

 下载:
下载:

-
The prevalence of obesity and type 2 diabetes mellitus (T2DM) has been increasing throughout the world over the past 20 years. Environmental chemicals known to regulate the endocrine system have been considered as a risk factor for the development of metabolic diseases[1]. Several people are exposed to environmental chemicals during their lives.
Di-(2-ethylhexyl) phthalate (DEHP) is currently the most highly produced phthalate. It is commonly used to add flexibility to plastic consumer products and polyvinyl chloride medical products. It can enter the human body and is metabolized by gut lipases[2]. Perinatal exposure to DEHP could induce adverse effects on glucose homeostasis in adulthood[3, 4]. These findings support the hypothesis that early exposure to DEHP can affect hormone biosynthesis and promote the onset of metabolic derangement in later life.
Because peroxisome proliferator-activated receptor gamma (PPARγ) can regulate adipocyte differentiation, the influence of PPARγ after DEHP exposure might represent the biological mechanism involved in the development of metabolic disorders. Therefore, we investigated whether exposure to DEHP in early life might influence the development of metabolic derangement in adulthood. To determine the effects of DEHP on metabolism, we measured fasting blood glucose and serum insulin levels and analyzed PPARγ expression in white adipose tissue (WAT).
Eight-week-old specific pathogen-free Sprague-Dawley pregnant female rats (n = 12) were purchased from the Charles River Orient Experimental Animal Breeding Center (Gyeonggi-do, Korea) and divided into the following three groups (n = 4): control, early life, and lifelong. The dosing solutions were prepared by dissolving 0.75 mg/(kg·day) DEHP in corn oil, and the administered dosage was adjusted daily based on weight (2.0 mL/kg). The amount of DEHP in our study was set 10 times lower than that used in previous study[5]. For control, corn oil was administered to the rat dams from gestation day (GD) 6 to postnatal day (PND) 21. The male pups received corn oil from PND 22 to PND 70. The rat dams in the early life group were administered 0.75 mg/(kg·day) DEHP from GD 6 to PND 21, and then corn oil was administered to the male pups from PND 22 to PND 70. The rat dams in the lifelong group were administered 0.75 mg/(kg·day) DEHP from GD 6 to PND 21, and then the male pups received 0.75 mg/(kg·day) DEHP from PND 22 to PND 70. The male pups were sacrificed on PND 70. Animal experiments were performed according to the Good Laboratory Practice guidelines of the Korea Testing and Research Institute.
Fasting blood glucose and serum triglyceride levels were simultaneously measured using a Hitachi 7060 chemistry analyzer (Hitachi Ltd., Japan). Serum insulin (Mercodia AB, Uppsala, Sweden), leptin (IBL, Gunma, Japan), and adiponectin (Adipogen Inc, Incheon, Korea) levels were measured using enzyme-linked immunosorbent assay according to the manufacturer protocols.
PPARγ mRNA expression was analyzed using a method described in a previous study[6]. The amplified PPARγ PCR products were normalized to rat glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The PPARγ primers were 5′-CCCTGGCAAAGCATTTGTAT-3′ (forward) and 5′-ACTGGCACCCTTGAAAAATG-3′ (reverse). The GAPDH primers were 5′-GGCACAGTCAAGGCTGAGAATG-3′ (forward) and 5′-ATGGTGGTGAAGACGCCAGTA-3′ (reverse). The PPARγ protein expression in WAT was also analyzed as described previously[6]. Images were obtained using the LAS 4000 instrument (GE Healthcare Bio-Sciences Corp., Piscataway, NJ, USA). The density of each band was quantified using the ImageQuant TL software.
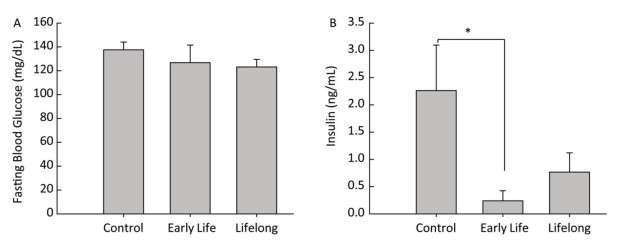
Data are expressed as mean ± standard error. Data analysis was performed using STATA (version 14.0, StataCorp LP, College Station, TX, USA). Statistical differences between the control and the treated groups were evaluated using the Wilcoxon rank sum test. P values < 0.05 were considered to represent a statistically significant difference. Blood glucose levels are increased when food enters the body. Increased blood glucose levels induce the secretion of insulin from the pancreas. Therefore, metabolic disturbances occur when the insulin or glucose feedback pathways are impaired. In our study, the average fasting blood glucose level was similar between the control and the DEHP-treated groups, with the levels being 37.6 ± 6.4 mg/dL in the control group, 126.8 ± 14.7 mg/dL in the early life group, and 123.2 ± 6.3 mg/dL in the lifelong group (Figure 1A). However, serum insulin levels were significantly decreased in the early life group (0.19 ± 0.15 ng/mL) compared to those in the control group (2.26 ± 0.83 ng/mL, P = 0.0143) (Figure 1B). An absence (or insufficient amount) of insulin after DEHP exposure might be associated with the development of hyperglycemia in which the conversion of glucose into glycogen would be inhibited. Hyperinsulinemia is associated with various metabolic disorders, including obesity, hypertension, and glucose intolerance. Although the present study observed the changes only in adulthood, perinatal exposure to DEHP could influence the balance of glucose and insulin in the developmental period and maintain up to young adulthood.
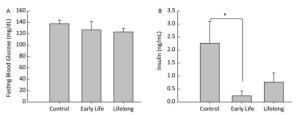
Figure 1. Effects of DEHP exposure in both the early life group and the lifelong group on (A) fasting blood glucose levels and (B) insulin levels in serum of male offspring (n = 8 per group). *Serum insulin levels were significantly lower in the early life group than in the control group (P = 0.0143). Values are mean and standard errors.
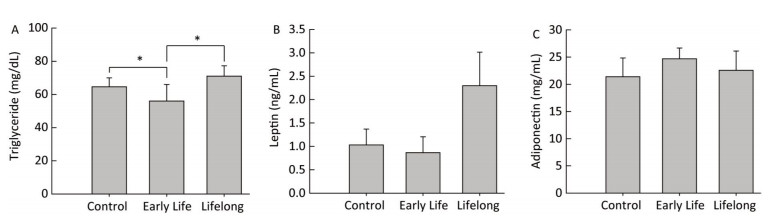
Triglycerides, the major elements of body fat, in serum were significantly decreased in the early life group (56.0 ± 9.9 mg/dL) compared to those in the control group and the lifelong group (64.6 ± 5.3 and 71.6 ± 6.2 mg/dL, P = 0.0408 and P = 0.0190, respectively) (Figure 2A). However, serum triglyceride levels were higher in the lifelong group than in the early life group (P = 0.0190, Figure 2A). Previous studies have reported a decrease in the triglyceride levels of adult male rats after exposure to DEHP [7.5 and 75 mg/(kg·day)] during the lactation period[3] and after exposure to orally administered DEHP (2% w/w) for 21 day[7]. Decreased triglyceride levels in serum might be related to the elevation of PPARγ expression after DEHP exposure. This is because the activation of PPARγ may regulate lipoprotein lipase, which can transport lipid molecules into the blood and reduce serum triglyceride levels[7, 8]. However, an increase in serum triglyceride levels has also been reported in male mice at PND 60 after DEHP [0.25 mg/(kg·day)] or MEHP [0.05 mg/(kg·day)] exposure during the perinatal period[9, 10]. It appears that the exposed dosages of DEHP might produce inconsistent results.
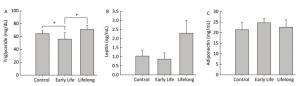
Figure 2. Effects of DEHP exposure in the early life group and the lifelong group on (A) triglyceride, (B) leptin, and (C) adiponectin levels in serum (n = 8 per group). Serum triglyceride levels in the early life group were significantly decreased compared to those in the control group (P = 0.0408). The lifelong group showed higher triglyceride levels in serum than the early life group (P = 0.0190). Values are mean and standard errors (*P < 0.05: significantly different from the control group).
Serum leptin (0.8 ± 0.3 and 2.2 ± 0.2 ng/mL, respectively) and adiponectin (24.6 ± 1.9 and 22.5 ± 3.5 mg/mL, respectively) concentrations in the early life group and the lifelong group did not show any statistical difference compared with the control group (1.0 ± 0.3 ng/mL and 21.3 ± 3.3 mg/mL, respectively) (Figure 2B and 2C). Leptin levels (2.2 ± 0.2 ng/mL) tended to be higher than those in the control group. However, the difference was not statistically significant because of the large variation within groups.
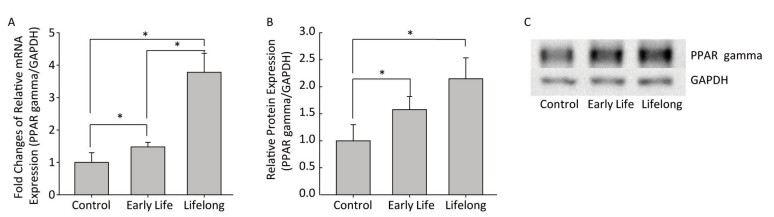
The early life and the lifelong groups showed 1.5- and 3.8- fold higher relative PPARγ mRNA expression levels than those in the control group, respectively (P = 0.0409 and P = 0.0100, respectively) (Figure 3A). The difference in the PPARγ mRNA expression between the early life and the lifelong group was statistically significant (P = 0.0156). The PPARγ protein expression in WAT was also increased in the early life and the lifelong groups compared to that in the control group (P = 0.0495 and P = 0.0495, respectively) (Figure 3B and 3C). In the lifelong group, the PPARγ protein expression tended to be higher than that in the early life group, although the difference was not statistically significant. It has been reported that increased expression of PPARγ following DEHP exposure could induce metabolic derangement by promoting oxidative stress and inhibiting the expression of insulin receptors and GLUT4 in L02 cells[11]. This suggests that perinatal exposure to DEHP increases PPARγ expression, leading to the development of metabolic disorders in adulthood.

Figure 3. Effects of DEHP exposure in the early life and the lifelong groups on PPARγ (A) mRNA and (B) protein expression (C) immunoblot images in white adipose tissue (n = 8 per group). The expression level of PPARγ mRNA was determined by real-time RT-PCR and normalized with GAPDH level, and the protein expression was determined by Western blotting. The PPARγ mRNA expression levels were significantly increased in the early life group (P = 0.0409) and the lifelong group (P = 0.0100). The lifelong group showed a statistically significant increase in PPARγ mRNA expression compared to that in the early life group (P = 0.0156). The immunoblot analysis showed similar significant results of PPARγ mRNA expression (P = 0.0495 in the early life and the lifelong groups). Values are mean and standard errors of three replicates. (*P < 0.05)
In conclusion, perinatal exposure to DEHP could affect the normal process of metabolism, and the alteration could be maintained in later life. Thus, this study suggests that there is a need for particular attention to DEHP exposure among infants and toddlers, which could induce the onset of metabolic syndrome in their adult period.
doi: 10.3967/bes2018.071
The Development of Metabolic Derangement in Male Offspring after Perinatal Exposure to Di-(2-Ethylhexyl) Phthalate
-
-
Figure 1. Effects of DEHP exposure in both the early life group and the lifelong group on (A) fasting blood glucose levels and (B) insulin levels in serum of male offspring (n = 8 per group). *Serum insulin levels were significantly lower in the early life group than in the control group (P = 0.0143). Values are mean and standard errors.
Figure 2. Effects of DEHP exposure in the early life group and the lifelong group on (A) triglyceride, (B) leptin, and (C) adiponectin levels in serum (n = 8 per group). Serum triglyceride levels in the early life group were significantly decreased compared to those in the control group (P = 0.0408). The lifelong group showed higher triglyceride levels in serum than the early life group (P = 0.0190). Values are mean and standard errors (*P < 0.05: significantly different from the control group).
Figure 3. Effects of DEHP exposure in the early life and the lifelong groups on PPARγ (A) mRNA and (B) protein expression (C) immunoblot images in white adipose tissue (n = 8 per group). The expression level of PPARγ mRNA was determined by real-time RT-PCR and normalized with GAPDH level, and the protein expression was determined by Western blotting. The PPARγ mRNA expression levels were significantly increased in the early life group (P = 0.0409) and the lifelong group (P = 0.0100). The lifelong group showed a statistically significant increase in PPARγ mRNA expression compared to that in the early life group (P = 0.0156). The immunoblot analysis showed similar significant results of PPARγ mRNA expression (P = 0.0495 in the early life and the lifelong groups). Values are mean and standard errors of three replicates. (*P < 0.05)
-
[1] Diamanti-Kandarakis E, Bourguignon JP, Giudice LC, et al. Endocrine-disrupting chemicals:an Endocrine Society scientific statement. Endocrine reviews, 2009; 30, 293-342. doi: 10.1210/er.2009-0002 [2] Schmid P, Schlatter C. Excretion and metabolism of di(2-ethylhexyl)phthalate in man. Xenobiotica, 1985; 15, 251-6. doi: 10.3109/00498258509045356 [3] Venturelli AC, Fischer SV, de Morais RN, et al. Effects of exposure to Di-(2-ethylhexyl) phthalate (DEHP) during lactation and puberty on sexual maturation and glycemic homeostasis in males rats. Clinical Nutrition ESPEN, 2015; 10, 8. http://europepmc.org/abstract/MED/28531447 [4] Lin Y, Wei J, Li Y, et al. Developmental exposure to di(2-ethylhexyl) phthalate impairs endocrine pancreas and leads to long-term adverse effects on glucose homeostasis in the rat. Am J Physiol Endocrinol Metab, 2011; 301, E527-38. doi: 10.1152/ajpendo.00233.2011 [5] Gayathri NS, Dhanya CR, Indu AR, et al. Changes in some hormones by low doses of di (2-ethyl hexyl) phthalate (DEHP), a commonly used plasticizer in PVC blood storage bags & medical tubing. Indian J Med Res, 2004; 119, 139-44. http://www.ncbi.nlm.nih.gov/pubmed/15147118 [6] Yang YJ, Kim SY, Hong YP, et al. Environmentally Relevant Levels of Bisphenol A May Accelerate the Development of Type Ⅱ Diabetes Mellitus in Adolescent Otsuka Long Evans Tokushima Fatty Rats. Toxicol Environ Health Sci, 2014; 6, 41-7. doi: 10.1007/s13530-014-0186-9 [7] Mocchiutti NO, Bernal CA. Effects of chronic di(2-ethylhexyl) phthalate intake on the secretion and removal rate of triglyceride-rich lipoproteins in rats. Food Chem Toxicol, 1997; 35, 1017-21. doi: 10.1016/S0278-6915(97)87270-5 [8] Feige JN, Gelman L, Rossi D, et al. The endocrine disruptor monoethyl-hexyl-phthalate is a selective peroxisome proliferator-activated receptor gamma modulator that promotes adipogenesis. J Biol Chem, 2007; 282, 19152-66. doi: 10.1074/jbc.M702724200 [9] Hao C, Cheng X, Guo J. Perinatal exposure to diethyl-hexyl-phthalate induces obesity in mice. Front Biosci, 2013; 5, 725-33. http://europepmc.org/abstract/MED/23277027 [10] Hao C, Cheng X, Xia H, et al. The endocrine disruptor mono-(2-ethylhexyl) phthalate promotes adipocyte differentiation and induces obesity in mice. Biosci Rep, 2012; 32, 619-29. doi: 10.1042/BSR20120042 [11] Zhang W, Shen XY, Zhang WW, et al. Di-(2-ethylhexyl) phthalate could disrupt the insulin signaling pathway in liver of SD rats and L02 cells via PPARgamma. Toxicol Appl Pharmacol, 2017; 316, 17-26. doi: 10.1016/j.taap.2016.12.010 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 1718
- HTML全文浏览量: 427
- PDF下载量: 36
- 被引次数: 0

 Quick Links
Quick Links