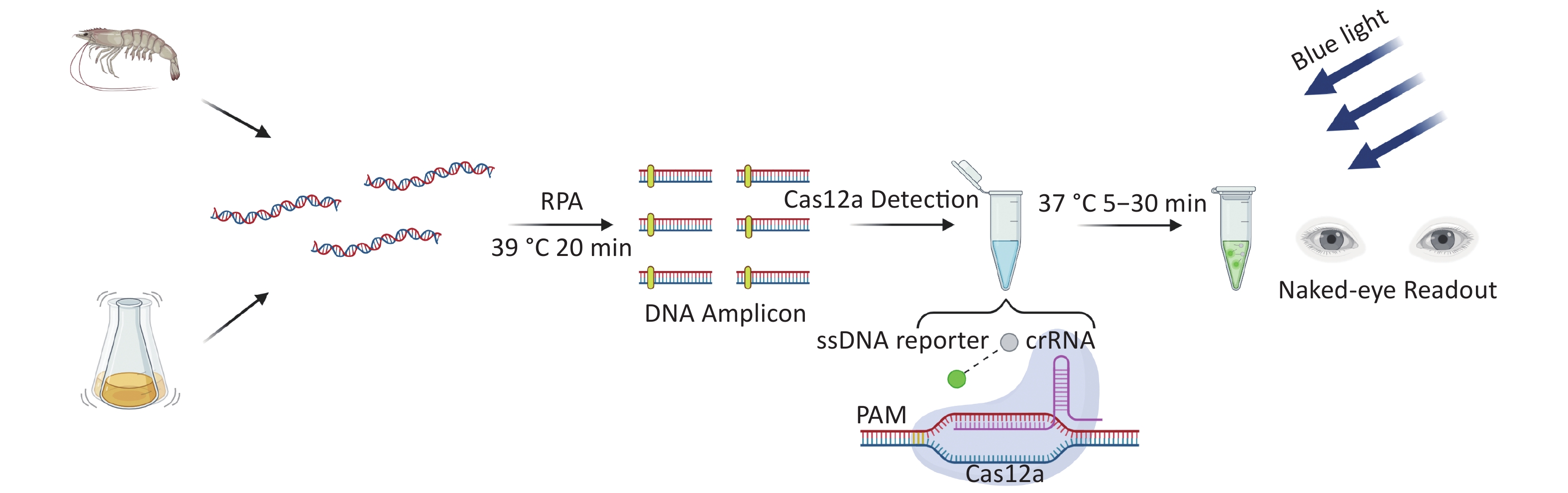

-
In recent years, Vibrio parahaemolyticus (VP) has been listed as one of the most important bacteria causing foodborne diseases. VP is a dynamic facultative anaerobe, free of spores and capsules, suitable for growth at 30−36 °C, and has the ability to withstand a salt concentration of 3%−8%[1,2]. VP is found in seawater, silt, fish, shrimps, crabs, shellfish, and other seafood[3,4]. Accidental consumption of seafood carrying VP can lead to foodborne poisoning, which often manifests as vomiting, diarrhoea, gastrointestinal spasm, fever, and even septicaemia in severe cases[5]. In addition, outbreaks of VP can kill large numbers of aquaculture animals in a short time, resulting in huge economic losses[6,7]. Since it was first discovered in Japan in 1950, VP has become a global public health problem, and poses a serious threat to human health and life[8].
It is necessary to detect pathogens with high sensitivity and specificity in the early stages of infection to effectively inhibit spread[9,10]. The traditional detection methods for VP include classic cultures, immunoassays, and nucleic acid detection[11,12]; however, classic cultures are limited due to the long detection cycle, labour-intensiveness, and a requirement for experienced technologists. Immunoassays have high specificity, but lack high selectivity for reagents and low sensitivity. Traditional PCR amplification requires precision instruments and equipment due to the need to set different temperature stages to achieve amplification. Therefore, PCR is generally used for laboratory research[13]. There is a growing demand for on-site application with visual results to provide portable, rapid testing in non-laboratory and resource-poor settings. With the development of science and technology, isothermal amplification technology led by recombinase polymerase amplification (RPA), has been used to detect pathogens because RPA does not require temperature control equipment and has great application potential in point-of-care (POC) detection[14,15]. Due to the low sensitivity and lack of convenient results visualization methods, further optimization is needed to apply RPA to on-site detection[16,17].
In recent years, due to advantages (simple operation, accurate targeting, and high efficiency), clustered regularly interspaced short palindromic repeats/CRISPR-associated protein (CRIRPR/Cas) technology has grown in popularity[18,19]. Due to trans-cleavage activity, CRISPR/Cas12a is used in gene knockout, gene therapy, gene modification[20-24], and various novel nucleic acid detection techniques[25-29]. In 2018, Chen et al. developed a new DNA detection method [DNA endonuclease targeted CRISPR trans reporter (DETECTR)] by combining CRISPR/Cas with isothermal amplification[30]. DETECTR uses RPA to amplify targeted double-stranded DNA (dsDNA) at a constant temperature. When Cas12a specifically recognises and cleaves the amplified product under the control of a programmable specific CRISPR RNA (crRNA), non-specific trans-cleavage activity is activated, which cleaves the single-stranded DNA (ssDNA) reporter in the system, generating a fluorescence signal. DETECTR is used for the rapid detection of viruses, and the sensitivity can reach the attomole level[30].
There are two important virulence factors in VP [thermostable direct hemolysin (TDH) and TDH-related hemolysin (TRH)], which are encoded by tdh and trh genes, respectively. Most clinical isolates carry one or both genes. To reduce the threat to human health of pathogenic VP, we developed a new detection method for VP based on CRIRPR/Cas12a with tdh and trh as targets, combined with RPA and visual detection (CRISPR/Cas12a-VD). The method is sensitive, specific, rapid, and the results can be interpreted by the unaided eye under blue light (Figure 1).
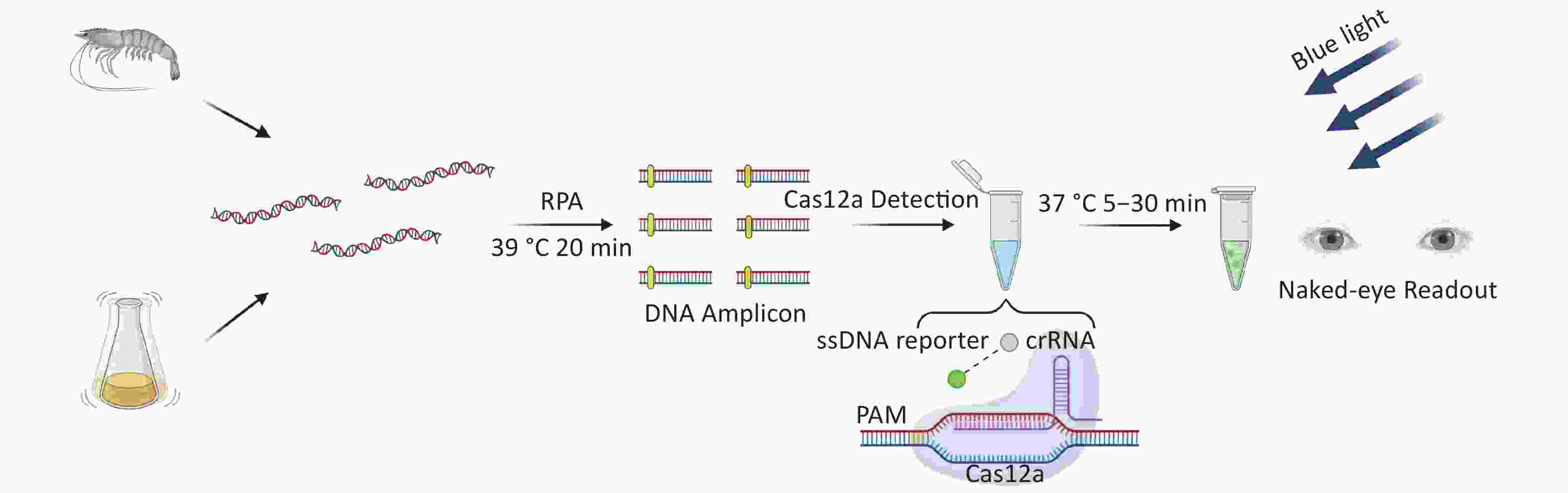
Figure 1. Schematic of CRISPR/Cas12a-VD for Rapid and Visual Nucleic Acid Detection. After 20 min of RPA reaction, the targeted cleavage of Cas12a enzyme for an additional 5–30 min. The green fluorescent signals can be observed by the naked eye under blue light.
-
A total of seven VP strains, three other Vibrio spp., and five other important foodborne pathogens were used in this study. The details of the strains are listed in Table 1. Each strain was separately cultured in 5 mL of LB broth at 37 °C overnight. The number of proliferative cells in each bacterial culture was enumerated by plate counting in the LB agar.
Stains Origin No. of strains Vibrio parahaemolyticus Isolated in our lab 7 Vibrio vulnificus ATCC 27562 1 Vibrio cholera ATCC 51394 1 Vibrio mimicus ATCC 33653 1 Listeria monocytogenes ATCC 19115 1 Salmonella typhimurium ATCC 14028 1 Bacillus cereus ATCC 11778 1 Escherichia coli O157:H7 ATCC 43888 1 Staphylococcus aureus ATCC 6538 1 Table 1. Bacterial strains used in this study
-
EnGenLba Cas12a was purchased from New England Biolabs (Ipswich, MA, USA). A TwistAmp Basic Kit was purchased from TwistDx Ltd. (Maidenhead, UK). A MiniBEST Bacteria Genomic DNA Extraction Kit (version 3.0) and a MiniBEST DNA Fragment Purification Kit (version 4.0) were purchased from TaKaRa Bio, Inc. (Dalian, China). FastStart Universal Probe Master Mix (ROX) was purchased from F. Hoffmann-La Roche Ltd. (Basel, Switzerland). Other regents used in this study were purchased from Thermo Fisher Scientific Inc. (Waltham, MA, USA).
-
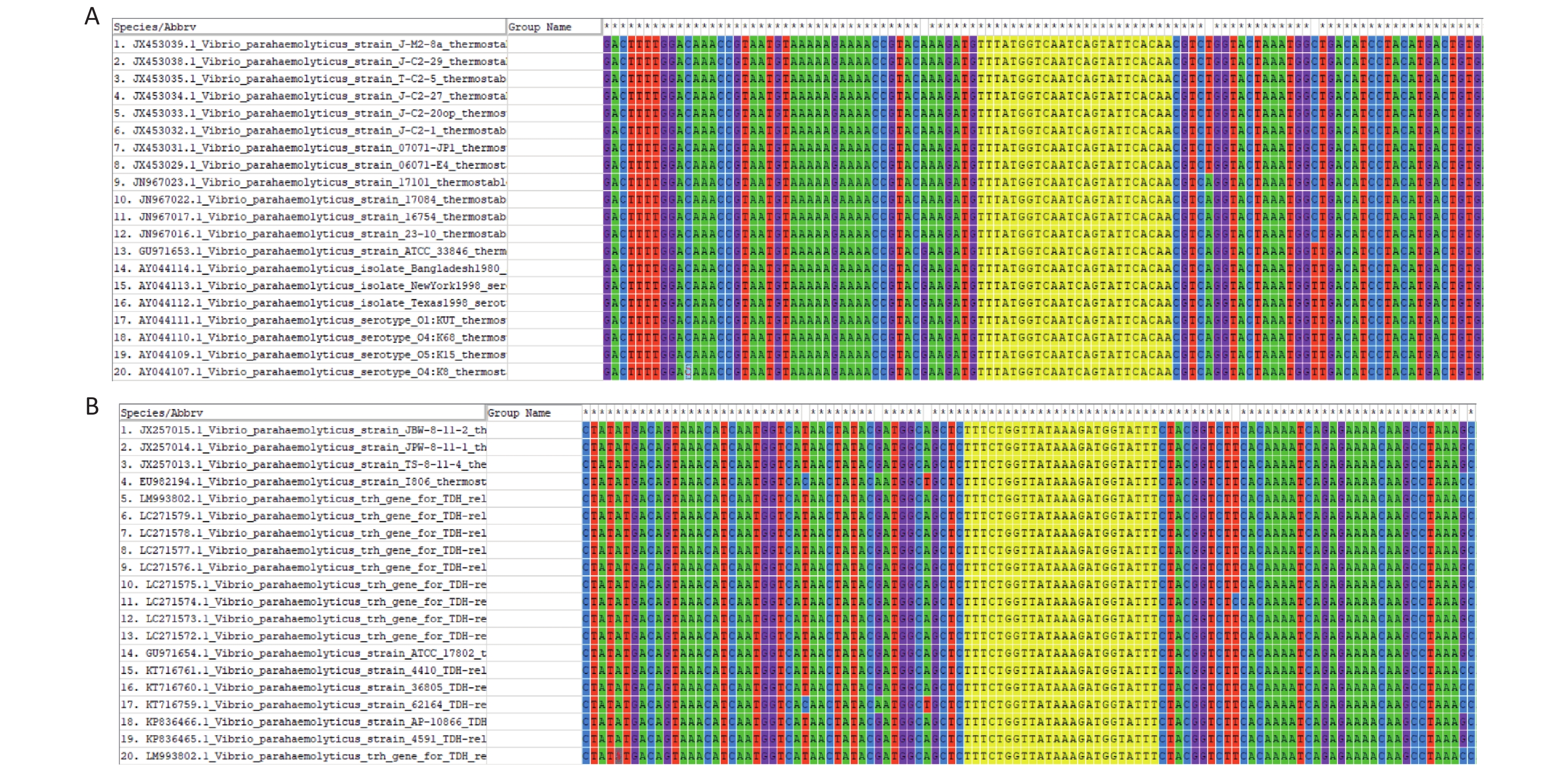
The FASTA sequences of the tdh and trh genes from different strains were searched and downloaded from the NCBI database. MEGA 7.0 sequence alignment software was used to identify conserved regions in the two genes that could serve as targets (Supplementary Figure S1, available in www.besjournal.com).
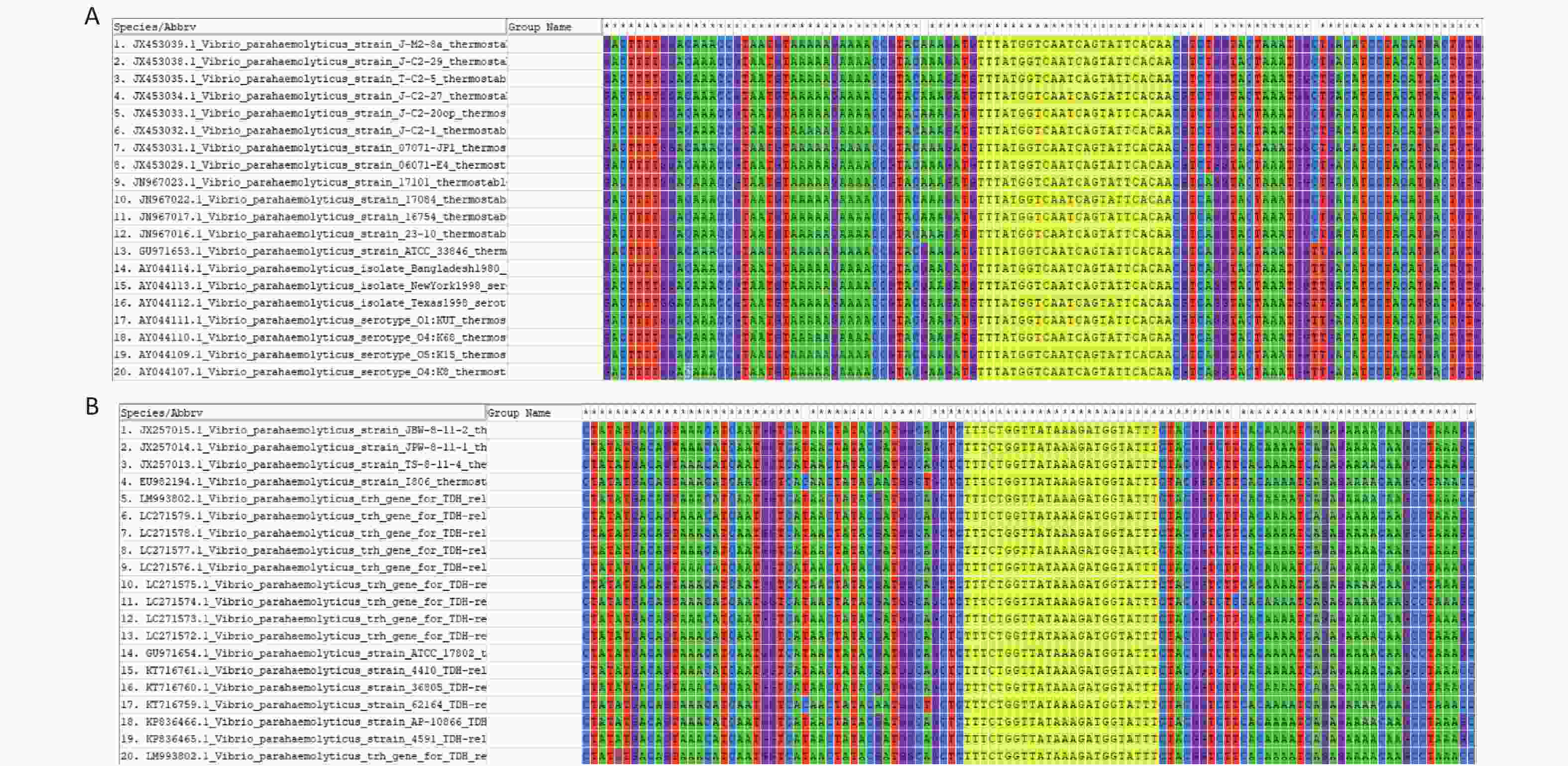
Figure S1. Sequence alignment of tdh and trh genes from 20 V. parahaemolyticus genotypes: (A) tdh genes (B) trh genes. The conserved sequences targeted by crRNA were highlighted with yellow color.
-
All primers were synthesised by Sangon Biotech (Shanghai, China), as was the ssDNA reporter (FAM-TTATT-BHQ1) and the tdh and trh gene fragments of VP. The genes were cloned into the pUC57 vector, and crRNAs were synthesised by Sangon Biotech. All sequences used in this study are shown in Supplementary Tables S1 and S2, available in www.besjournal.com.
Name Sequence (5' to 3') tdh gene fragment TATTTTGCAAAAAAATCATTTTTATTTATATCCATGTTGGCTG
CATTCAAAACATCTGCTTTTGAGCTTCCATCTGTCCCTTTTCC
TGCCCCCGGTTCTGATGAGATATTGTTTGTTGTTCGAGATAC
AACTTTTAATACCCAAGCTCCGGTCAATGTAAAGGTCTCTGA
CTTTTGGACAAACCGTAATGTAAAAAGAAAACCGTACGAAG
ATGTTTATGGTCAATCAGTATTCACAACGTCAGGTACTAAAT
GGTTGACATCCTACATGACTGTGAACATTAATGATAAAGACT
ATACAATGGCAGCGGTGTCTGGCTATAAGAGCGGTCATTCTG
CTGTGTTCGTAAAATCAGATCAAGTACAACTTCAACATTCCT
ATAATTCTGTAGCTAACTTTGTTGGTGAAGATGAAGGTTCTA
TTCCAAGTAAAATGTATTTGGATGAAACTCCAGAATATTTTG
TTAATGTAGAAGCATATGAGAGTGGTAGTGGTAATATATTG
GTAATGTGTATATCCAACAAAGAATCGTTTTTTGAATGTtrh gene fragment ATGAAACTAAAACTCTACTTTGCTTTCAGTTTGCTATTGGTTT
CAATGTTTTCAGTATCTAAATCATTCGCGATTGATCTGCCATC
AATACCTTTTCCTTCTCCAGGTTCGGCTGAACTGTTATTTGTT
GTTAGAAATACAACAATCAAAACTGAATCCCCGGTTAAGGCA
ATTGTGGAGGACTATTGGACAAACCGAAACATAAAAAGAAAA
CCATACAAAGATGTATACGGTCAATCGGTTTTCACAACAGCAG
GTTCAAAGTGGTTAAGCGCCTATATGACAGTAAACATCAATGG
TCATAACTATACGATGGCAGCTCTTTCTGGTTATAAAGATGGT
ATTTCTACGGTCTTCACAAAATCAGAGAAAACAAGCCTAAAGC
AAGACTATTCCTCGGTAAAGTCTTTTGTTGATGACAGCGAAGA
ATCAATACCAAGTATAACTTATTTAGATGAAACATCAGAATAC
TTTGTTACTGTCGAGGCATATGAGAGCGGCAATGGACATATGT
TTGTTATGTGCATTTCCAACAAATTATCATTTGGTGAATGTAAA
TCACAAATTTAATable S1. Oligonucleotides used to construct plasmid templates
Name Sequence (5' to 3') tdh-RPA-F1 CAACTTTTAATACCCAAGCTCCGGTCAATG tdh-RPA-R1 CAGCAGAATGACCGCTCTTATAGCCAG tdh-RPA-F2 CCGGTTCTGATGAGATATTGTTTGTTGTTC tdh-RPA-R2 GCAGAATGACCGCTCTTATAGCCAGACACC trh-RPA-F3 CAATTGTGGAGGACTATTGGACAAACCGAAAC trh-RPA-R3 CTTTAGGCTTGTTTTCTCTGATTTTGTGAAGACC trh-RPA-F4 GTGGAGGACTATTGGACAAACCGAAACATA trh-RPA-R4 TACCGAGGAATAGTCTTGCTTTAGGCTTGT trh-RPA-F5 GTGGAGGACTATTGGACAAACCGAAACATA trh-RPA-R5 GACTTTACCGAGGAATAGTCTTGCTTTAGG tdh-q-F AAACATCTGCTTTTGAGCTTCCA tdh-q-R CTCGAACAACAAACAATATCTCATCAG tdh-q-P FAM-TGTCCCTTTTCCTGCCCCCGG-TAMRA trh-q-F ATTCGCGATTGATCTGCCAT trh-q-R ATAGTCCTCCACAATTGCCT trh-q-P FAM-CCTTCTCCAGGTTCGGCTGAACTGT-TAMRA tdh-crRNA UAAUUUCUACUAAGUGUAGAUUGGUCAAUCAGUAUUCACAA trh-crRNA UAAUUUCUACUAAGUGUAGAUUGGUUAUAAAGAUGGUAUUU ssDNA reporter FAM-TTATT-BHQ1 Table S2. Primers, CrRNA, and SsDNA reporter used in this study
-
According to the manufacturer’s manual, primer pairs were designed for tdh and trh genes of VP. The specific amplification process was carried out in accordance with the instructions of the TwistAmp Basic Kit. The rehydration mixture for each reaction contained 2.4 μL of forward primer (10 μmol/L), 2.4 μL of reverse primer (10 μmol/L), 29.5 μL of primer-free rehydration buffer, and 13.2 μL of template and sterile water (total volume = 47.5 μL). This solution was mixed with dehydration reagent, and 2.5 μL of 280 mmol/L magnesium acetate (MgOAc) was added to start the reaction. The mixture was incubated in a PCR machine at 37 °C for 20 min. Amplification products were purified and separated by 1.5% agarose gel electrophoresis.
-
The Cas12a-mediated cleavage assay was carried out in reaction buffer and contained 20 mmol/L Tris-HCl (pH 7.5), 100 mmol/L KCl, 5 mmol/L MgCl2, 1 mmol/L dithiothreitol (DTT), and 1% glycerol. Each reaction system contained 50 nmol/L Cas12a, 62.5 nmol/L crRNA, 50 nmol/L ssDNA reporter, and 2 μL template in a total reaction volume of 20 μL. Reactions were carried out at 37 °C for 120 min, and the fluorescence signal was measured every minute using a ViiA 7 system (Applied Biosystems, Waltham, MA, USA).
-
Cas12a-mediated cleavage assay was combined with RPA for CRISPR/Cas12a-VD. After the plasmid template was pre-amplified by RPA, the amplified products were added to the Cas12a-mediated cleavage system for detection. Each reaction system contained 125 nmol/L Cas12a, 156.25 nmol/L crRNA, 625 nmol/L ssDNA reporter, and supplementary amplification products to 20 μL. After incubation at 37 °C for 5–30 min, strong green fluorescence signals were observed in positive samples by the unaided eye using a Blue Light Gel Imager (440–485 nm; Sangon Biotech, Shanghai, China).
-
Fresh cultures were prepared using the aforementioned method. VP suspensions of different cell densities were prepared in phosphate-buffered saline (PBS) for inoculation. According to the instructions of the TaKaRa MiniBEST Bacteria Genomic DNA Extraction Kit (version 3.0), genomic DNA was extracted from VP strains, and tdh and trh genes were detected by CRISPR/Cas12a-VD.
-
Fresh shrimps purchased from a local supermarket in Tianjin were de-headed and decontaminated by immersing in 75% ethanol for 2 min to eliminate interference from other microorganisms. After washing in sterile water, the shrimps were incubated in a biosafety cabinet under UV light for 30 min. Sampling and testing of the treated shrimps were carried out to rule out the possibility of pre-contamination by VP. Sterile shrimps were incubated in VP suspensions for 30 min, then transferred into clean plates and incubated in the biosafety cabinet for another 30 min to promote bacterial attachment at ambient temperature. The surfaces of shrimps were wiped with disposable Q-tips, which were then placed in 1.5-mL centrifuge tubes and 200 μL of sterile water was added to elute VP[31,32]. After DNA extraction using a TaKaRa MiniBEST Bacteria Genomic DNA Extraction Kit (version 3.0), tdh and trh genes were detected by CRISPR/Cas12a-VD.
-
TaqMan real-time PCR detection was performed using the ViiA 7 system. Each 20-μL reaction volume was made up of 10 μL of FastStart Universal Probe Master Mix (ROX), 0.5 μL of forward primer (10 μmol/L), 0.5 μL of reverse primer (10 μmol/L), 0.5 μL of TaqMan probe (10 μmol/L), 2 μL of template, and 6.5 μL of sterile water. The amplification conditions included initial denaturation steps of 50 °C for 2 min and 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. Fluorescence information was collected at the 60 °C annealing extension per cycle.
-
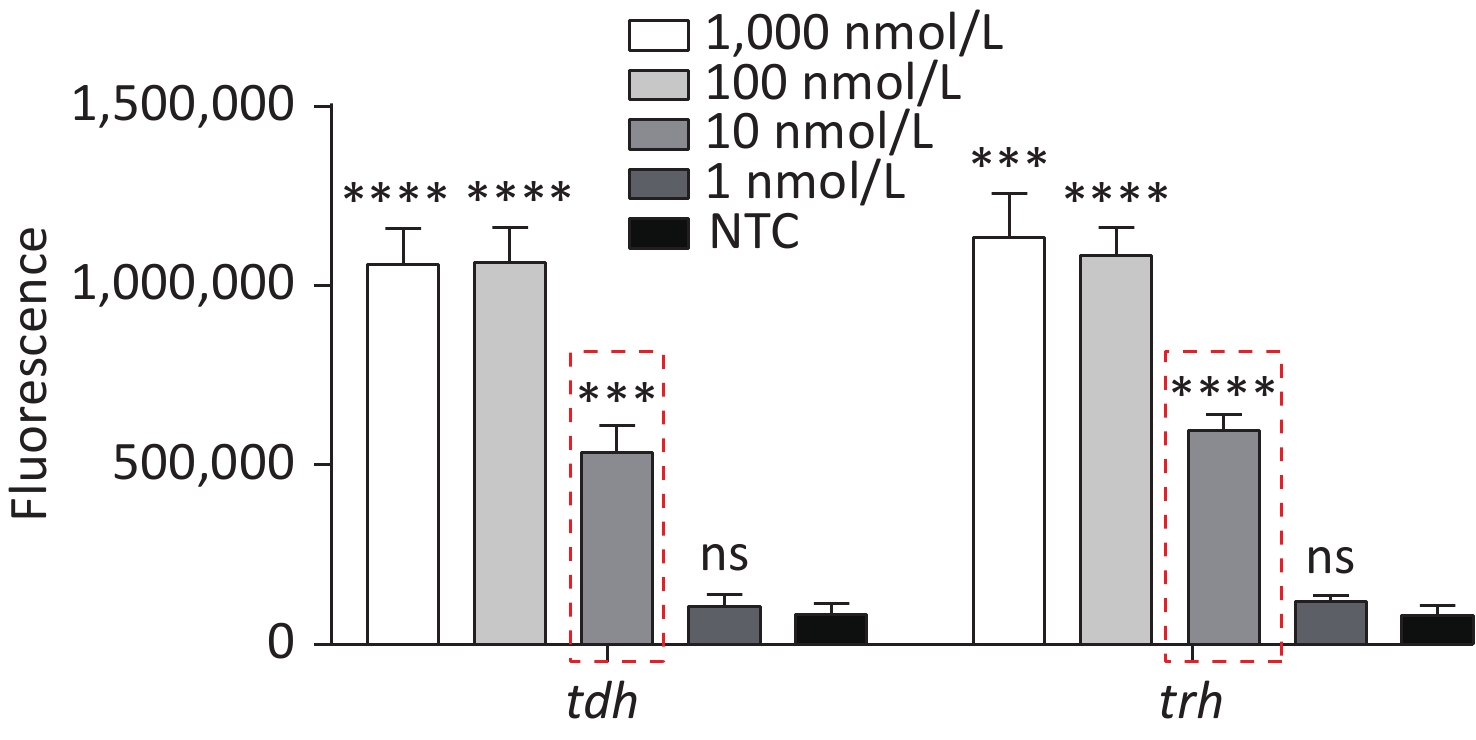
To detect VP genomic DNA with sensitivity and specificity, we first constructed a Cas12a-based cleavage system for tdh and trh genes. The system consisted of the following three main components: Cas12a; a specific crRNA; and an ssDNA reporter. Because Cas12a recognises the protospacer adjacent motif (PAM) sequence of TTTN, we selected the qualified conserved sequences as targets for tdh and trh genes, and designed the corresponding crRNAs. We serially diluted the plasmid template and performed the experiment. When Cas12a specifically recognised and cleaved the target plasmid via guidance from the crRNA, the non-specific trans-cleavage activity was activated and the ssDNA reporter was cleaved, generating a fluorescent signal. The limit of detection (LOD) of the Cas12a cleavage system for tdh and trh genes was 10 nmol/L (Figure 2), which is consistent with the results published in 2018 by Chen et al.[30].
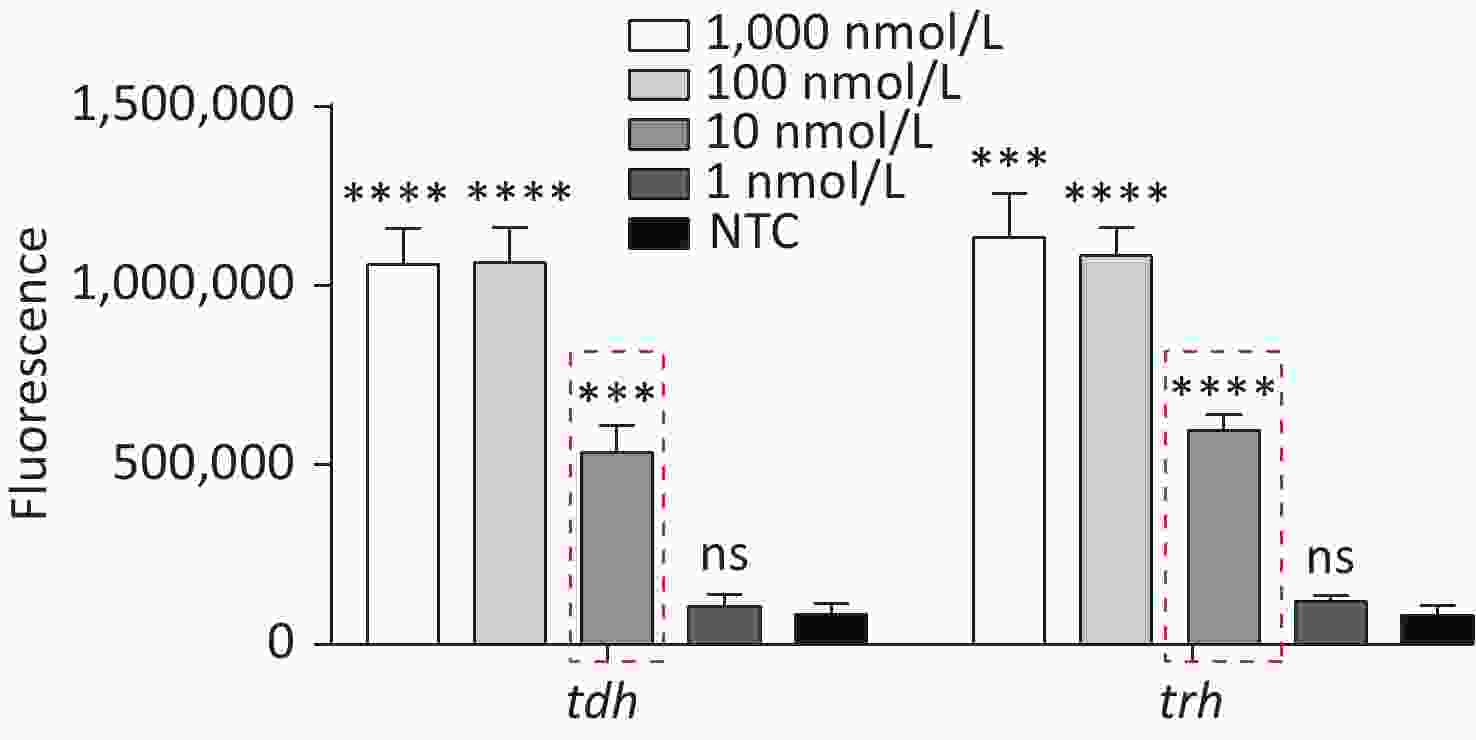
Figure 2. The sensitivity of the Cas12a-mediated cleavage assay to detect tdh and trh genes from 1,000 nmol/L to 1 nmol/L. n = 3, two-tailed Student’s t test; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 and ns not significant; NTC nontarget control; bars represent mean ± SD.
-
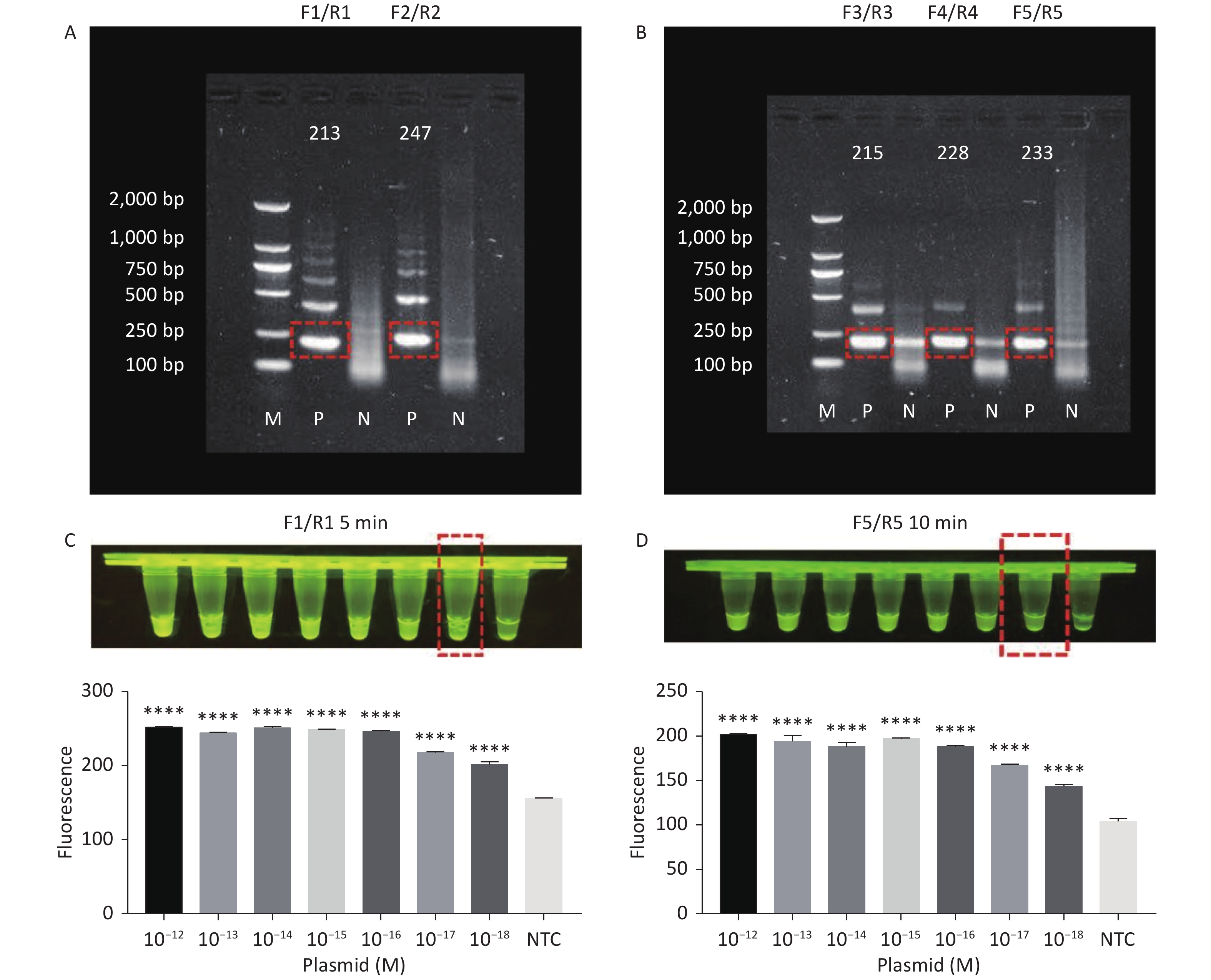
RPA amplifies trace nucleic acid templates to a detectable level in a short time[14,15]. The optimum reaction temperature is ~37 °C, and complex temperature control equipment is not needed[14,15]. Compared with traditional PCR, RPA is more suitable for field detection[33]. When combined with RPA, the Cas12a LOD can be greatly improved. Therefore, we designed several pairs of primers for tdh and trh genes. The results of agarose gel electrophoresis showed that two pairs of primers successfully amplified the expected bands for the tdh gene (Figure 3A), and three pairs of primers successfully amplified the expected bands for the trh gene (Figure 3B). We screened all five pairs of primers in combination with the Cas12a cleavage system to select the best-performing primer pairs.
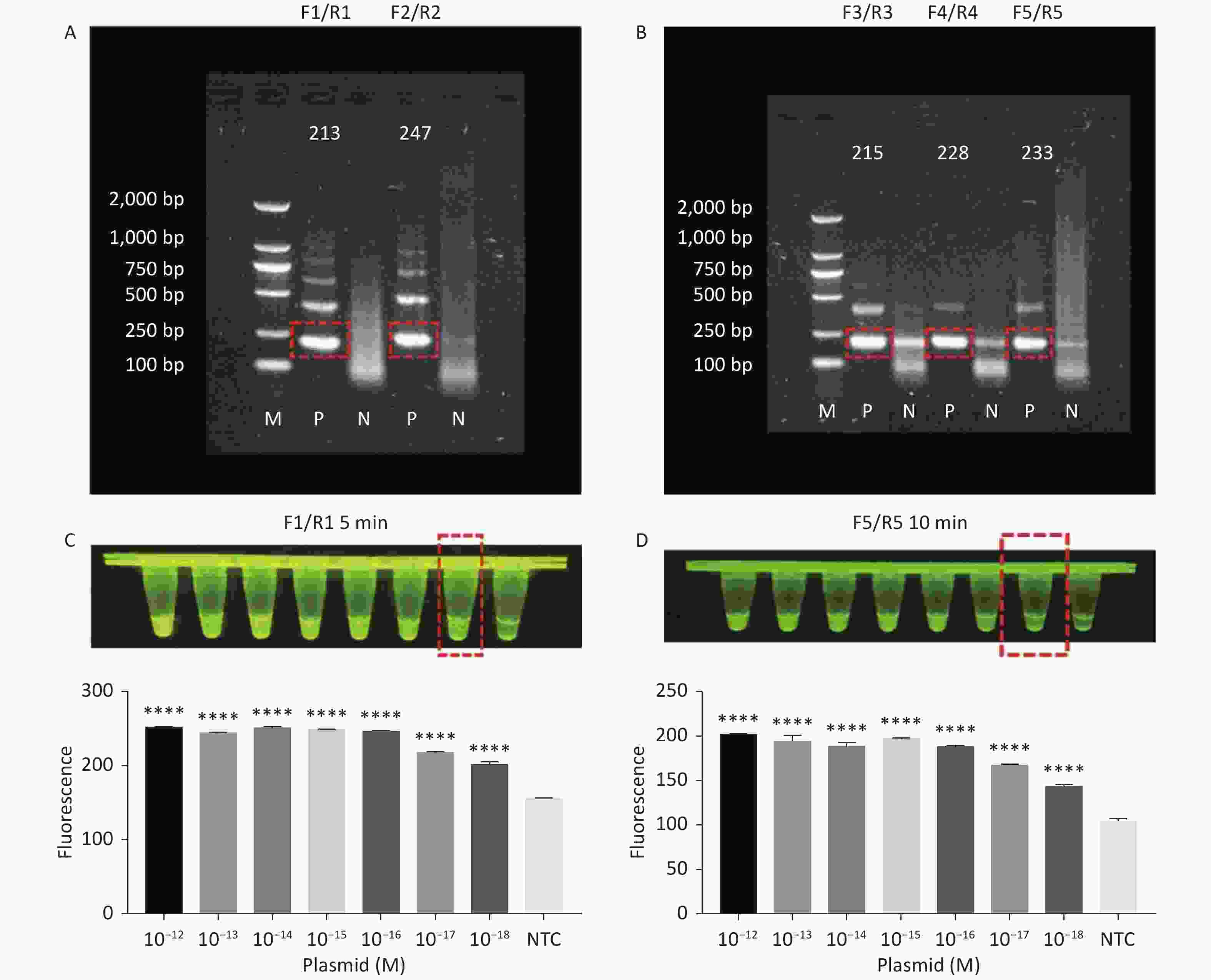
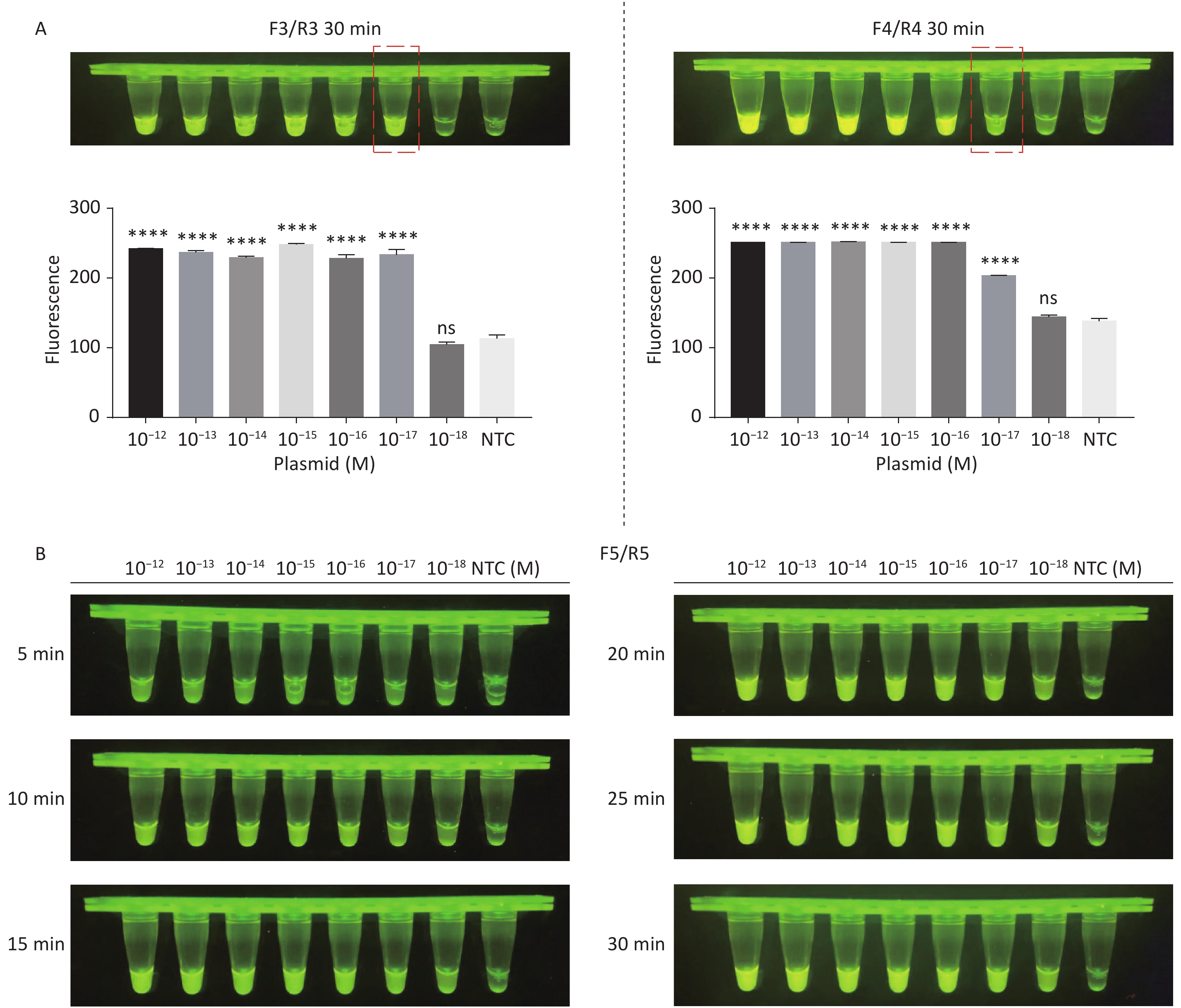
Figure 3. (A) Agarose gel electrophoresis of the RPA products of the primer pairs F1/R1 and F2/R2 for the tdh gene. (B) Agarose gel electrophoresis of the RPA products of the primer pairs F3/R3, F4/R4, and F5/R5 for the trh gene. The target plasmid and sterile water were used as templates. M: DL2000 Marker; P: Positive; N: Negetive. (C) The sensitivity of the CRISPR/Cas12a-VD method to detect tdh gene from 10−12 M to 10−18 M. The amplified products of F1/R1 were added to the Cas12a-mediated cleavage system of incubation for 5 min. (D) The sensitivity of the CRISPR/Cas12a-VD method to detect trh gene from 10−12 M to 10−18 M. The amplified products of F5/R5 were added to the Cas12a-mediated cleavage system of incubation for 10 min. The green fluorescent signals were quantified with ImageJ. n = 3, two-tailed Student’s t test; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 and ns not significant; NTC nontarget control; bars represent mean ± SD.
-
To facilitate direct interpretation of the test results with the unaided eye, we increased the concentration of ssDNA reporter. When the trans-cleavage activity of Cas12a is activated, many ssDNA cleavages occur in the system, and the fluorescence groups are far away from the quenching groups[29], hence we observed strong green fluorescence signals with the unaided eye under blue light. To enable CRISPR/Cas12a-VD to quickly and accurately detect VP, we used the above method to further screen the five pairs of primers that had been screened, and optimised the detection time.
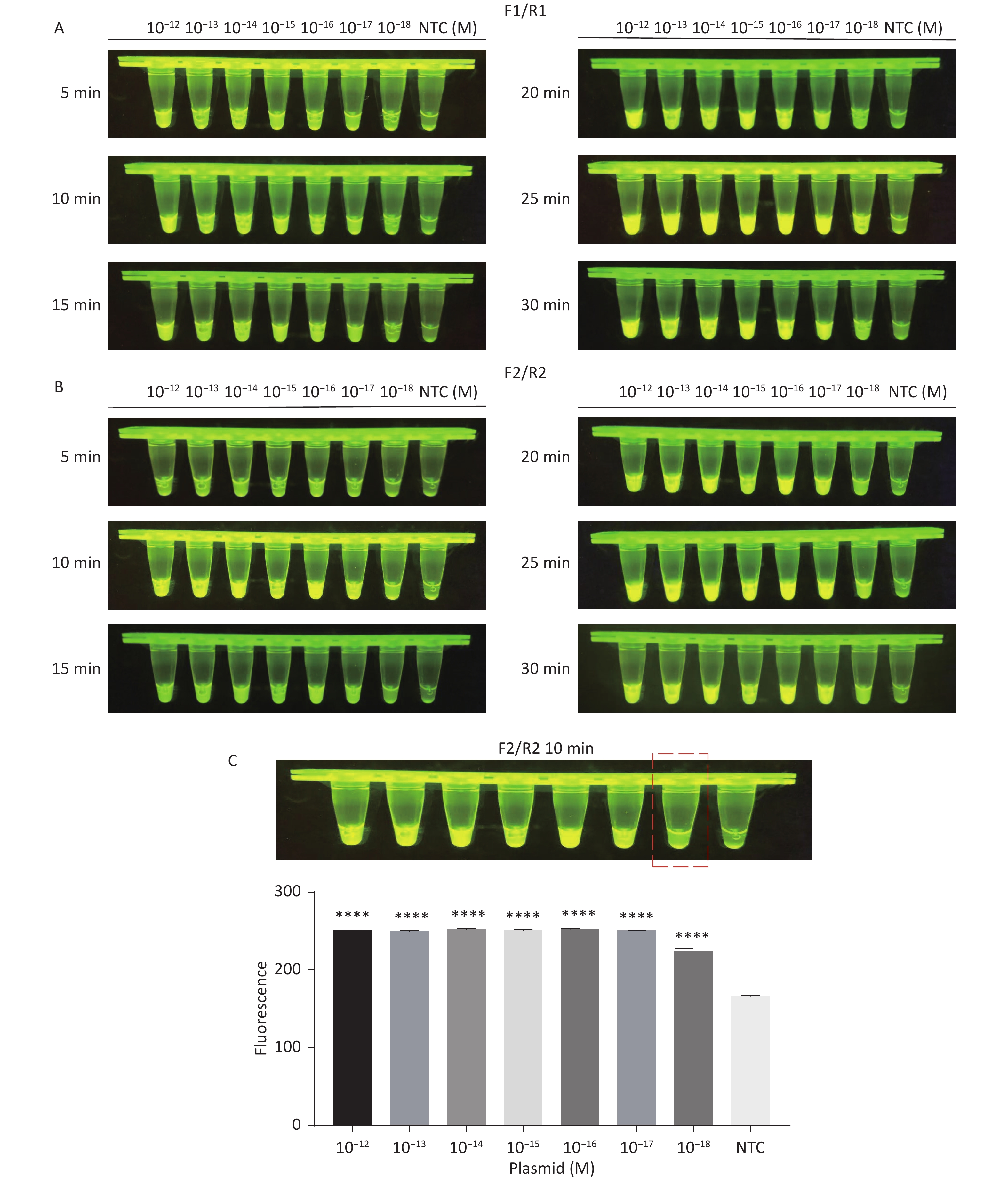
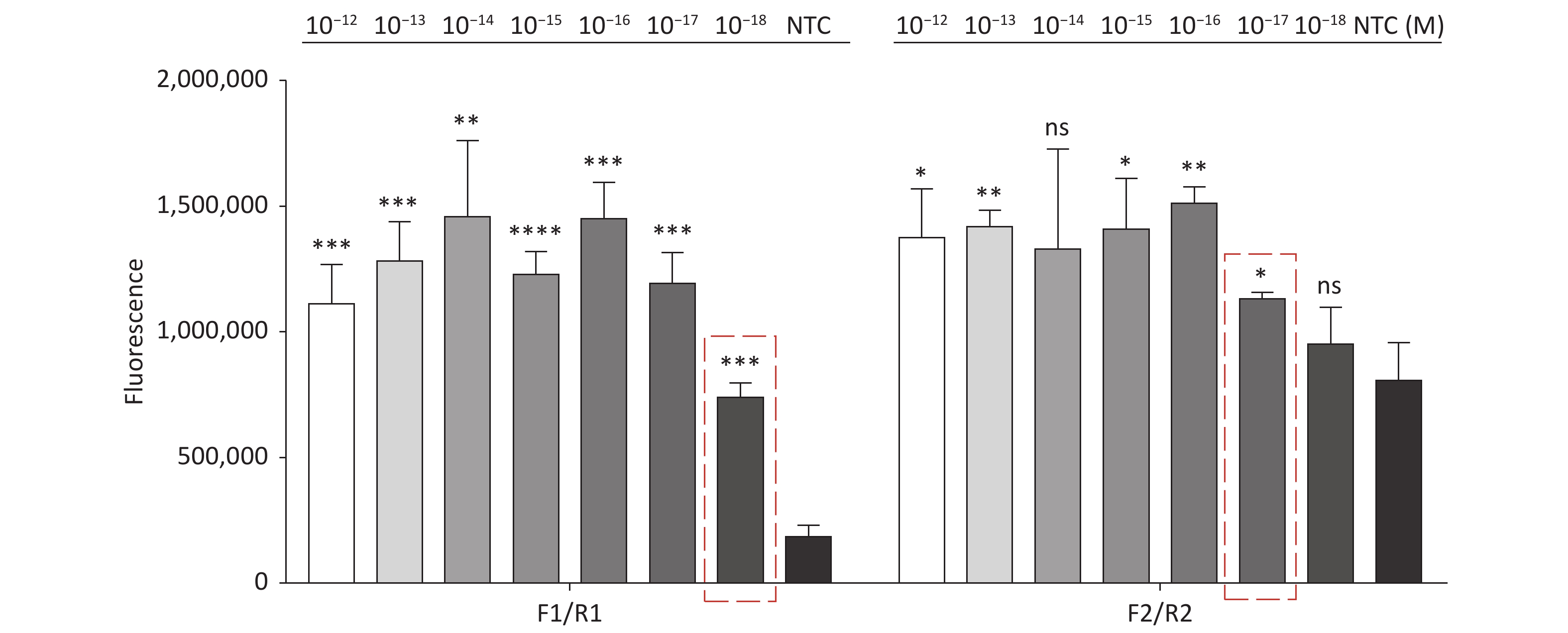
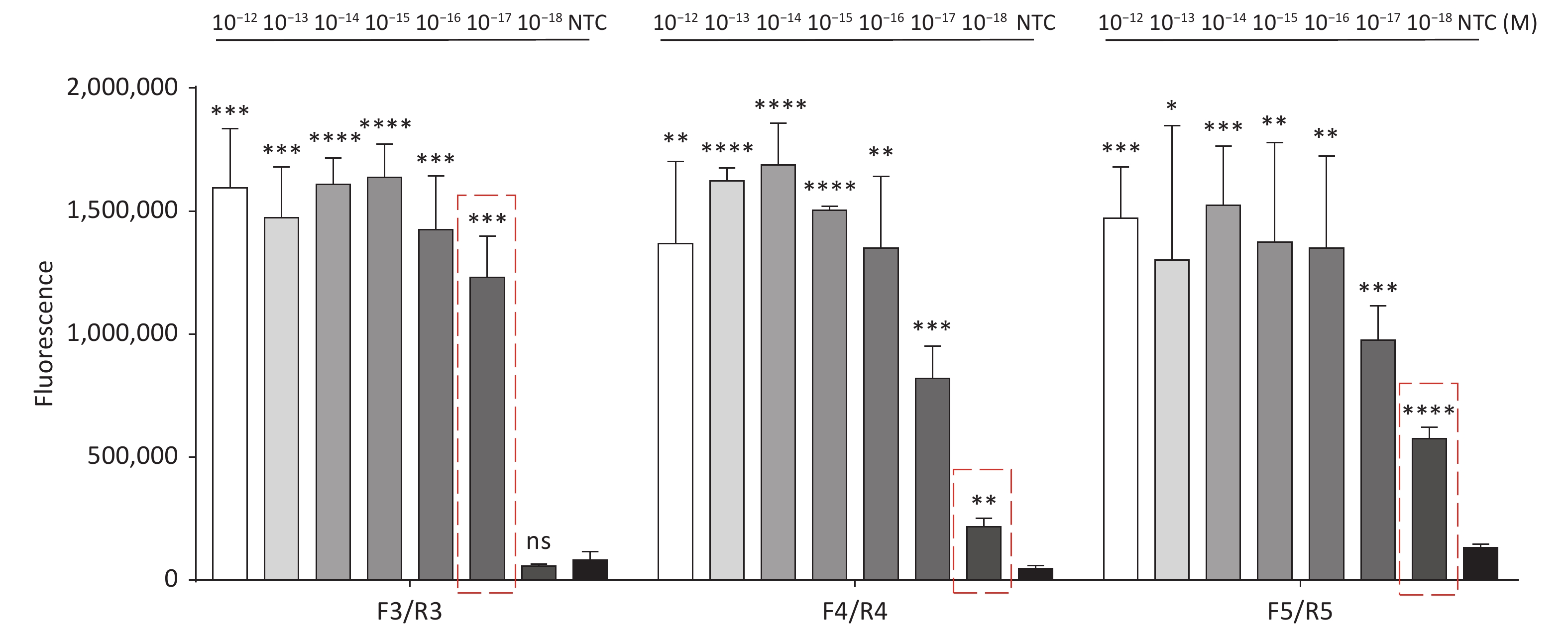
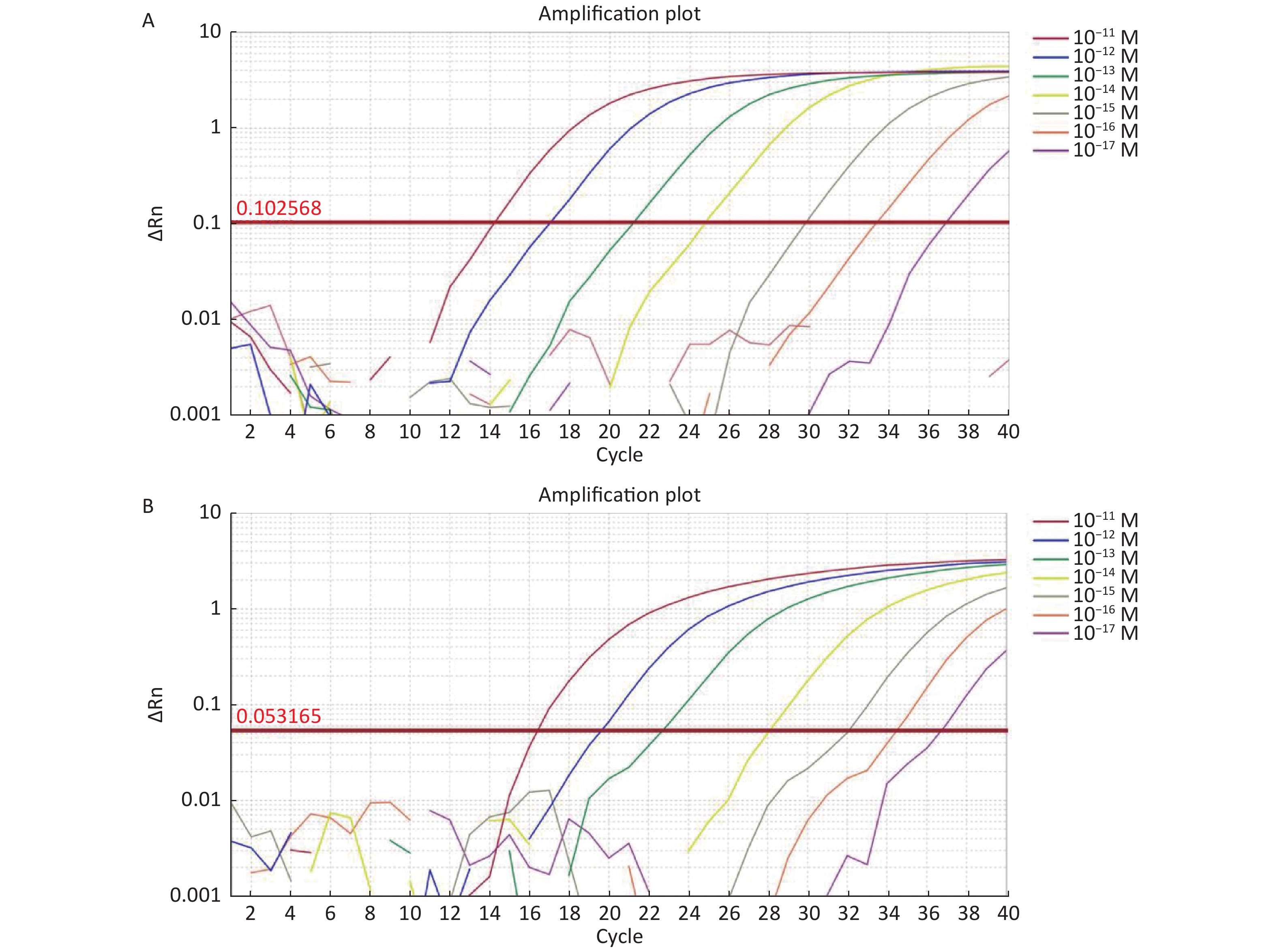
For the tdh gene, plasmid template could be detected at concentrations as low as 10−18 M using primer pairs (F1/R1 and F2/R2; Supplementary Figure S2A, S2B, available in www.besjournal.com). After the amplification products from the primer pair F1/R1 were added to the system, a green fluorescence signal different from the blank control was observed after 5 min (Figure 3C), while primer pair F2/R2 required 10 min for the signal to appear (Supplementary Figure S2C). For the trh gene, a plasmid template concentration of 10−17 M was successfully detected by primer pairs F3/R3 and F4/R4 (Supplementary Figure S3A, available in www.besjournal.com), while 10−18 M plasmid template could be detected by F5/R5 after 10 min (Figure 3D and Supplementary Figure S3B). In addition, we verified the above results by fluorescence detection using a real-time PCR machine. For both target genes, a higher amplification efficiency was achieved for primer pairs F1/R1 and F5/R5 (Supplementary Figures S4 and S5, available in www.besjournal.com), and both primer pairs achieved complete detection of the target nucleic acid within 30 min. There was no significant difference in fluorescence intensity between different concentrations of positive tubes because once the crRNA and Cas12a complex captures the target plasmid, trans-cleavage activity was activated and the surrounding ssDNA reporter was quickly cleaved, resulting in a sharp non-linear increase in fluorescence intensity. In contrast, TaqMan real-time PCR for both genes were performed on the ViiA 7 system with detection limits of 10−17 M (Supplementary Figure S6, available in www.besjournal.com).
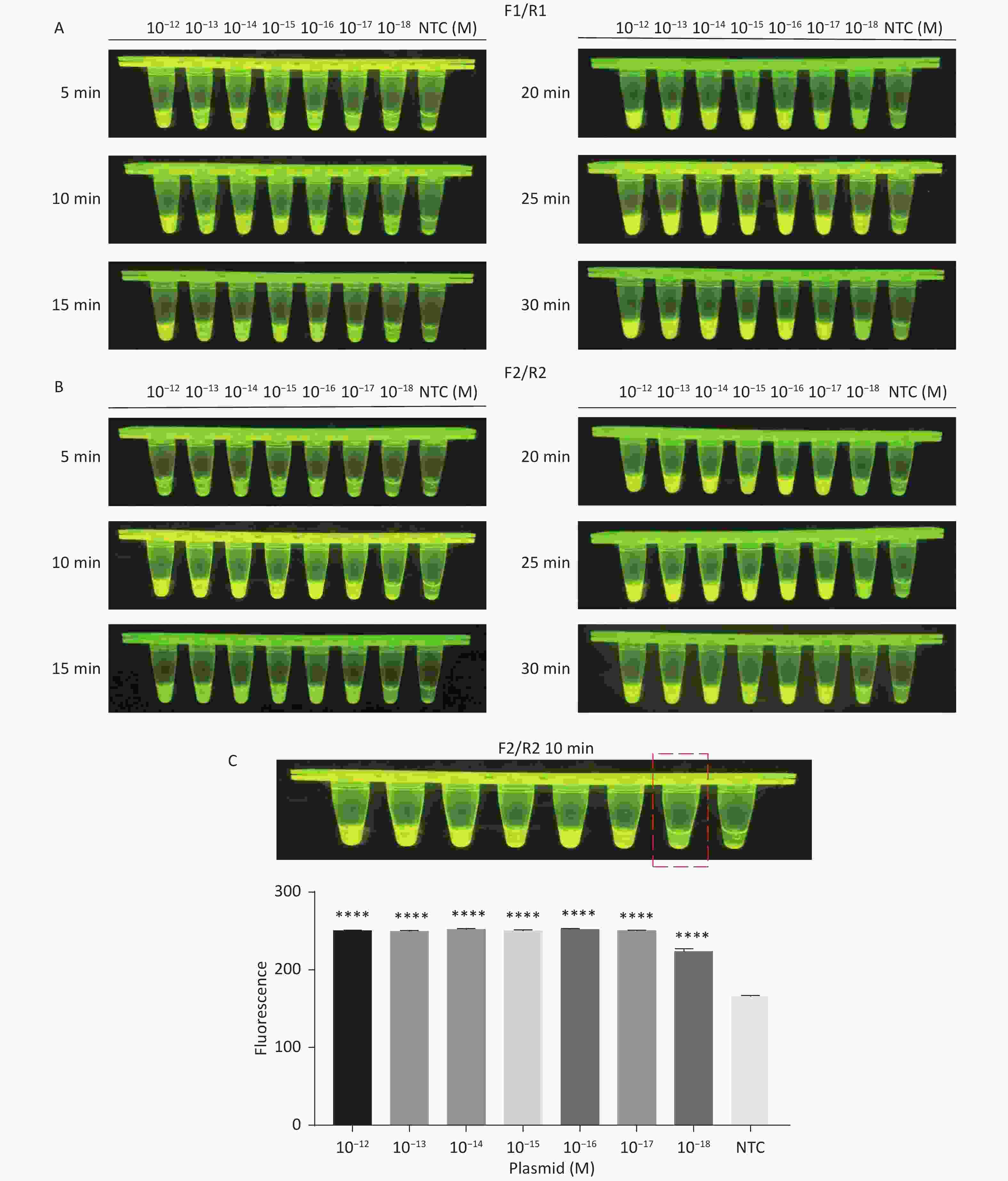
Figure S2. Sensitivity of the CRISPR/Cas12a-VD method for detecting the tdh gene from 10−12 M to 10−18 M. (A) Amplified products obtained using F1/R1 were added to the Cas12a-mediated cleavage system and incubated for 5−30 min. (B) Amplified products obtained using F2/R2 were added to the Cas12a-mediated cleavage system and incubated for 5−30 min. (C) Amplified products obtained using F2/R2 were added to the Cas12a-mediated cleavage system and incubated for 10 min. The green fluorescent signal was quantified by ImageJ. n = 3, two-tailed Student’s t test; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 and ns not significant; NTC nontarget control; bars represent mean ± SD.
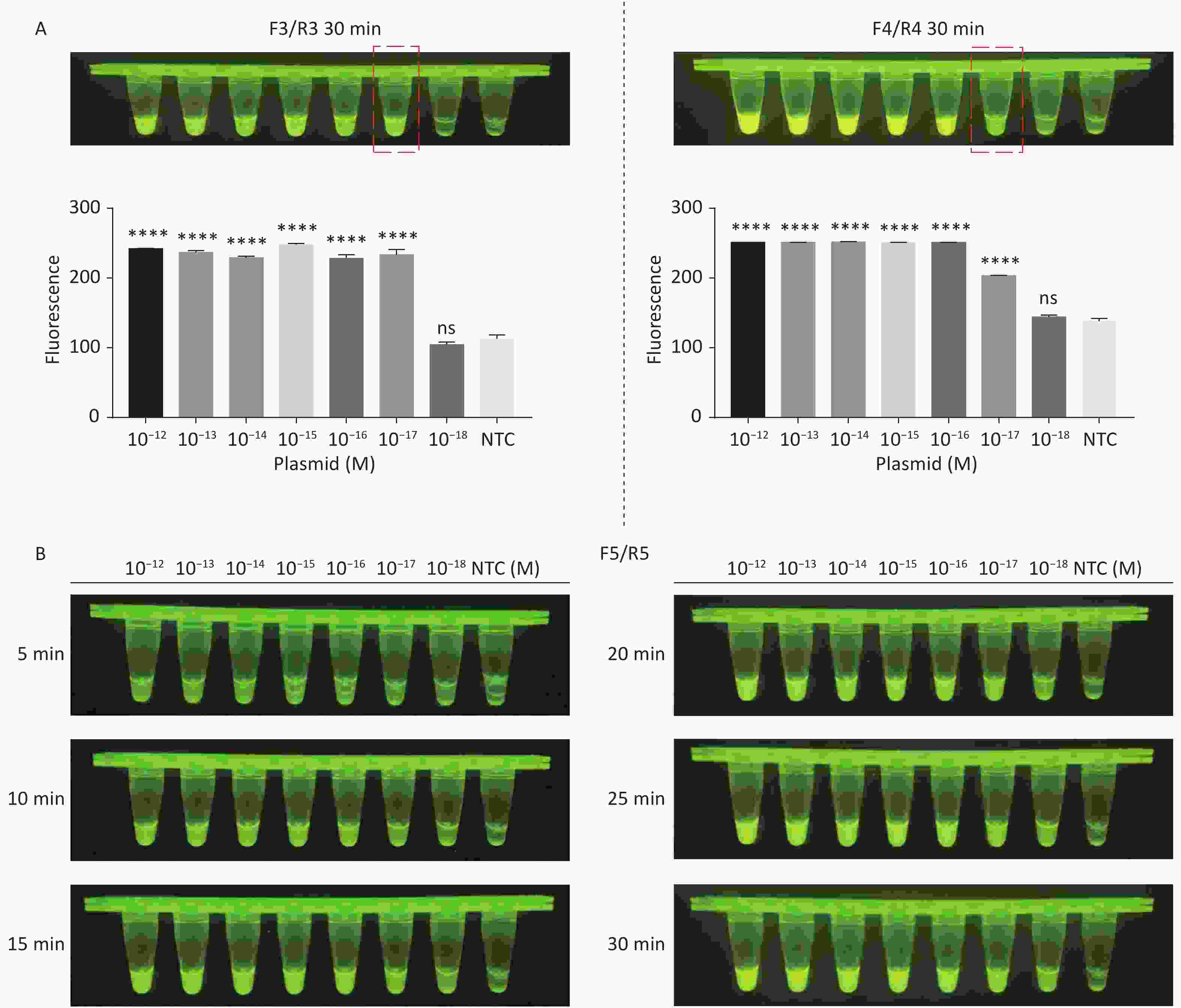
Figure S3. Sensitivity of the CRISPR/Cas12a-VD method for detecting the trh gene from 10−12 M to 10−18 M. (A) Amplified products obtained using F3/R3 and F4/R4 were added to the Cas12a-mediated cleavage system and incubated for 30 min. The green fluorescent signal was quantified with ImageJ. (B) Amplified products obtained using F5/R5 were added to the Cas12a-mediated cleavage system and incubated for 5−30 min. n = 3, two-tailed Student’s t test; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 and ns not significant; NTC nontarget control; bars represent mean ± SD.
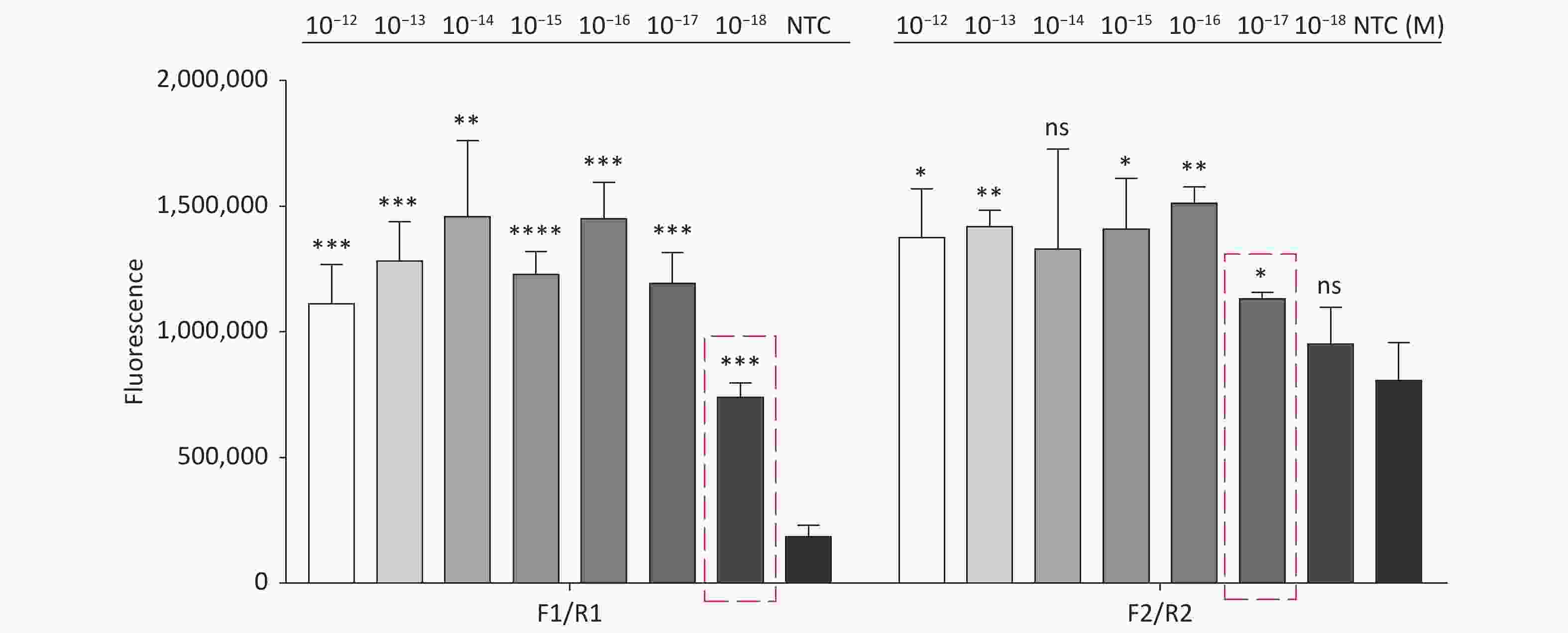
Figure S4. Sensitivity of the CRISPR/Cas12a coupled with fluorescence readout for detecting the tdh gene from 10−12 M to 10−18 M using primer pairs F1/R1 and F2/R2. n = 3, two-tailed Student’s t test; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 and ns not significant; NTC nontarget control; bars represent mean ± SD.
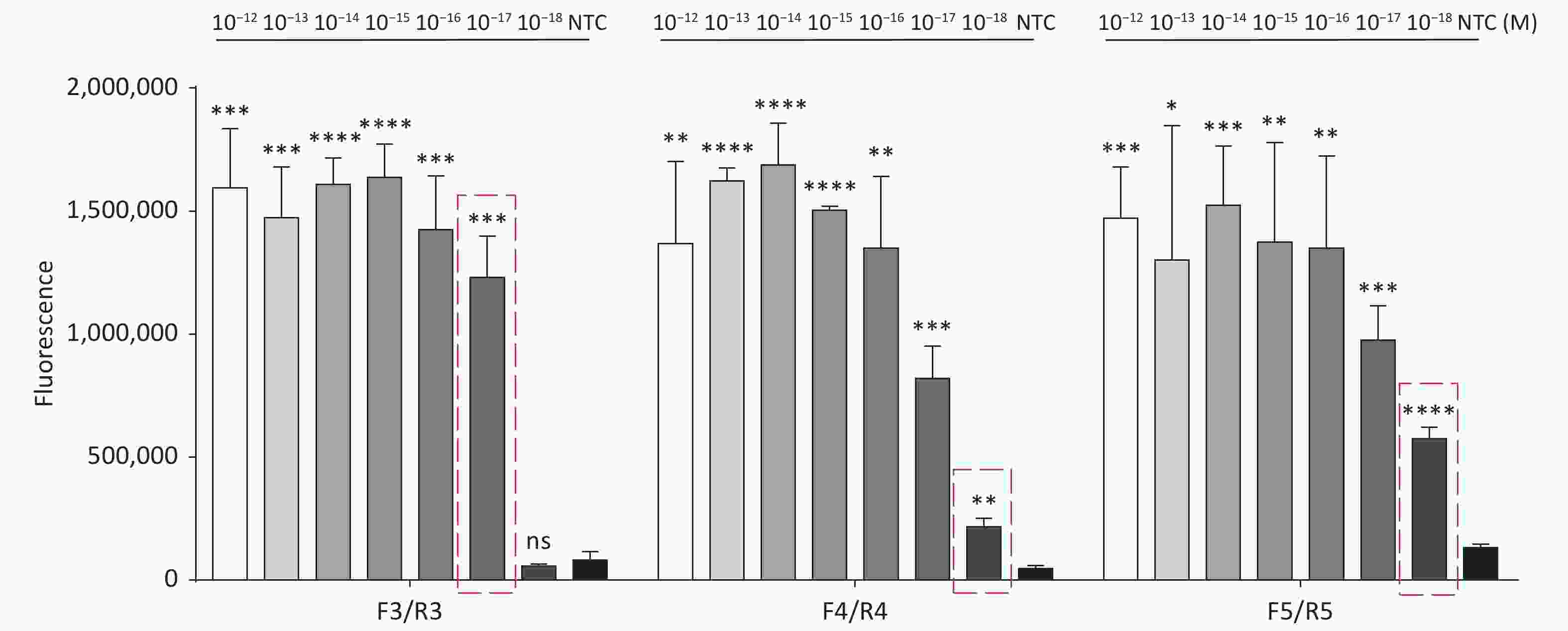
Figure S5. Sensitivity of the CRISPR/Cas12a coupled with fluorescence readout for detecting the trh gene from 10−12 M to 10−18 M using primer pairs F3/R3, F4/R4, and F5/R5. n = 3, two-tailed Student’s t test; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 and ns not significant; NTC nontarget control; bars represent mean ± SD.
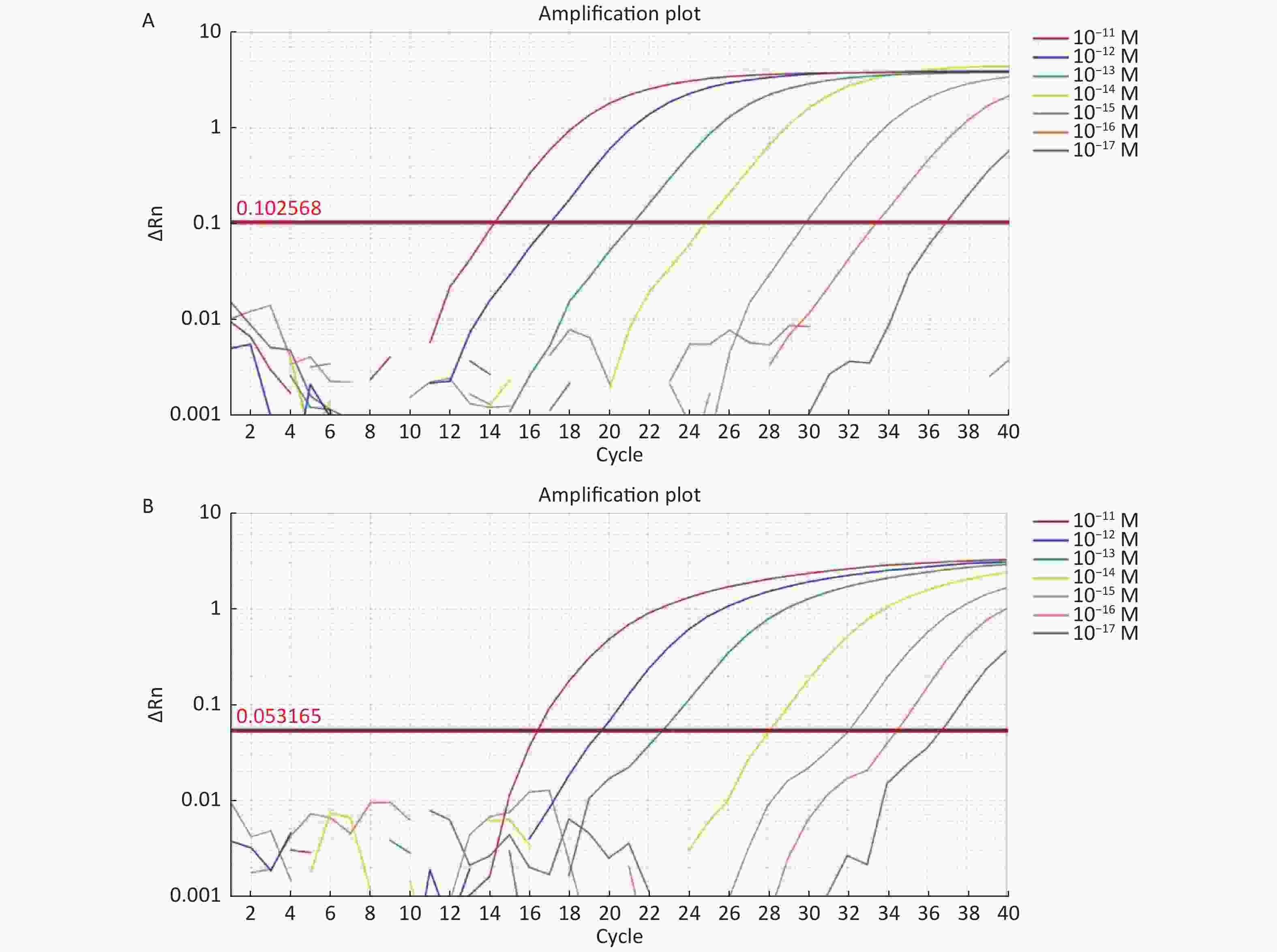
Figure S6. (A) The sensitivity of Real-time PCR to detect tdh gene from 10−11 M to 10−17 M. (B) The sensitivity of Real-time PCR to detect trh gene from 10−11 M to 10−17 M.
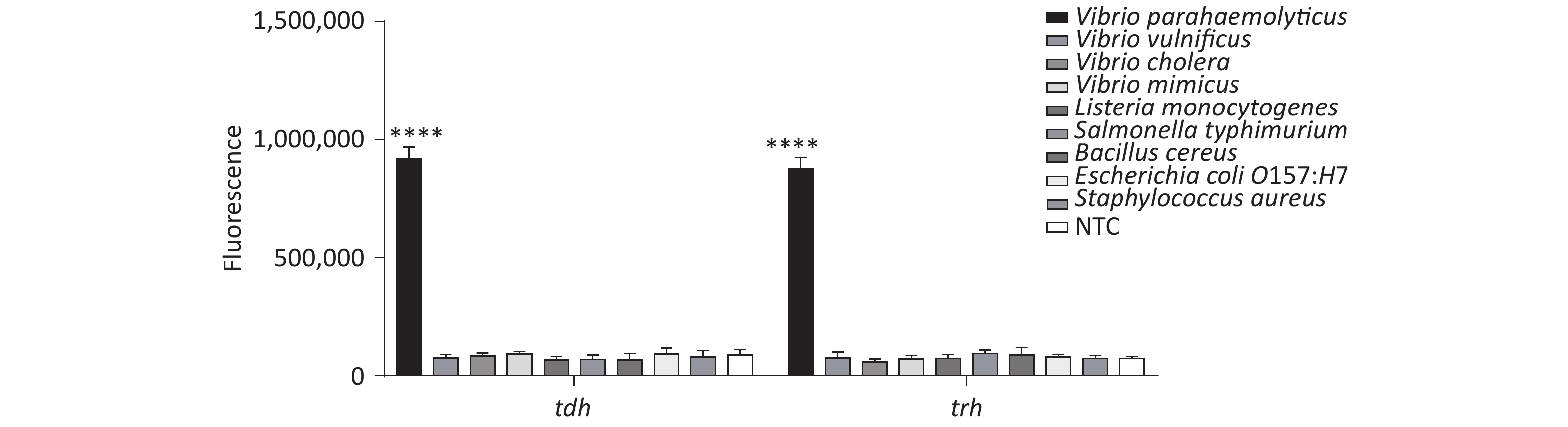
In addition, we tested the specificity of the method, and the results revealed good specificity and no cross-reactivity with other foodborne pathogens (Supplementary Figure S7, available in www.besjournal.com).
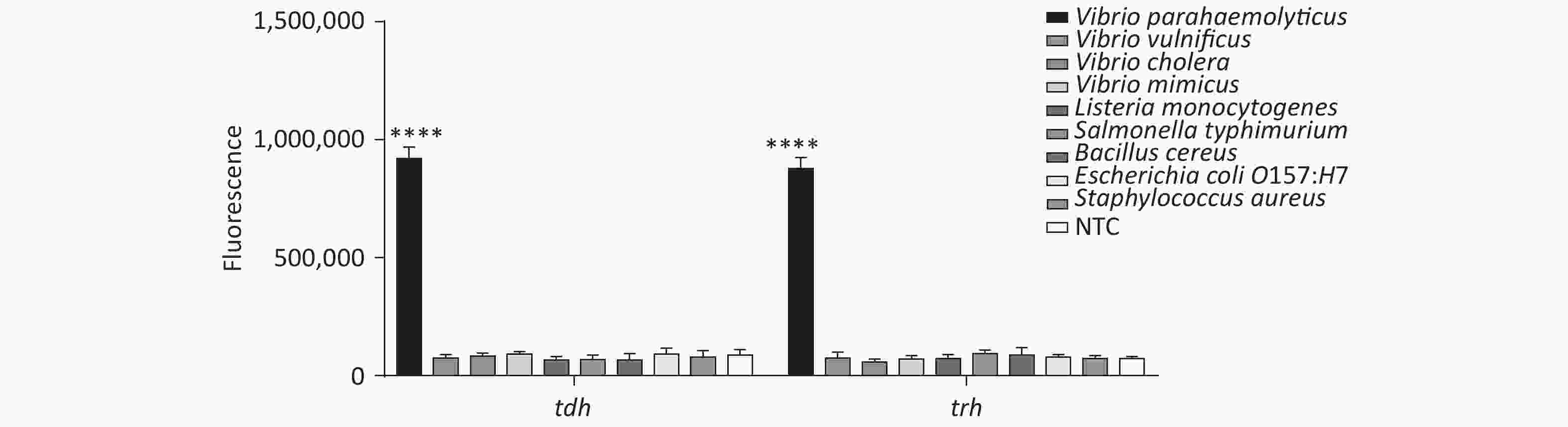
Figure S7. Specificity of the CRISPR/Cas12a coupled with fluorescence readout for detecting tdh and trh genes. n = 3, two-tailed Student’s t test; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 and ns not significant; NTC, nontarget control; bars represent mean ± SD.
-
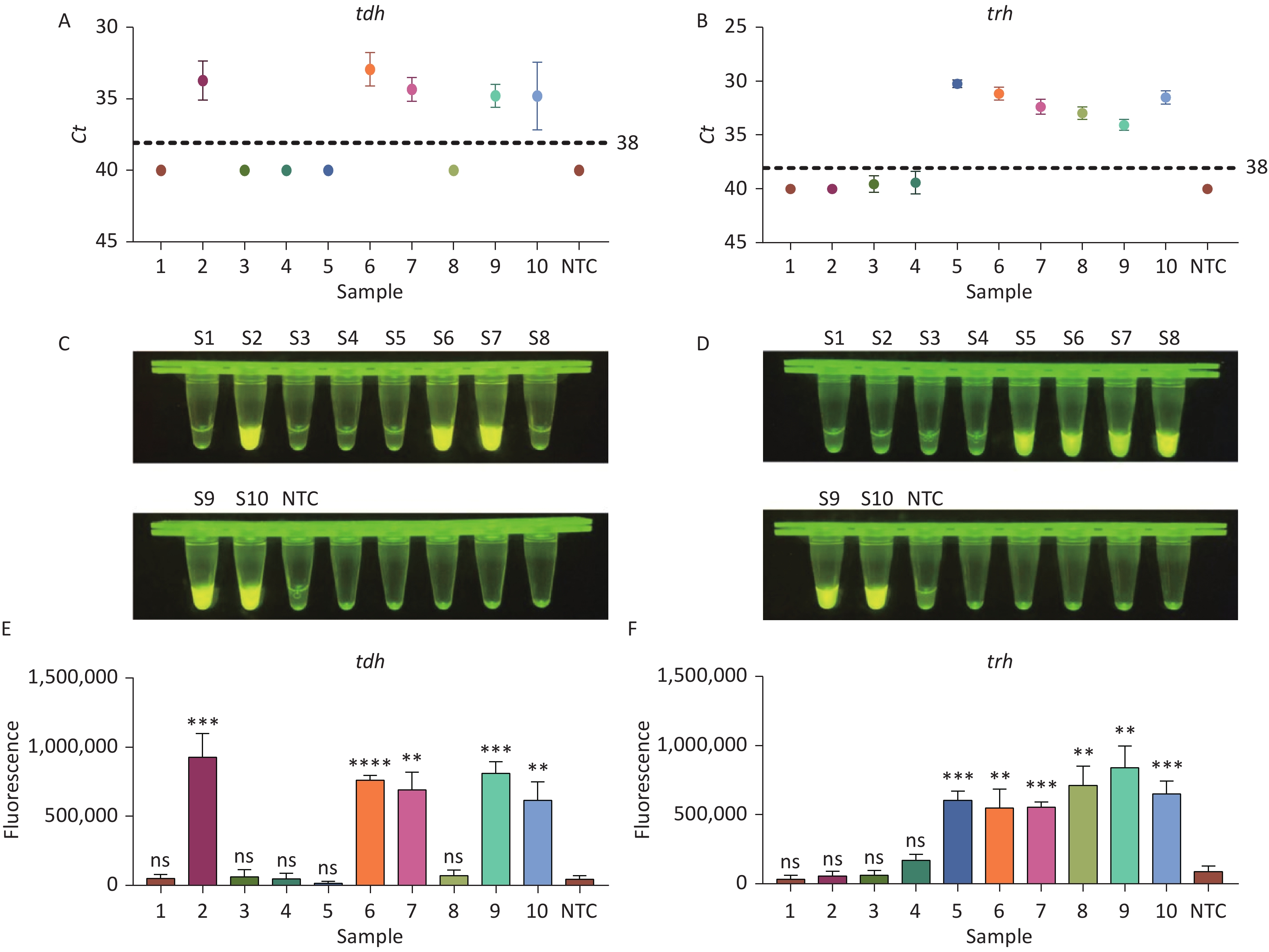
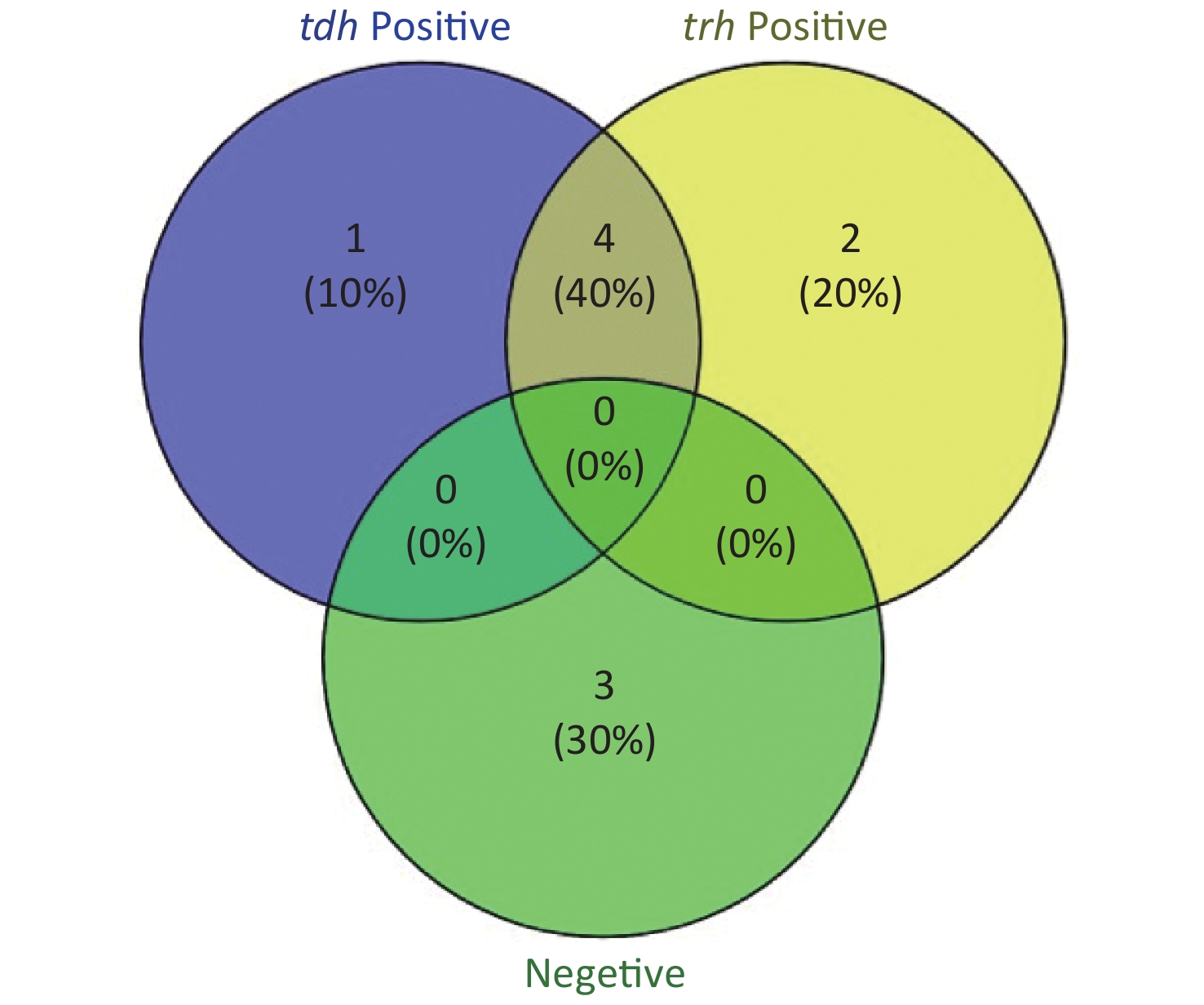
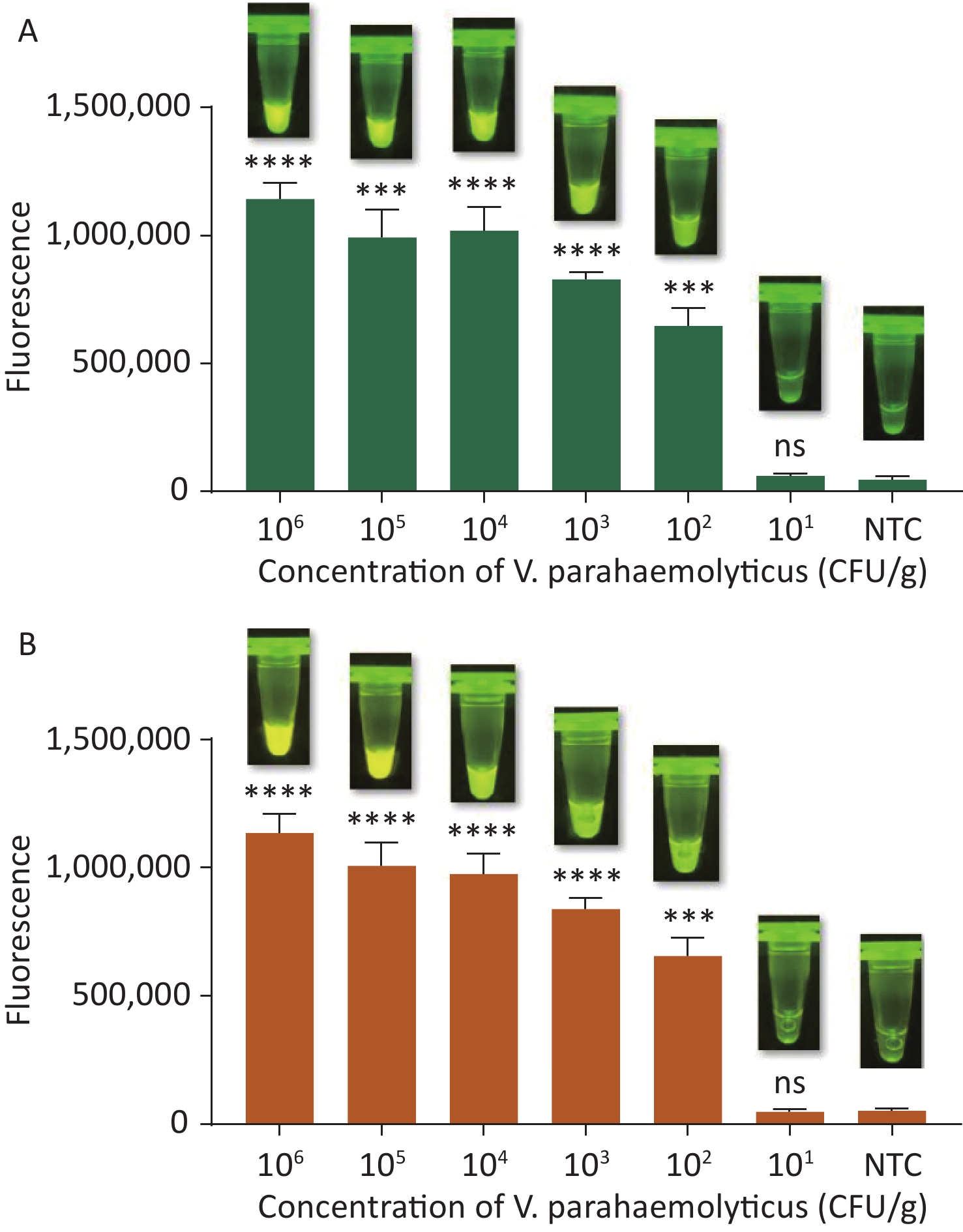
To evaluate whether CRISPR/Cas12a-VD could be applied to detection in pure culture, we extracted DNA samples from 10 suspected VP-positive strains stored in our laboratory, and detected tdh and trh genes by real-time PCR and CRISPR/Cas12a-VD. The results of CRISPR/Cas12a-VD showed that among the 10 bacterial samples, seven were positive and three were negative, of which five were positive for tdh and six were positive for trh (Figures 4C, 4D, and 5). The results of real-time PCR and CRISPR/Cas12a fluorescence detection were consistent with those of CRISPR/Cas12a-VD (Figure 4A, 4B, 4E, 4F and Table 2). Next, we selected sample No.6, which was positive for both genes, prepared a series of gradient concentrations of VP suspensions, and used CRISPR/Cas12a-VD to detect the tdh and trh genes, respectively. The results showed that suspensions with a concentration as low as 102 CFU/g could be detected for both genes (Figure 6). This finding indicated that CRISPR/Cas12a-VD also detected low concentrations of VP in pure culture.
Methods CRISPR/Cas12a-VD Comparison of two methods real-time PCR Positive Negative Total Sensitivity (%) Specificity (%) Kappa Positive 7 0 7 Negative 0 3 3 100 100 1.00 Total 7 3 10 Table 2. The Performance comparison between CRISPR/Cas12a-VD and real-time PCR for detection in pure culture
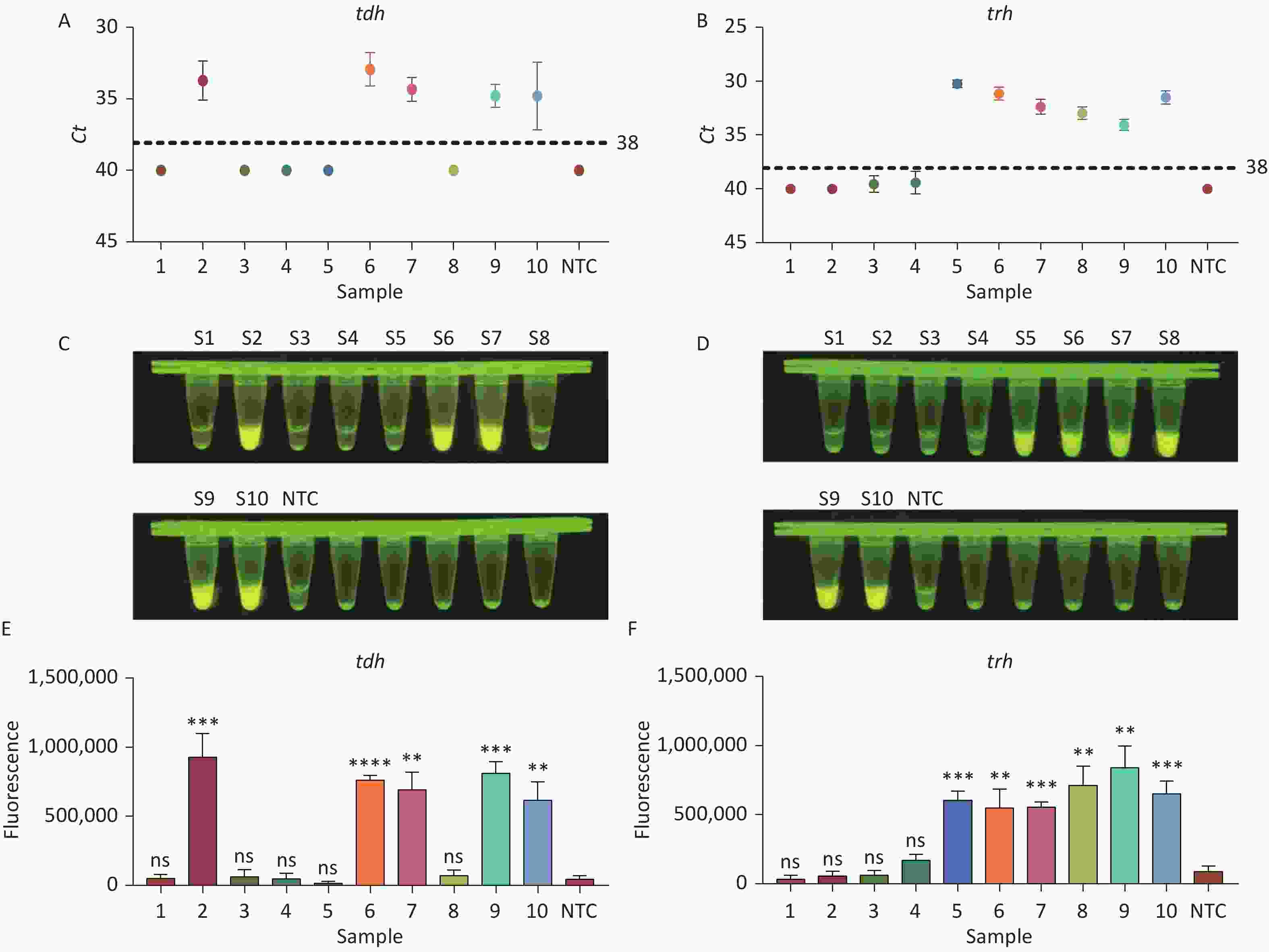
Figure 4. (A) Detection of tdh gene of V. parahaemolyticus in pure culture with Real-time PCR. (B) Detection of trh gene of V. parahaemolyticus in pure culture with Real-time PCR. When Ct value below 38, the sample is positive. (C) Detection of tdh gene of V. parahaemolyticus in pure culture with the CRISPR/Cas12a-VD. (D) Detection of trh gene of V. parahaemolyticus in pure culture with the CRISPR/Cas12a-VD. (E) Detection of tdh gene of V. parahaemolyticus in pure culture using CRISPR/Cas12a coupled with fluorescence readout. (F) Detection of trh gene of V. parahaemolyticus in pure culture using CRISPR/Cas12a coupled with fluorescence readout. n = 3, two-tailed Student’s t test; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 and ns not significant; NTC nontarget control; bars represent mean ± SD.
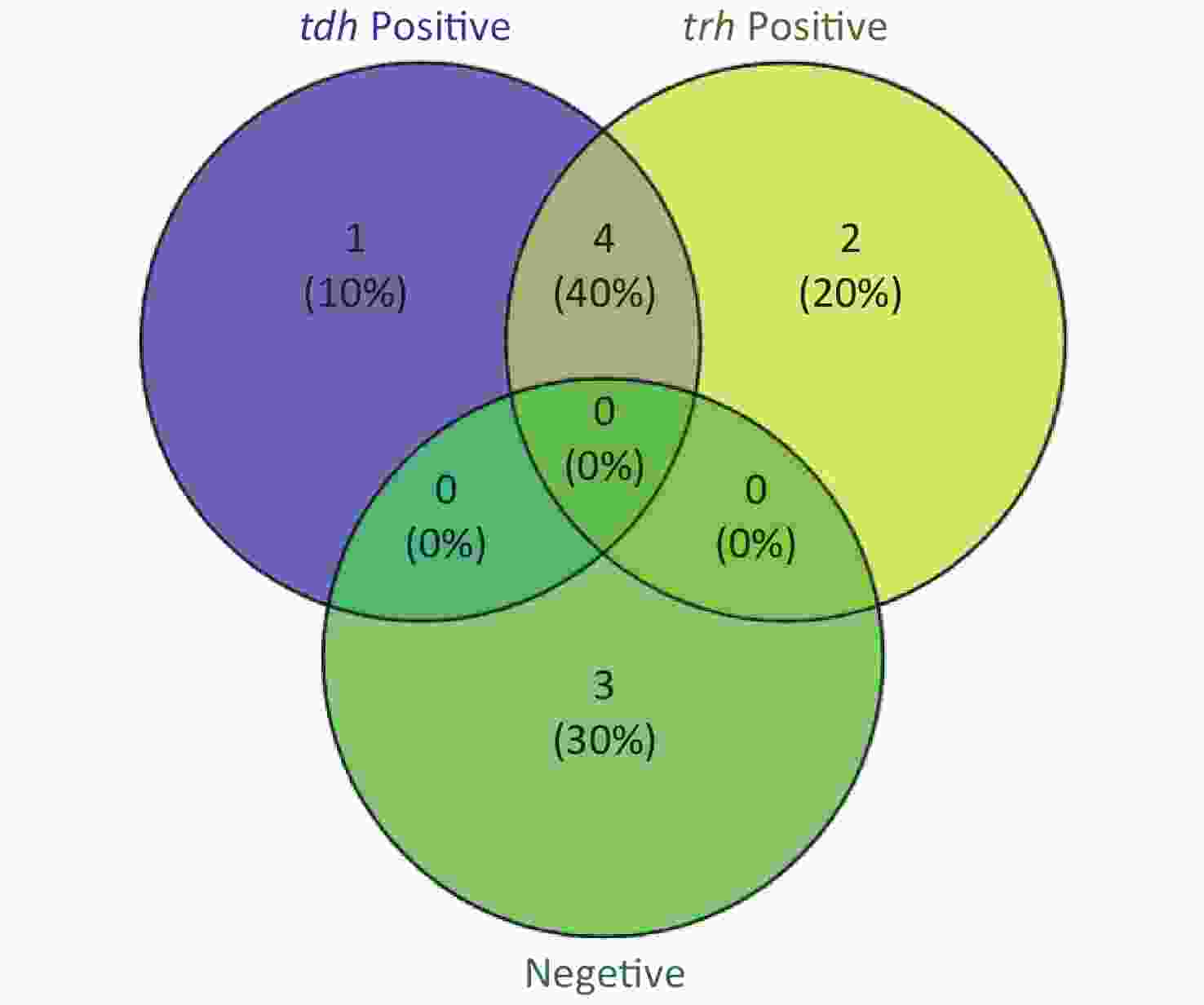
Figure 5. The Venn diagram shows the detection results of tdh and trh genes in pure culture by CRISPR/Cas12a-VD and Real-time PCR.
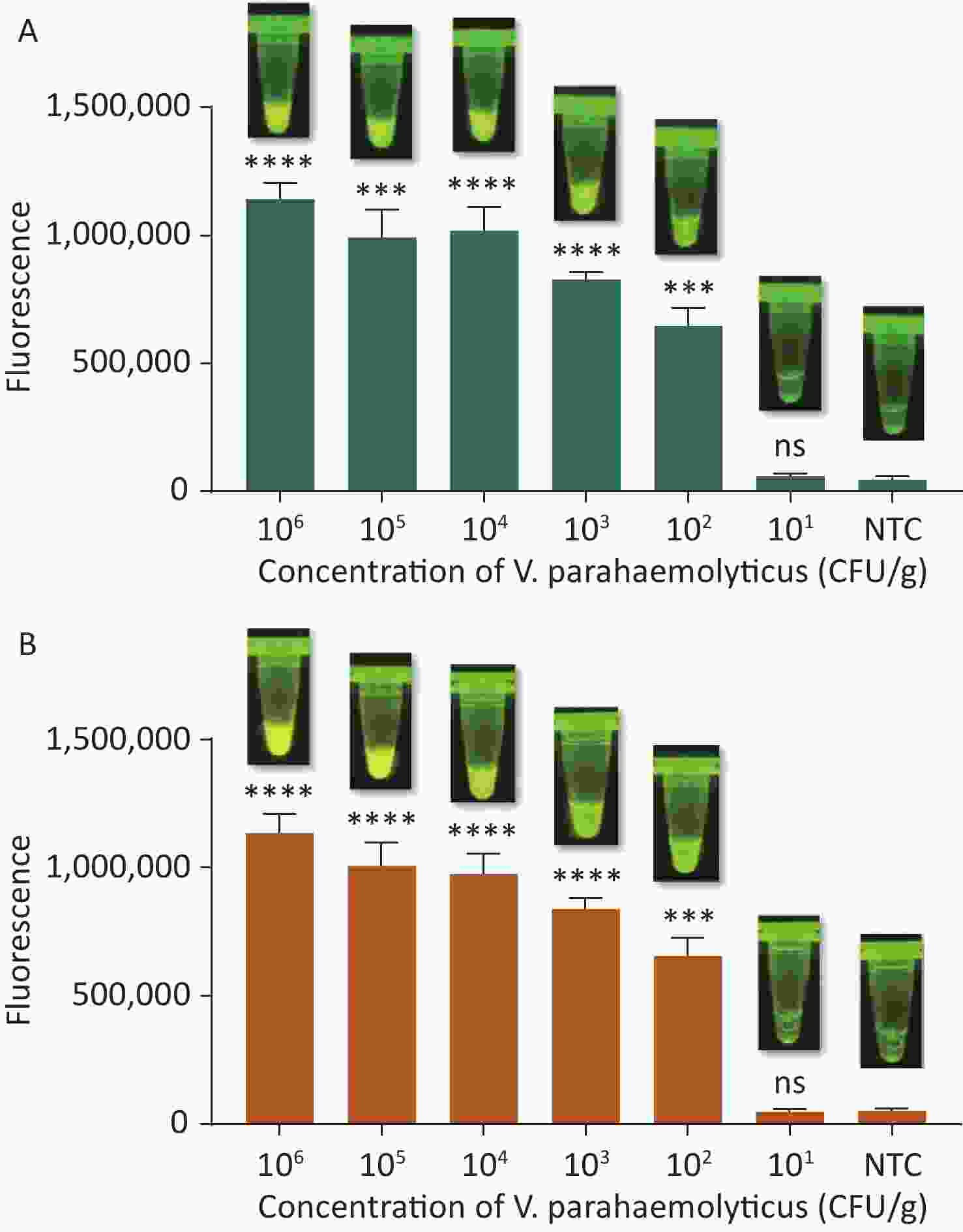
Figure 6. (A) The sensitivity of the CRISPR/Cas12a-VD method to detect tdh gene in pure culture from 106 CFU/g to 101 CFU/g. (B) The sensitivity of the CRISPR/Cas12a-VD method to detect trh gene in pure culture from 106 CFU/g to 101 CFU/g. Meanwhile, the CRISPR/Cas12a coupled with fluorescence readout was used as a contrast. n = 3, two-tailed Student’s t test; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 and ns not significant; NTC nontarget control; bars represent mean ± SD.
-
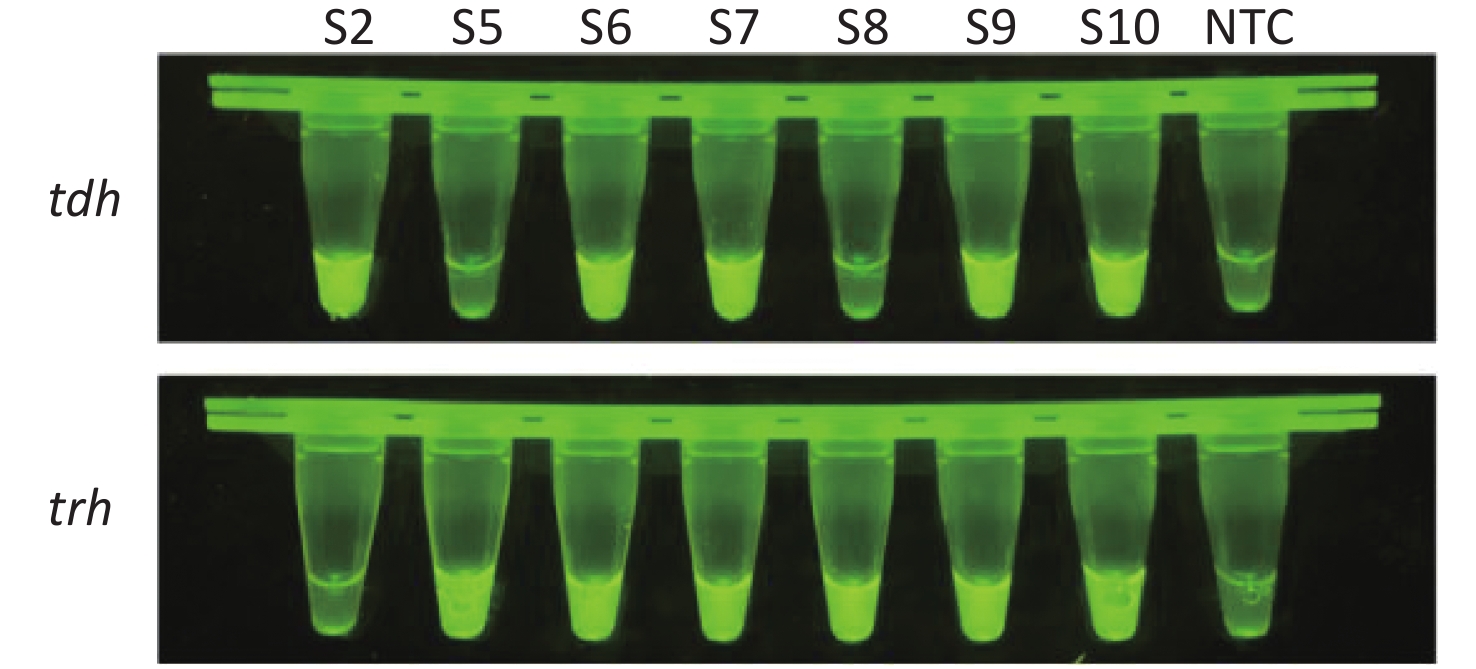
We used CRISPR/Cas12a-VD to detect spiked food samples to evaluate the practicability of CRISPR/Cas12a-VD. According to the detection results of the pure cultures mentioned above, we prepared suspensions of VP with a cell density of 102 CFU/g for the seven positive samples to contaminate the sterile shrimps. After extracting DNA, we used the CRISPR/Cas12a-VD method to detect tdh and trh genes. CRISPR/Cas12a-VD accurately detected all positive-labelled samples, and there was no difference between these results and the detection results in pure culture (Figure 7). Therefore, CRISPR/Cas12a-VD detected VD in food samples in a practical manner.
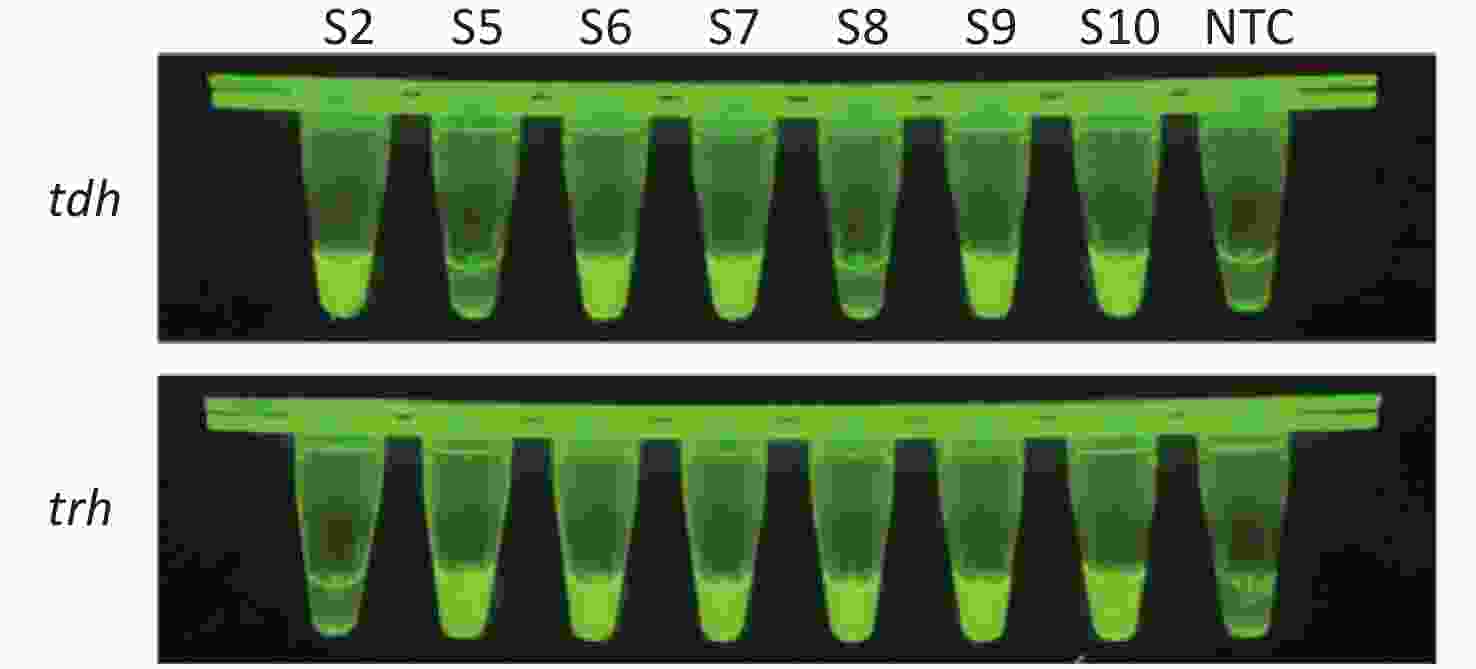
Figure 7. Detection of tdh and trh genes of V. parahaemolyticus in spiked shrimp samples with the CRISPR/Cas12a-VD.
-
VP is widespread in seawater, seafloor sediments, and seafood due to its halophilic nature, hence VP is distributed widely and spreads rapidly. Cases of food poisoning and aquaculture seafood deaths caused by VP have been reported worldwide, thus VP poses a serious threat to human life and property. Therefore, the demand for rapid detection of pathogens is becoming more and more urgent. In recent years, a variety of isothermal nucleic acid amplification techniques have been developed for rapid detection of pathogens in the field. Indeed, isothermal nucleic amplification techniques are simple to operate and do not require complex equipment, thus are gradually replacing traditional PCR amplification.
In 2010, Chen et al.[34] used loop-mediated isothermal amplification (LAMP) for rapid detection of VP in seafood samples, while Wang et al.[35] established a multiple endonuclease restriction real-time loop-mediated isothermal amplification (Mert-LAMP) technique that detects multiple different targets in real time, which overcomes the limitations of traditional LAMP techniques. Due to the complexity of primer design and the need to react at 62 °C for 60 min, Mert-LAMP is not ideal for rapid detection in the face of large-scale outbreaks.
Developed by TwistDx in 2006, RPA mimics the nucleic acid replication machinery in T4 phage[14,15]. The amplification process relies on three core enzymes [recombinase, single-stranded DNA-binding protein (SSB), and strand-displacement polymerase]. Recombinase pairs primer with a homologous sequence in the target DNA. The SSB binds to the DNA strand replaced by the primer and stabilizes the D-loop that has been formed to prevent dissociation of the primer. Finally, strand-displacement polymerase adds bases to the 3' end of the primer and primer extension occurs. RPA achieves rapid amplification of trace nucleic acids under a constant temperature of 25–42 ℃, and the reaction time is only 5–20 min. Compared with LAMP, the primer design of RPA is simple and the reaction can be triggered at room temperature, thus RPA is more suitable for on-site detection of pathogens. In recent years, RPA has become one of the most convenient nucleic acid detection methods for point-of-care diagnosis.
Geng et al.[36] established a real-time RPA technique for the gyrB gene of VP, which can accurately detect genomic DNA at concentrations as low as 102/reaction in 20 min. This method achieves relatively rapid detection, but lacks sensitivity and requires a fluorescence detector that may increase the burden for operators in the field. Yang et al.[37] established a set of new detection methods to overcome these shortcomings. By modifying primers and probes, the amplification results from RPA can be displayed using flow test strips, which provides a new platform for detection of pathogens without specialist equipment[37]. Zhang et al.[31] detected the tlh gene of VP by combining CRISPR/Cas12a with PCR. Accurate temperature control is essential for PCR. Herein, we optimised this method by replacing PCR with RPA. While maintaining high sensitivity, this not only simplifies the experimental equipment needed, but also greatly shortens the amplification time.
The CRISPR/Cas system is an acquired immune system of prokaryotes present in almost all archaea and ~40% of bacteria[38,39]. The CRISPR/Cas system resists the invasion of foreign nucleic acids, such as bacteriophages and plasmids[38,39]. Cas12a (Cpf1), a member of the class 2 CRISPR/Cas system, was first reported in 2013[40]. This novel effector protein was identified by bioinformatics analysis, and Zhang et al.[41] performed the first preliminary characterisation of the properties of Cas12a in 2015. This protein dimerises through its RuvC-like nuclease domain, which is similar to Cas9[42-44]. Cas12a interferes with exogenous DNA by identifying T-rich PAM sequences, cleaves DNA with guidance from a single crRNA, and produces a 5’ prominent sticky end far from the PAM site[42-44]. In 2018, Chen et al.[30] reported that Cas12a can target and bind dsDNA under mediation of crRNA, thereby activating specific dsDNA cleavage and non-specific ssDNA trans-cleavage activities. In nucleic acid detection, combining RPA and CRISPR/Cas12a effectively doubles the amplification signal, thus increasing detection sensitivity. In addition, due to the high specificity of CRISPR/Cas12a for target recognition, binding downstream of RPA also evaluates amplification products, thus eliminating the possibility of false-positive results.
Using this knowledge, we herein developed a new detection method for VP (CRISPR/Cas12a-VD) by combining CRISPR/Cas12a with isothermal amplification, and achieved visual detection with the unaided eye under blue light by incorporating a high concentration of ssDNA reporter in the cleavage system. This method accurately detects target DNA at concentrations as low as 10−18 M (single molecule detection) without cross-reactivity with other types of bacteria. By screening RPA primers and optimisation of the reaction time, we shortened the detection time to 30 min and < 1 h if sample pre-treatment and nucleic acid extraction steps were included. Nucleic acid release may be accelerated by heat treatment or using pyrolysis buffer, which would further shorten the detection time[14,15]. In detection of pure cultures, the detection results of this method were completely consistent with real-time PCR, and the results of the sensitivity test showed that this method could detect suspensions with a concentration as low as 102 CFU/g. In the detection of spiked samples, the detection results of the two genes in the positive samples by this method were also consistent with the abovementioned results. This finding showed that the new method has good practical ability. In addition, we designed RPA primers and crRNAs for both tdh and trh genes of VP, which not only made the test results more reliable, but also proved useful for virulence analysis of the strains.
In summary, CRISPR/Cas12a-VD has advantages for VP detection, including high sensitivity and specificity, rapid operation, and the results can be read by the unaided eye without complex equipment, making CRISPR/Cas12a-VD suitable for fast detection in the field. This novel method could contribute to the prevention and control of VP.
-
The authors declare no competing financial interests.
-
LI Jun Wen and YANG Dong conceived and designed the study. JIANG Han Ji performed experiments. TAN Rong and JIN Min performed statistical analysis. YIN Jing and LI Hai Bei collected samples. JIANG Han Ji and ZHOU Shu Qing wrote the manuscript. GAO Zhi Xian, SHI Dan Yang, and CHEN Tian Jiao reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
HTML
Bacterial Strains and Culture Media
Materials
Target Sequence Screening
Nucleic Acid Preparation
RPA Assay
Cas12a-mediated Cleavage Assay
The CRISPR/Cas12a-VD Method
Detection of VP in Pure Culture by CRISPR/Cas12a-VD
Analysis of Spiked Shrimp Samples by CRISPR/Cas12a-VD
Real-time PCR Detection
Sensitivity of the Cas12a-mediated Cleavage Assay
Preliminary Screening of RPA Primers
Sensitivity and Specificity of CRISPR/Cas12a-VD
Analysis of CRISPR/Cas12a-VD in Pure Culture
Analysis of CRISPR/Cas12a-VD in Spiked Shrimp Samples
 21424Supplementary Materials.pdf
21424Supplementary Materials.pdf
|

|

 Quick Links
Quick Links
 DownLoad:
DownLoad: