

 下载:
下载:

-
Human adenoviruses (HAdVs) belong to the genus Mastadenovirus, family Adenoviridae, and can cause respiratory tract and gastrointestinal tract infections, as well as ocular infections in humans[1]. Till date, a total of 84 unique genotypes of AdVs have been identified and classified as human Mastadenovirus species A to G, and specific types are often associated with certain clinical symptoms, epidemiological settings, and demographic risk groups[2]. Among these species, members of the species HAdV-B (HAdV-3, HAdV-7, HAdV-11, HAdV-16, HAdV-21, HAdV-34, HAdV-35, HAdV-50, etc), HAdV-C (HAdV-1, HAdV-2, HAdV-5, and HAdV-6), and HAdV-E (HAdV-4) have been generally associated with respiratory infections[3]. HAdV infections are mild and self-limited in healthy individuals, whereas they can result in high mortality rates in immunocompetent and immunocompromized patients[4].
Till date, HAdV infections have been well investigated in several countries[2]. However, there are limited data regarding the prevalence and genotypes of HAdVs causing infections in Qinghai Province, China. Hence, this study was conducted to perform a molecular characterization of the HAdVs prevalent in Xining City, the provincial capital comprising approximately half the population of Qinghai Province, which would provide a scientific basis for the control and prevention of respiratory AdV infections. This study was approved by the Ethics Committee of Qinghai Center for Disease Control and Prevention.
The influenza surveillance data indicate that a large number of influenza-like but influenza virus-negative cases have been detected. Since AdVs can also cause influenza-like illness, we intended to investigate the prevalence of AdV based on the influenza surveillance network. Nasopharyngeal swabs were collected from outpatients aged 2 months to 15 years with influenza-like illness (fever ≥ 38 ℃ accompanied by respiratory symptoms such as runny nose, sore throat, and cough) from Women's and Children's Hospital in Qinghai Province from November 2016 to October 2017. A total of 393 influenza virus-negative nasopharyngeal swab specimens were selected for HAdV screening in this study.
Real-time PCR platform (probe-based) was used for HAdV detection, which could provide greater efficiency and improved sensitivity and specificity compared with type-specific serum neutralization and traditional PCR assays (probeless)[5]. The viral DNA was extracted using the QIAamp DNA Mini Kit (QIAGEN, Germany) according to the manufacturer's instructions. HAdVs were detected by real-time PCR using TaqMan Universal Master Mix Ⅱ, with UNG (Applied Biosystems, USA). HAdV forward primer 5'-GGACGCCTCGGAGTACCTGAG-3', reverse primer 5'-CACAGTGGGGTTTCTGAACTTGTT-3', and probe 5'-CTGGTGCAGTTTGCCCGCGCCA-3' labeled with FAM on the 5'-end as a fluorescent dye and with BHQ-1 on the 3'-end as a fluorescence quencher dye were used. According to the manufacturer's instructions, 2 μL of DNA template was added to each 18 μL of the PCR mixture, which included 10 μL of PCR buffer, 1 μL each of primer and probe, and 5 μL of RNase-free water. The PCR was performed on ABI 7500 Real-Time PCR (Applied Biosystems, USA), with the following program: 50 ℃ for 2 min, 95 ℃ for 10 min, and then 40 cycles at 95 ℃ for 15 s and 60 ℃ for 1 min.
Subsequently, 200 μL of each HAdV-positive sample was inoculated into human epidermoid cancer cells (Hep-2) for virus isolation. Cell cultures were examined for cytopathic effect for 7-21 days from the time of inoculation. For further characterization, the hexon and fiber genes of HAdV isolates were amplified using primers designed by Sarantis et al.[6] and Xu et al.[7], respectively. Then, 5 μL of DNA was added to 20 μL of PCR mixture consisting of 12.5 μL of GoTaq Green MasterMix 2× (Promega, USA), 1 μL of each (forward and reverse) primer (10 μmol/L), and 5.5 μL of RNase-free water. The PCR was performed as follows: 94 ℃ for 3 min, followed by 35 cycles at 94 ℃ for 30 s, 55 ℃ for 30 s, and 72 ℃ for 45 s, with a final extension step at 72 ℃ for 10 min. The PCR products were identified using 1.5% agarose gel electrophoresis and then sent to Sangon Biotech (Shanghai, China) for Sanger sequencing.
The HAdV nucleotide sequences were analyzed using BioEdit version 7.0.4.1 and the NCBI BLAST software (http://blast.ncbi.nlm.nih.gov/). MEGA 6.06 software was used for phylogenetic analysis of the aligned sequences. The phylogenetic tree was generated using the neighbor-joining algorithm. The credibility of the phylogenetic trees was tested by applying a bootstrap test with 1, 000 replications.
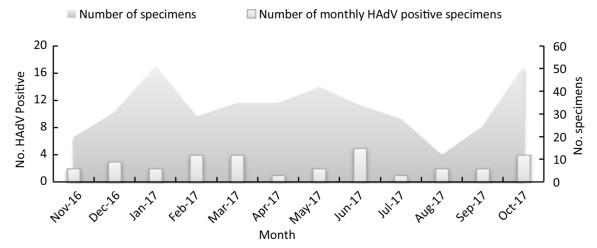
HAdV infections were detected in 32 of 393 influenza-like cases from November 2016 to October 2017 in Xining City, with a positivity rate of 8.14%, which is similar to the positivity rate of HAdV infection reported earlier in Guangzhou[8]. In addition, HAdV infections were observed throughout the year, but with no obvious peak periods (Figure 1).
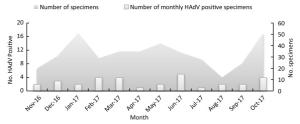
Figure 1. Monthly HAdV-Positivity for nasopharyngeal specimens from November 2016 to October 2017 in Xining city (China).
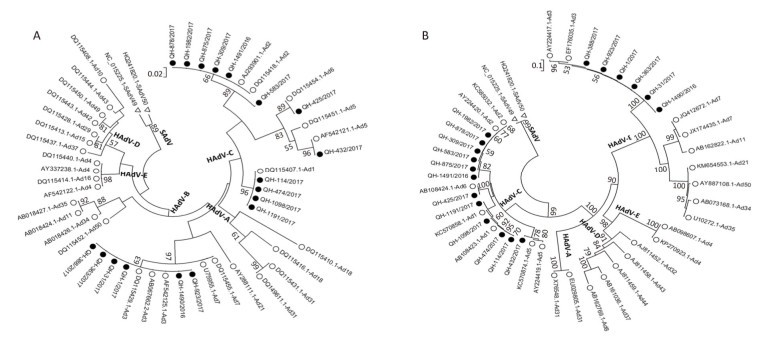
The 32 samples that were PCR-positive for HAdV were inoculated into Hep-2 cells for virus isolation, and 18 HAdV strains were isolated through 7-21 days from the time of inoculation. For further characterization, the hexon gene of the 18 HAdV isolates was amplified and sequenced, and the nucleotide sequences were submitted to GenBank (accession numbers MF985709-MF985726). Molecular typing assignments were based on the identity of the closest matching sequences after both BLAST and phylogenetic analysis. Results showed that 6 HAdV isolates of the 18 (33.3%; all HAdV-3) belonged to species B and 12 of the 18 (66.7%; 4 HAdV-1, 6 HAdV-2, 1 HAdV-5, and 1 HAdV-6) belonged to species HAdV-C (Figure 2A). Furthermore, the partial fiber gene sequences (GenBank: MF985727–MF985744) and their phylogenetic analysis (Figure 2B) confirmed the clustering of the newly detected HAdV types obtained using the hexon-based phylogenetic analysis. The observed HAdV prevalence was slightly different from that reported in other areas of China; HAdV-3 and -7 were reported to be the predominant genotypes in Beijing[9], and HAdV-3 was found to be predominant in Hangzhou and Guangzhou[8, 10].
Figure 2. Phylogenetic tree based on partial (A) hexon and (B) fiber gene sequences of HAdV Strains. Sequences originating from this study are indicated with black dots, HAdV sequences from GenBank are indicated with black circles, and SAdV sequences from GenBank are indicated with hollow black triangles as outgroup. Bootstrap proportions (1, 000 replications) are indicated as a percentage in each node.
In conclusion, our study demonstrated that HAdV infections were prevalent throughout the year but without apparent epidemic seasonal in Xining City, which indicated that corresponding prevention and control strategies should be taken into consideration in this area. Previous studies have shown that the antiviral drug response was distinct for different HAdV genotypes, and that certain new types may acquire different pathogenicity and have strong potential for widespread and epidemic outbreaks[11]. Therefore, genotype diagnosis may be beneficial for the effective treatment of HAdV infections in the future. In this study, for the first time, we have performed a molecular characterization of HAdV infections in Xining City by sequencing the partial hexon and fiber genes of the detected HAdVs and identified the members of the species HAdV-B and HAdV-C as the prevalent HAdV types in Xining City, with HAdV-3 (HAdV-B), HAdV-1, and HAdV-2 (HAdV-C) being the most prominent isolates. These results will be useful for understanding the prevalence of HAdV infections in Xining City and will provide a scientific basis for implementing preventive measures of controlling future outbreaks. Because of the limited number of collected samples and only partial sequences were obtained from the hexon and fiber genes, we plan continuous surveillance of HAdV infections in the entire Qinghai Province in the future, including a comprehensive molecular investigation of the circulating HAdV genotypes by whole genome sequencing.
We wish to express our gratitude to LU Zhuo Zhuang, PhD, of China CDC for providing the HAdV hexon primer sequences for qPCR and the staff who participated in the nasopharyngeal swab specimen collection.
doi: 10.3967/bes2019.005
Molecular Characterization of Human Respiratory Adenovirus Infection in Children from November 2016 to October 2017 in Xining City, China
-
-
Figure 1. Monthly HAdV-Positivity for nasopharyngeal specimens from November 2016 to October 2017 in Xining city (China).
Figure 2. Phylogenetic tree based on partial (A) hexon and (B) fiber gene sequences of HAdV Strains. Sequences originating from this study are indicated with black dots, HAdV sequences from GenBank are indicated with black circles, and SAdV sequences from GenBank are indicated with hollow black triangles as outgroup. Bootstrap proportions (1, 000 replications) are indicated as a percentage in each node.
-
[1] The Gene Ontology project in 2008. Nucleic Acids Res, 2008; 36(Database issue), D440-4. [2] Ferreyra LJ, Giordano MO, Martinez LC, et al. Tracking novel adenovirus in environmental and human clinical samples:no evidence of endemic human adenovirus type 58 circulation in Cordoba city, Argentina. Epidemiol Infect, 2015; 143, 1427-31. doi: 10.1017/S0950268814002192 [3] Luksic I, Kearns PK, Scott F, et al. Viral etiology of hospitalized acute lower respiratory infections in children under 5 years of age——a systematic review and meta-analysis. Croat Med J, 2013; 54, 122-34. doi: 10.3325/cmj.2013.54.122 [4] Lion T. Adenovirus infections in immunocompetent and immunocompromised patients. Clin Microbiol Rev, 2014; 27, 441-62. doi: 10.1128/CMR.00116-13 [5] Jones MS, Hudson NR, Gibbins C, et al. Evaluation of type-specific real-time PCR assays using the LightCycler and J. B. A. I. D. S. for detection of adenoviruses in species HAdV-C. PLoS One, 2011; 6, e26862. doi: 10.1371/journal.pone.0026862 [6] Sarantis H, Johnson G, Brown M, et al. Comprehensive detection and serotyping of human adenoviruses by PCR and sequencing. J Clin Microbiol, 2004; 42, 3963-9. doi: 10.1128/JCM.42.9.3963-3969.2004 [7] Xu W, McDonough MC, Erdman DD. Species-specific identification of human adenoviruses by a multiplex PCR assay. J Clin Microbiol, 2000; 38, 4114-20. http://d.old.wanfangdata.com.cn/OAPaper/oai_pubmedcentral.nih.gov_87550 [8] Zou L, Zhou J, Li H, et al. Human adenovirus infection in children with acute respiratory tract disease in Guangzhou, China. APMIS, 2012; 120, 683-8. doi: 10.1111/apm.2012.120.issue-8 [9] Li QG, Zheng QJ, Liu YH, et al. Molecular epidemiology of adenovirus types 3 and 7 isolated from children with pneumonia in Beijing. J Med Virol, 1996; 49, 170-7. doi: 10.1002/(ISSN)1096-9071 [10] Xie L, Yu XF, Sun Z, et al. Two adenovirus serotype 3 outbreaks associated with febrile respiratory disease and pharyngoconjunctival fever in children under 15 years of age in Hangzhou, China, during 2011. J Clin Microbiol, 2012; 50, 1879-88. doi: 10.1128/JCM.06523-11 [11] Morfin F, Dupuis-Girod S, Mundweiler S, et al. In vitro susceptibility of adenovirus to antiviral drugs is species-dependent. Antivir Ther, 2005; 10, 225-9. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=b08fe6f3dd1dfd7ac250bd1880b8d6a1 -

 点击查看大图
点击查看大图
图(2)
计量
- 文章访问数: 2109
- HTML全文浏览量: 485
- PDF下载量: 59
- 被引次数: 0

 Quick Links
Quick Links