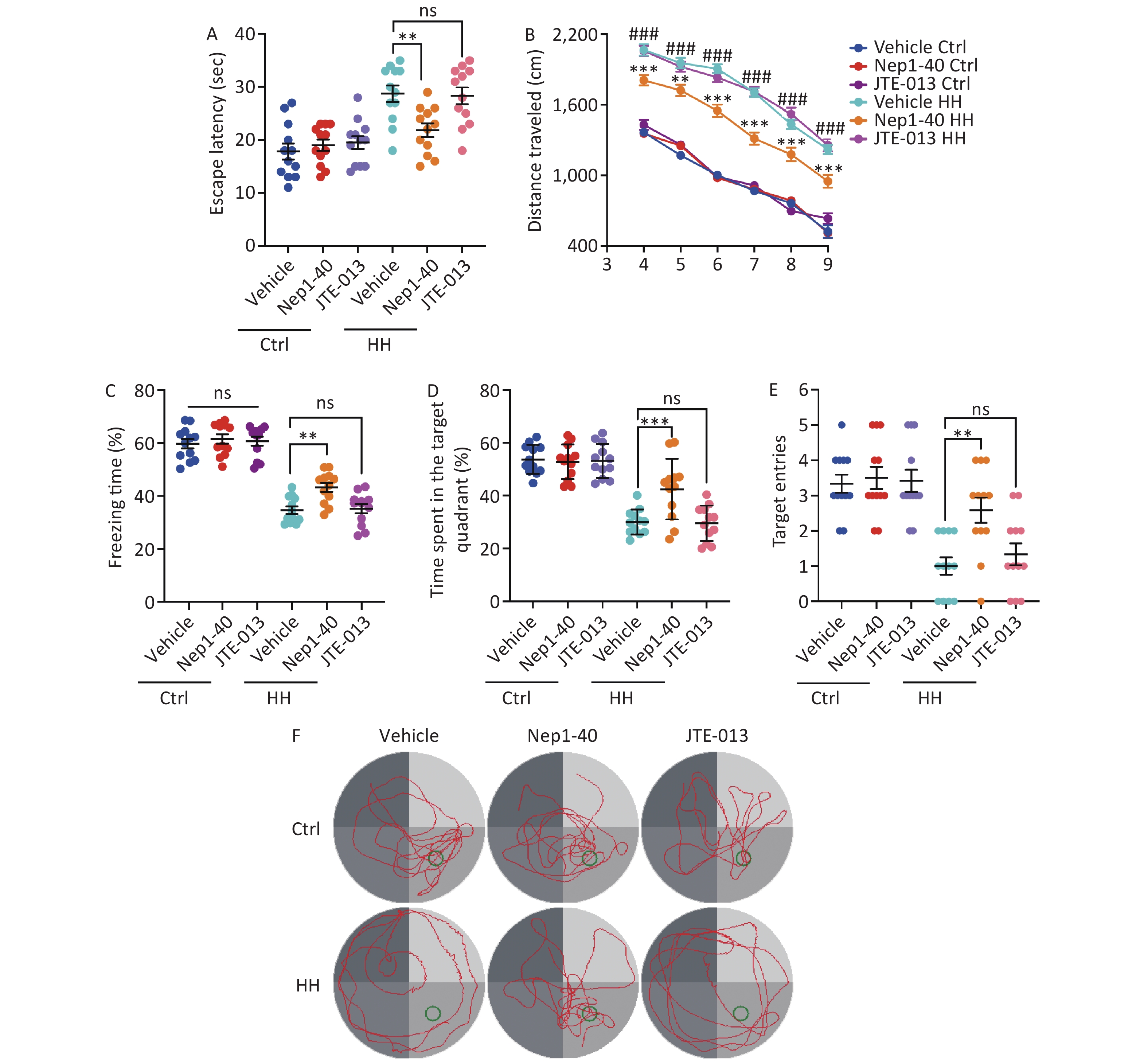
We treated rats with the antagonists, NEP1-40 and JTE-013. These two compounds selectively bind to the receptors NgR1 and S1PR2, which correspond to the functional domains Nogo66 and NogoΔ20 of Nogo-A. In contrast to those in the control group, rats treated with NEP1-40 (10 μg/μL, 2 μL/day) showed substantial increase in the freezing behavior during the contextual fear test after HH exposure (P < 0.01) (Figure 8A). NEP1-40 rats also showed improved learning and spatial memory after HH exposure in the MWM test, with better performance than that of the vehicle group during probe testing (P < 0.001, P < 0.01, P < 0.001, P < 0.01, respectively) (Figure 8B–F). No difference was observed in the JTE-013 (10 μg/μL, 2 μL) group compared to the vehicle control group (ns, ns, ns, and ns, respectively) (Figure 8B–F).
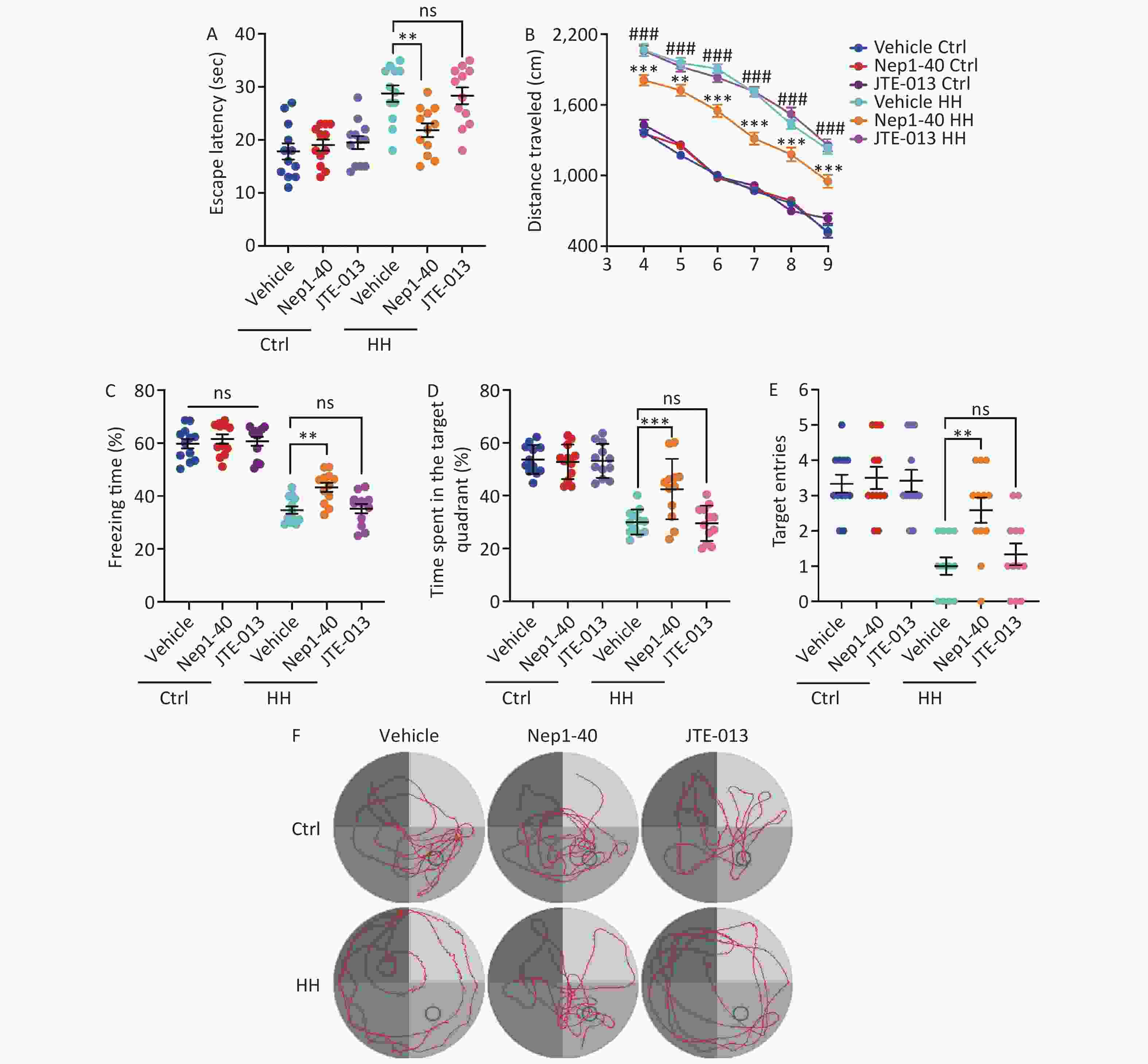
Figure 8. The effects of Nogo66 and NogoΔ20 antagonists on rat behavior after exposure to HH. (A) Freezing time (%) in the context test was measured after electric shock training. Statistical significance was assessed using one-way ANOVA followed by Tukey post hoc tests. **P < 0.01, ns, not significant (N = 12). (B) Swimming distance to the platform. Statistical significance was analyzed using one-way ANOVA followed by Tukey post hoc tests. **P < 0.01 vs. Vehicle HH, ***P < 0.001 vs. Vehicle HH, ###P < 0.001 vs. Vehicle Ctrl (N = 12). (C) primary escape latency, (D) time spent in the target quadrant, and (E) number of target entries in the probe testing phase. (F) Representative swimming tracks in the Morris Water Maze (MWM) test. Ctrl, control; HH, high-altitude hypoxia. Statistical significance was analyzed using one-way ANOVA followed by Tukey post hoc tests. **P < 0.01, ***P < 0.001, ns, not significant (N = 12).
-
The potential harm of HH exposure on L&M behavior has been well-documented[28], but the underlying mechanism is not fully understood. To investigate the effects of high-altitude hypoxia on the structure and function of the hippocampus, we utilized a low-pressure oxygen chamber designed with altitude as the control parameter, which allowed the regulation of chamber pressure to simulate real high-altitude hypoxic environments for animal modeling. Currently, no unified method is available for constructing animal models of high-altitude hypoxia, and researchers often use different altitudes for animal modeling based on experimental purposes. Although regions above 2,500 m in altitude are referred to as plateau areas, the incidence of acute mountain sickness increases with altitude (from approximately 7% at 2,200 m to over 50% at 4,559 m), and cognitive impairments, such as learning and memory deficits, only appear above 4,000 m in altitude[29-30]. Moreover, there are differences in hypoxia tolerance between rodents and humans. Although few populations in the real world live above 5,000 m in altitude, we used this parameter to create a model of learning and memory impairment caused by hypoxia at low pressure.
In the present study, we found that HH exposure significantly increased the expression of Nogo-A and NgR1 in the hippocampus. Nogo-A and its receptor play crucial roles in modulating synaptic plasticity through the regulation of LTP and long-term depression (LTD)[31]. Nogo-A and its receptor NgR1 are present at synapses in pyramidal cells in the CA1 and CA3 regions of the hippocampus[31] and in neurons of the motor cortex[32]. An overexpression of NgR1 in hippocampal neurons is associated with a reduction in synaptic quantity, which is thought to stem from the inhibition of new synapse formation[33]. This finding aligns with the observation that NgR1 knockout rats display an increase in the number of synapses in hippocampal pyramidal neurons[33-34]. Because Nogo-A is overexpressed in the rat hippocampal region, we proposed that Nogo-A/NgR1 signaling is activated in the hippocampus following HH exposure. Our findings demonstrated that ablation of the Nogo-A gene and shRNA-mediated interference markedly ameliorated the L&M deficits induced by HH exposure.
It is important to consider that the functions of Nogo-A may extend beyond neurons, because the AAV injected into the hippocampus is not specific to neurons and interference from glial cells cannot be ruled out despite validation in SH-SY5Y cells. Nogo-A is expressed not only in hippocampal neurons but also in glial cells. Nogo-A knockdown in oligodendrocytes almost completely protected against oxidative stress induced by exogenous H2O2[17]. Considering the important role of Nogo-A in mediating oxidative stress and neuronal damage in other cells, further research is needed to use targeted Nogo-A knockout methods in neurons and glial cells to clarify its cell-specific effects.
Exposure to high-altitude hypoxia increases ROS levels[21]. As oxidative stress has been widely accepted as a considerable factor in alterations in L&M[35], changes in oxidative stress-related indicators were examined in this study. We observed that the shRNA-mediated knockdown of Nogo-A in the hippocampus markedly decreased oxidative stress levels and enhanced L&M after exposure.
We postulate that the synaptic damage in hippocampal neurons is intricately linked to the onset of mitochondrial oxidative stress in this specific brain region. In neurons, mitochondria are crucial for maintaining energy homeostasis in metabolically demanding cellular regions, such as axons and synapses[36]. Mitochondria provide the necessary ATP for synaptic functions, including synaptic assembly, action potential generation, synaptic vesicle mobilization, and Ca2+ buffering[37]. Our data showed that SH-SY5Y cells exhibit basal Nogo-A and NgR1 expression levels. In this study, we observed that mitochondrial volume contraction, increased membrane density, and loss of cristae in SH-SY5Y cells with low Nogo-A expression were improved after OGD/R. However, this improvement was reversed when Nogo-P4 was supplemented. These findings further confirm our previous hypothesis that the NogoA/NgR1 pathway is involved in the neuronal oxidative stress response. Oxidative stress-induced mitochondrial dysfunction is believed to impair synaptic plasticity[38]. The induction of hydrogen peroxide production in neurons could reduce LTP, which is consistent with our findings[39]. However, we did not conduct additional experiments using antioxidants to validate the relationship between mitochondrial oxidative stress and synaptic damage.
In summary, our results demonstrate that exposure to HH impairs the L&M of rats in contextual fear and MWM behavioral tests. Genetic knockout of Nogo-A, specific knockdown of Nogo-A in the hippocampal region, and pharmaceutical inhibition of NgR1 alleviated L&M impairments caused by HH. In addition, we found that the HH-induced activation of Nogo-A/NgR1 causes synaptic damage in neurons through the oxidative stress pathway. This study enhances our understanding of Nogo-A function and provides a new perspective for therapeutic interventions for cognitive impairments related to HH.
-
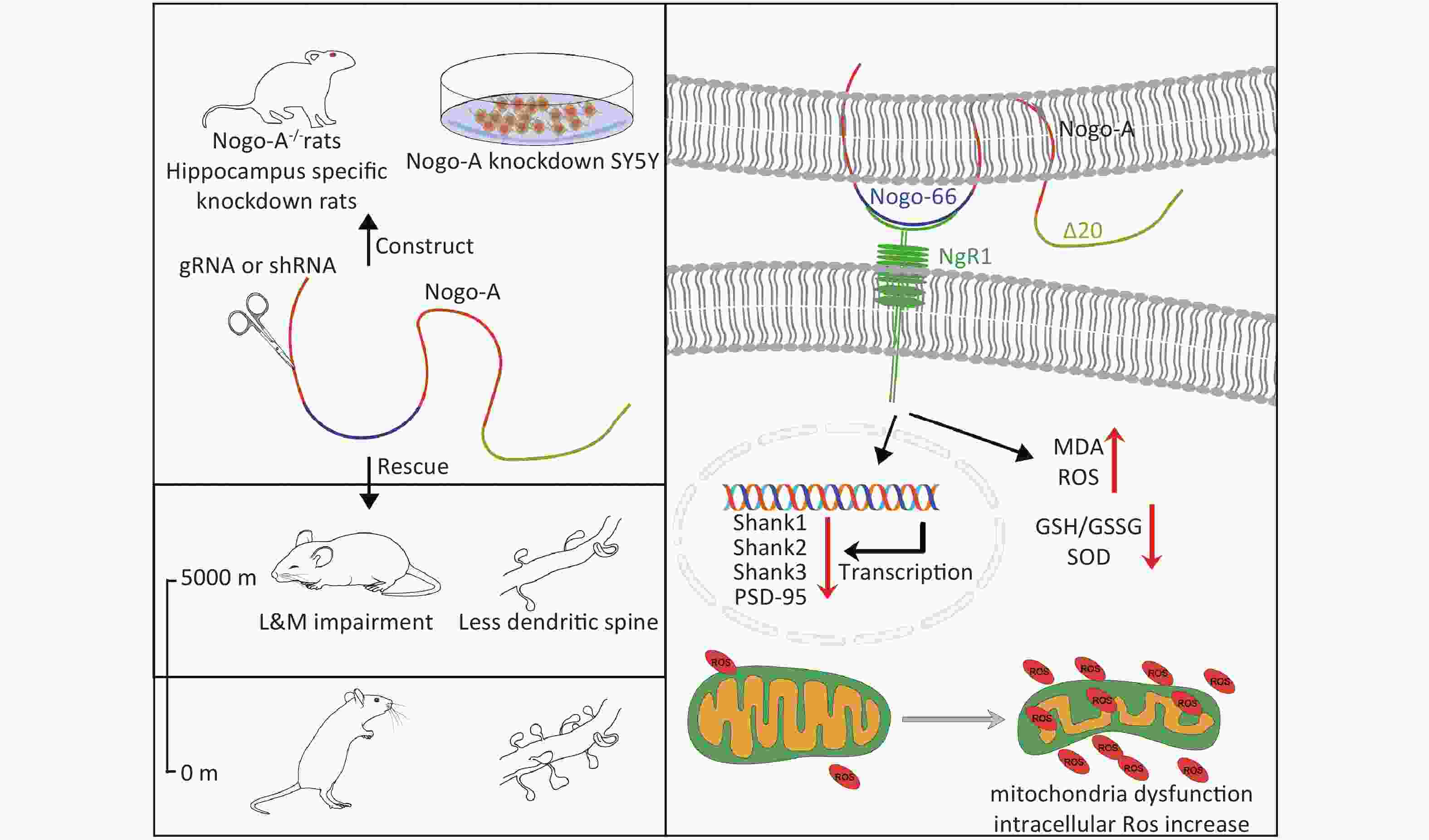
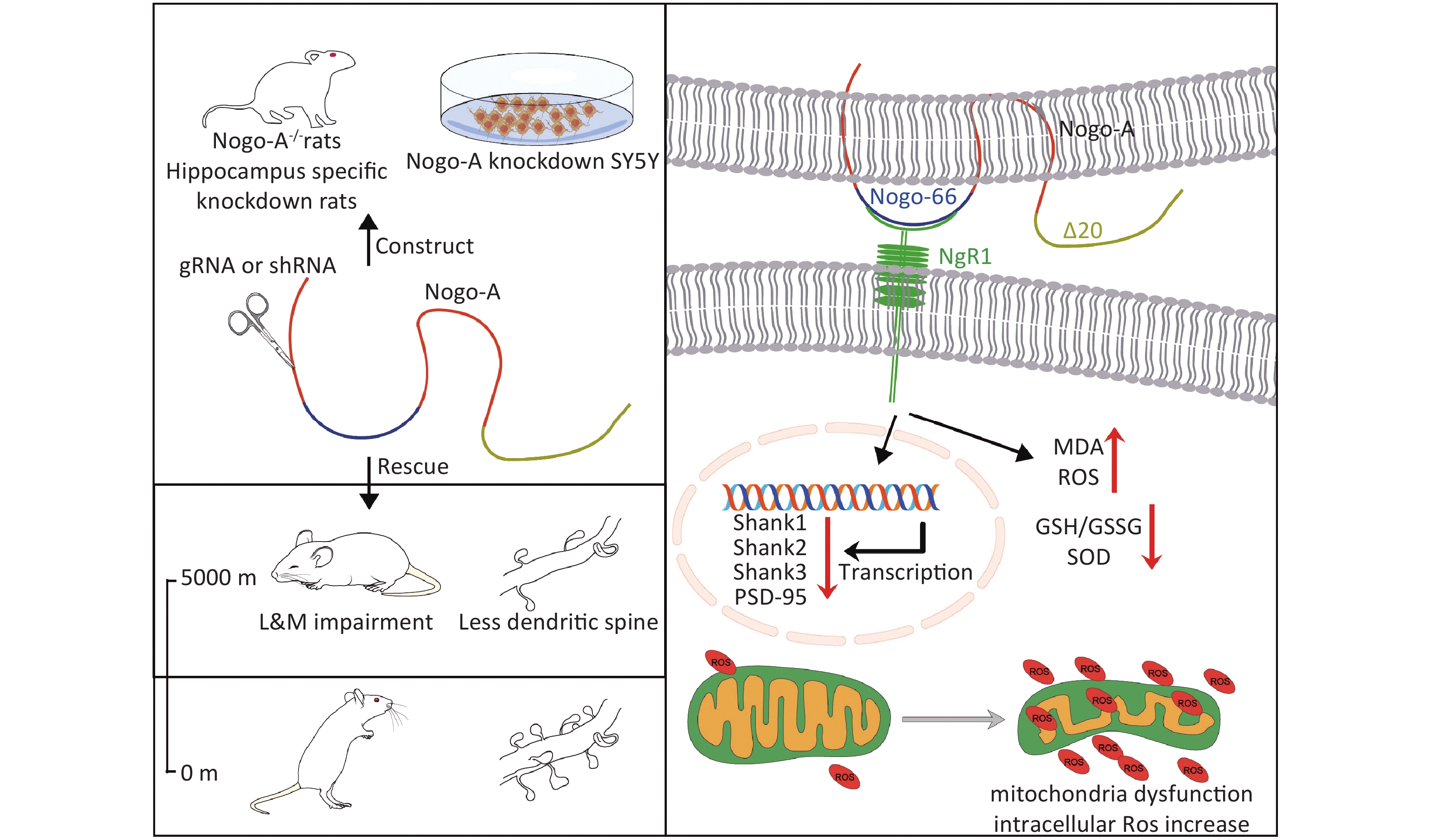
Figure S1. Graphical Abstract. SOD, superoxide dismutase; MDA, activity andmalondialdehyde; ROS, reactive oxygen species.
HTML
Animals
High-altitude Model Establishment
Contextual Fear Conditioning
Morris Water Maze (MWM)
Drugs and Administration
Quantitative Real-time PCR
Golgi Staining
Immunoblotting Analysis In Vivo Experiments
Measurement of Superoxide Dismutase (SOD) Activity and Malondialdehyde (MDA), Reactive Oxygen Species (ROS) Levels and GSH/GSSG Ratio in Tissues
Cell Culture
OGD/R Models
RNA Interference by shRNA for In Vitro Experiments
Nogo-P4 Administration
Immunofluorescence
Immunoblotting Analysis for In Vitro Experiments
Transmission Electron Microscopy (TEM)
Measurement of the Mitochondrial Membrane Potential
Examination of Intracellular ROS Generation
Statistics and Data Collection
Exposure to High-altitude Hypoxia Impaired L&M and increased Oxidative Stress in the Rat Hippocampus
High-altitude Hypoxia Exposure Increased the Expression of Nogo-A and NgR1 in the Hippocampus and Reduced Dendritic Spine Density in Hippocampal Neurons
Knockdown of Nogo-A Expression in the Hippocampus Ameliorated High-altitude Hypoxia-induced Learning and Memory Impairment and Reduced Oxidative Stress Levels
Knockout of Nogo-A Improves Learning and Memory in Rats Exposed to High-altitude Hypoxia
Knockdown of Nogo-A in SH-SY5Y Cells Ameliorated OGD/R-induced Changes in Oxidative Stress
Nogo-P4 Exacerbated Mitochondrial Morphology and Function Damage in SH-SY5Y Cells Treated with shRTN4 in the OGD/R Model
Learning and Memory Impairment Induced by High-altitude Hypoxia Exposure is Mediated through Nogo-A/NgR1
Competing Interests None of the authors has a financial interest related to this study to disclose.

 Quick Links
Quick Links
 DownLoad:
DownLoad: